Introduction
Cellular senescence, an irreversible state of cell
growth arrest, is an important physiological mechanism in
protecting cells against malignant transformation (1–3). In
primary mammalian cells, senescence can be triggered by diverse
cellular stressors (4). Although
senescent cells are non-dividing, they are still metabolically
active with several distinctive morphological changes, such as
enlarged and flatted cell shape and can be visualized with a widely
accepted and used marker, senescence-associated beta-galactosidase
(SA-β-gal) staining (5,6). In murine liver carcinoma, p53
restoration induces senescence and leads to tumor regression
(7). Therefore, tailored
pro-senescence therapies in tumor cells is an attractive approach
to cancer treatment.
Hepatocellular carcinoma (HCC) is the fifth most
common neoplasm, accounting for >90% primary liver cancer and
also the second leading cause of cancer death worldwide (8). More than 70% patients with HCC at the
time of diagnosis are not suitable for surgical resection due to
the lack of early symptoms. Conventional cytotoxic chemotherapy or
(and) radiotherapy dose not significantly prolong survival or
improve the quality of life for patients with advanced HCC. The
development of efficacious therapeutic strategies for the treatment
or chemoprevention of HCC would have a dramatic effect on those who
have been diagnosed with or are at risk for HCC.
Metformin (1,1-dimethylbiguanide hydrochloride), a
biguanide derivative, has emerged as potential anti-neoplastic
agent in the prevention and treatment of HCC (9). Moreover, several studies have
demonstrated that metformin has an inhibitory effect on hepatoma
cell growth both in vitro and in vivo (10–12).
Due to its potential as a promising candidate for cancer
therapeutics, metformin has garnered much interest and several
mechanisms of action have been identified such as induction of cell
cycle arrest and apoptosis as well as inhibition of epithelial to
mesenchymal transition in certain cancers (13–15).
However, to date little is known about the possible effect of
metformin on cellular senescence and the mechanism of action of
metformin.
In the present study, we investigated the effect of
metformin on hepatoma cell proliferation and mechanisms responsible
for its action. We discovered that low concentration of metformin
retarded proliferation of hepatoma cells and induced phenotypic
changes typically associated with cellular senescence. The
induction of senescence-like growth arrest is related to activation
of AMPK pathway. Importantly, these findings may help to explain
the differential impact of metformin and provide a potential
strategy of promoting senescence for adjuvant treatment of HCC.
Materials and methods
Materials and cells
Metformin was purchased from Sigma-Aldrich (St.
Louis, MO, USA). Compound C, an AMPK inhibitor, was supplied by
Calbiochem (La Jolla, CA, USA). Antibodies against AMPK, p-AMPK
(Thr172), p-ACC (Ser79), p53, acetyl-p53 (Lys382), p-p53 (Ser15),
p21waf1/cip1, p16INK4A, p-RB (Ser795), PARP and SIRT1
were provided by Cell Signaling Technology (Danvers, MA, USA). The
antibody specially recognizing Dec1 was obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). p53 and control siRNA
were purchased from Cell Signaling Technology. Human liver cancer
cell lines HepG2 and Bel-7402 (both bearing wild-type p53), a gift
from Professor X. Guo (Sun Yat-Sen University, Guangzhou, China)
were maintained in RPMI-1640 supplied with 10% fetal bovine serum
and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA).
Cultures were incubated at 37°C and 5% CO2.
Logarithmically growing cells were treated with the indicated
concentrations of metformin or compound C for 48 or 24 h and then
replaced with regular culture medium.
MTS assay
Hepatoma cells were seeded on 96-well plates at
5×103 cells per well. After different treatments, 20
μl of MTS reagent was added to each well using a multi-well
pipettor and incubated for 3 h. Cell proliferation was assessed by
measuring the absorbance at 490 nm using a Bio-Tek Synergy2
microplate reader. Each experiment was repeated 3 times.
BrdU incorporation assay
In cells treated with metformin, compound C or
siRNA, BrdU incorporation was assayed using a commercial kit (Cell
Signaling Technology). Briefly, cells were grown on 22-cm
coverslips and incubated with anti-BrdU primary antibody at 4°C
overnight. The cells were then washed with PBS and processed for
immunofluorescence. Cells were mounted with Prolong Gold Antifade
Reagent (Invitrogen, Eugene, OR, USA) and visualized under a
fluorescence microscope (Leica CTR6000) at ×100 or ×200
magnification.
Annexin V/propidium iodide staining
assays
To detect whether metformin induces cell apoptosis,
we stained the treated cells with an Annexin V kit (Sigma-Aldrich).
Briefly, 1×106 hepatoma cells were treated with
different concentration of metformin for 48 h, stained with Annexin
V/propidium iodide (PI) and then subjected to FACSCalibur cytometry
(BD Bioscience, Mountain View, CA, USA).
Cell cycle analysis
Cell cycle distribution was performed as previously
described (12). The cells were
harvested after treatment with metformin, then fixed with 70% cold
ethanol and stained with 50 mg/ml PI followed by RNase A treatment
for 30 min at room temperature. DNA content was analyzed with a
FACSCalibur cytometer (BD Bioscience). The population of cells in
each phase was determined by using ModFit LT software (BD
Bioscience).
Senescence-associated β-galactosidase
(SA-β-gal) staining
SA-β-gal staining was performed as previously
described (16). In brief,
hepatoma cells were seeded in 6-well plates and fixed with 4%
formaldehyde for 5 min at room temperature. The cells were then
washed with PBS and incubated with fresh SA-β-gal staining solution
containing 1.0 mg/ml X-galactosidase (Stratagene, La Jolla, CA,
USA) at 37°C for 16–18 h to visualize SA-β-gal staining.
Western blotting
Cell extracts were prepared by lysing the cells in
RIPA buffer and protein concentration was quantified through BCA
assay. Protein sample (50 μg) was subjected to 10%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred
onto polyvinylidene difluoride membrane (Millipore Corp.,
Billerica, MA, USA). The membranes were blocked with 5% non-fat
milk in PBS containing 0.05% Tween-20 for 1 h at room temperature
and then incubated with specific primary antibodies. Detection of
specific proteins was performed by enhanced chemiluminescence
reagent (Thermo Scientific, Rockford, IL, USA).
Transfection of siRNA
Transfection of hepatoma cells with siRNA was
carried out using Lipofectamine 2000, according to the
manufacturer’s instructions (Invitrogen). Briefly, aliquots of
1.5×104 cells were seeded on 6-well plates and grown to
50–70% confluence before transfection. Cells were then transfected
with 60 pmol p53 siRNA for 6 h plus 1 μl Lipofectamine in
Opti-MEM® I medium. Subsequently, the transfection
medium was replaced by fresh regular culture medium. After 48 h of
incubation, protein levels were detected by western blot
analysis.
Statistical analysis
The data are presented as the mean ± standard
deviation and statistical comparisons between groups were done
using one-way analysis of variance followed by Student’s t-test.
P<0.05 was considered statistically significant.
Results
Effect of metformin on hepatoma cell
proliferation and apoptosis
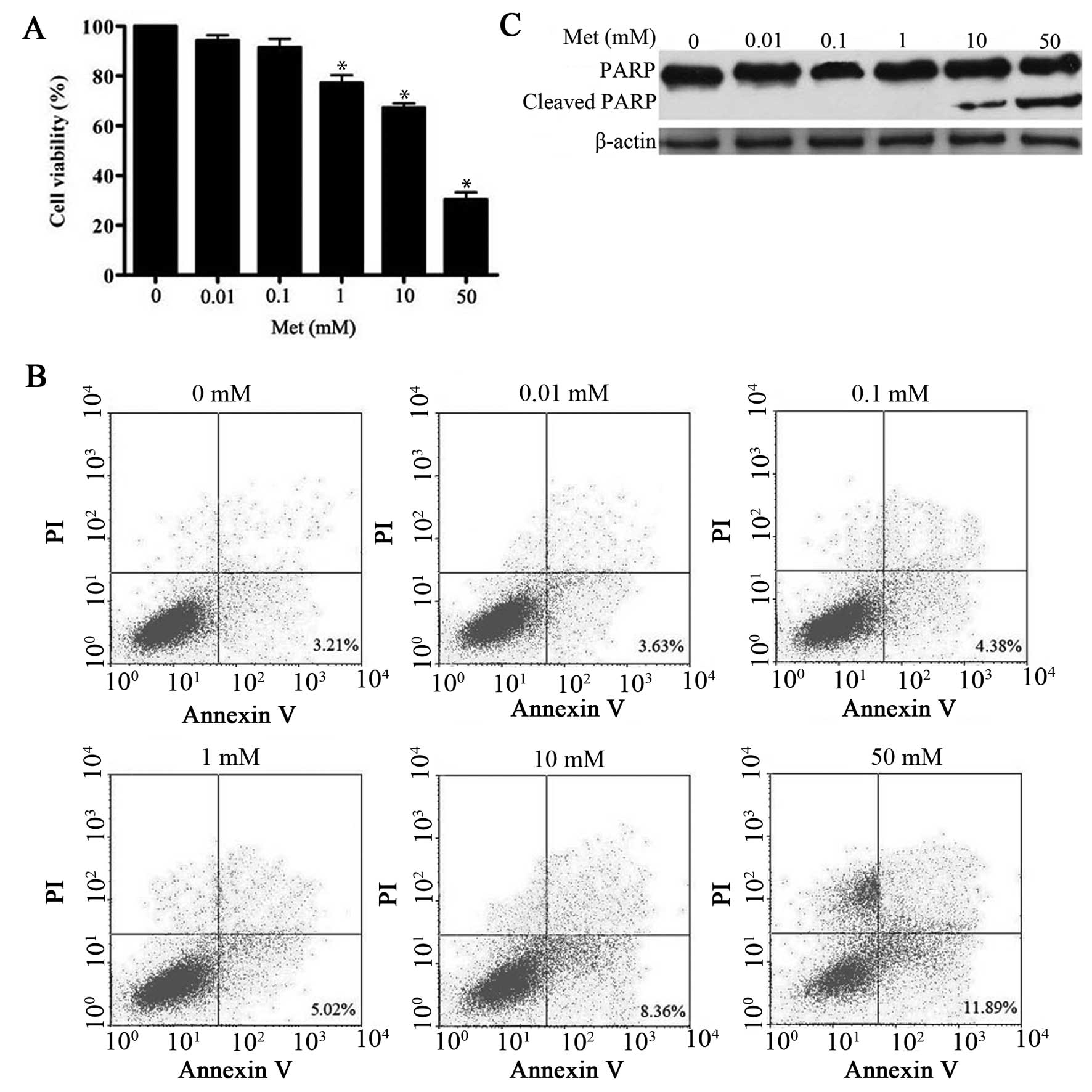
In this study, metformin caused inhibition of cell
proliferation in a dose-dependent manner (Fig. 1A), which is consistent with
previous observations (11,12).
In addition, several studies have shown that metformin has
pro-apoptotic effects on hepatoma cells at concentration of ≥5 mM
(11,12). In the present study, 10–50 mM
metformin caused a significant induction of apoptosis as evidenced
by Annexin V staining and accumulation of cleavage PARP protein.
However, low dose of metformin (0.01–1 mM) did not (Fig. 1B and C). Interestingly, 1 mM
metformin was sufficient to impair HepG2 cell proliferation without
apparent induction of apoptosis. Similar results were obtained in
another hepatoma cell line Bel-7402 (data not shown) following
metformin treatment, indicating that the effect is not cell line
specific. On the basis of these findings, we concluded that
metformin exerts a multiphase effect on hepatoma cells, with low
dose leading to a sustained decrease in proliferation, and
triggering of a higher rate of apoptotic cell death.
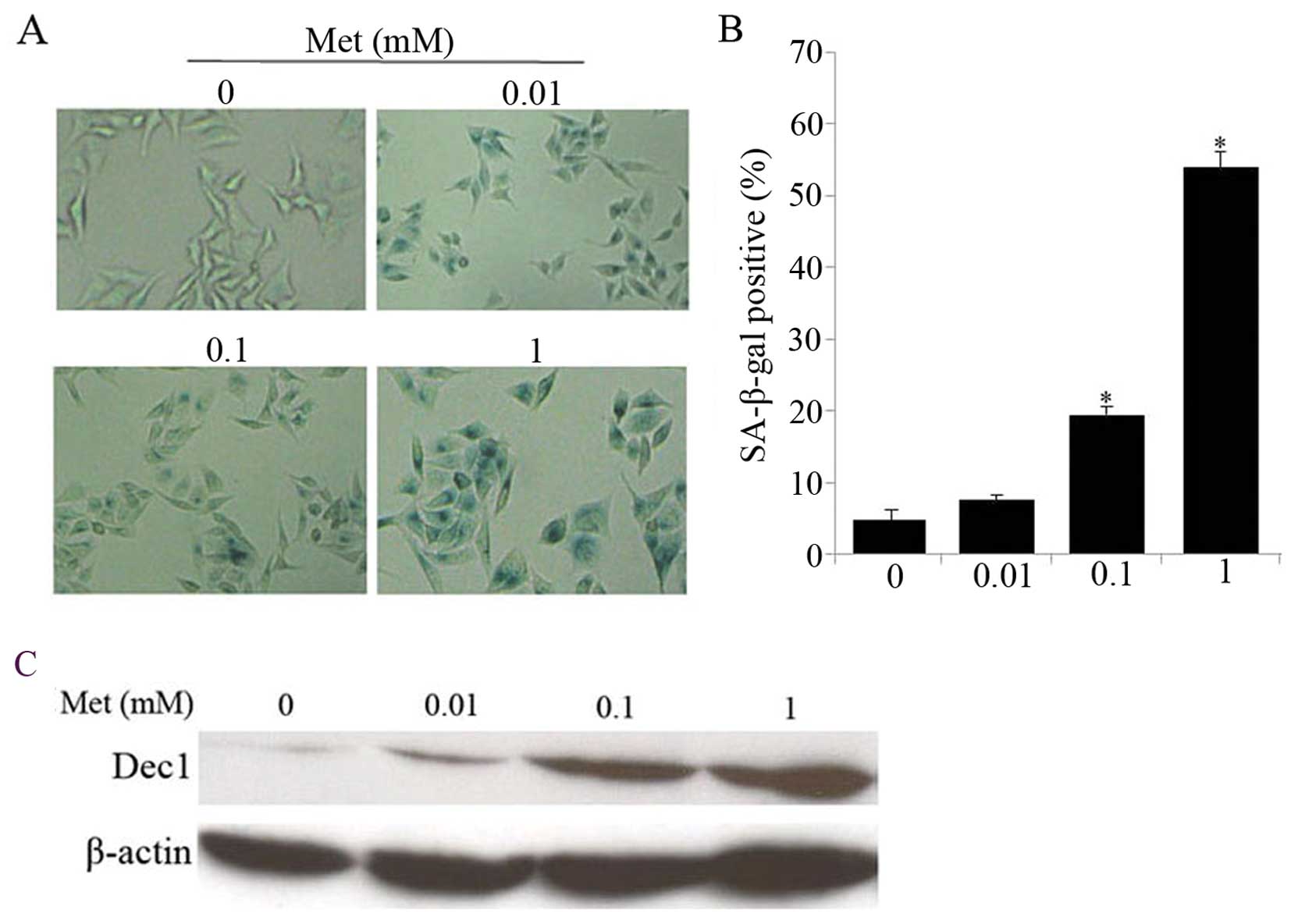
Low doses of metformin induces cellular
senescence in hepatoma cells
HepG2 cells exposed to low concentration of
metformin were characterized by enlarged and flatted cell phenotype
in our initial observation (data not shown), which is likely
associated with cellular senescence (17), therefore we sought to determine
whether low doses of metformin might induce senescence in hepatoma
cells. Indeed, low concentration of metformin induced HepG2 cell
senescence in a dose-dependent manner, as evident by increased
SA-β-gal activity, a universal marker of cellular senescence
(Fig. 2A and B). Moreover, protein
expression level of Dec1, one of the established markers for
senescence (18) was enhanced in
response to metformin compared with controls (Fig. 2C), which is in accordance with
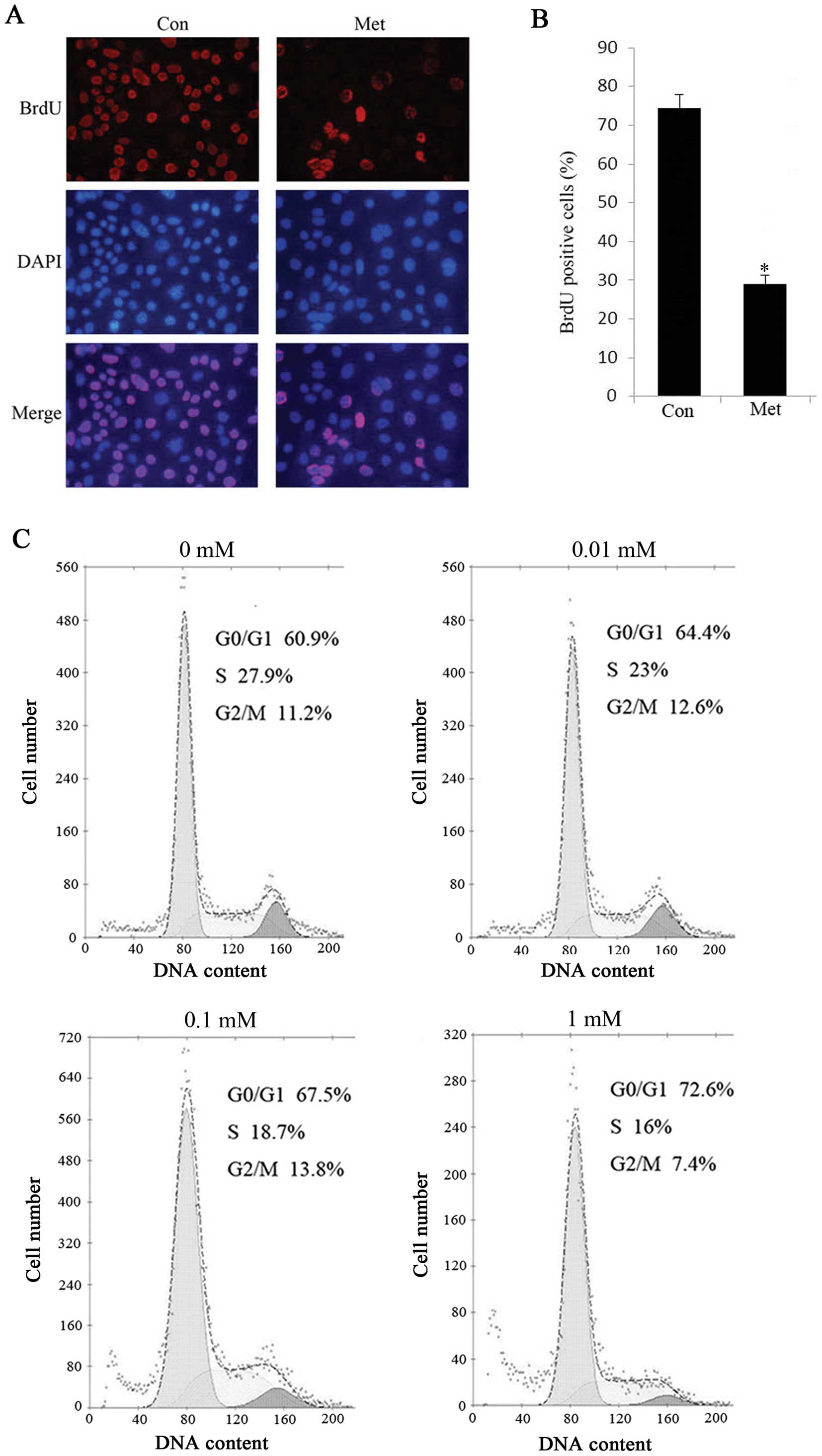
SA-β-gal staining results. Additionally, HepG2 cells treated with 1
mM metformin exhibited a marked decrease in BrdU incorporation, a
marker of cell proliferation (Fig. 3A
and B). Of note, when we treated HepG2 cells with metformin for
48 h and either metformin was withdrawn or continued the treatment
for another 72 h, there was no significant difference in cell
viability between the persistent metformin-treated cells and the
metformin-removed cells (data not shown), suggesting that
metformin-induced proliferation arrest is irreversible even when
the drug is withdrawn. To further clarify the effect of metformin
on cellular senescence, we investigated the cell cycle progression
in the absence or presence of metformin by flow cytometric
analysis. The results indicated that the percentage of
G0/G1 phase cells was significantly increased
in metformin treated cells and correspondingly, the cell population
of G2/M was declined compared with the controls
(Fig. 3C). Similarly, we observed
senescence in Bel-7402 cells with metformin treatment (data not
shown), indicating that low doses of metformin triggered senescence
in hepatoma cells.
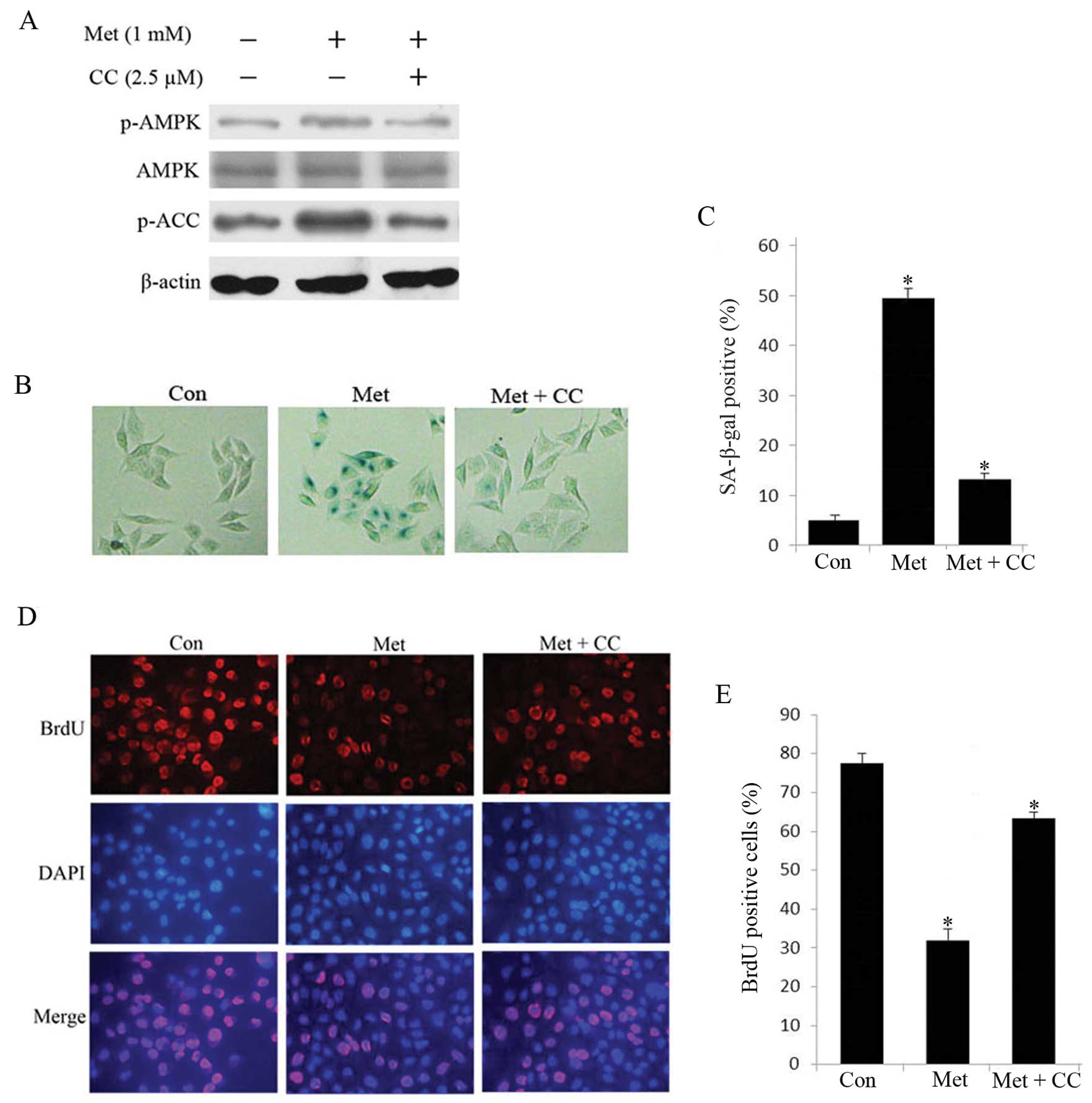
Metformin induces hepatoma cell
senescence by activating AMPK signaling
Next, we sought to determine the mechanism by which
metformin induced senescence in hepatoma cells. Since 1 mM
metformin caused significant induction of cellular senescence
without apparent induction of apoptosis, we treated HepG2 cells
with 1 mM metformin in the subsequent experiments. We detected the
expression of phosphorylation of AMPK and its downstream gene ACC
through western blot analysis. In metformin-treated senescent HepG2
cells, robust phosphorylation of AMPK and ACC was observed compared
with untreated cells (Fig. 4A),
indicating that activation of AMPK signaling cascade are relevant
to the senescence in HepG2 cells. To further confirm the
relationship between HepG2 cell senescence and AMPK activation, we
examined the effects of compound C, a specific AMPK inhibitor, on
senescence in HepG2 cells. As shown in Fig. 4B–E, compound C reduced
metformin-stimulated SA-β-gal-positive cells and restored
proliferation. In addition, the protein level of p-AMPK and p-ACC
was attenuated compared to untreated cells (Fig. 4A). These results suggest that AMPK
pathway plays a crucial role in metformin-induced senescence in
hepatoma cells.
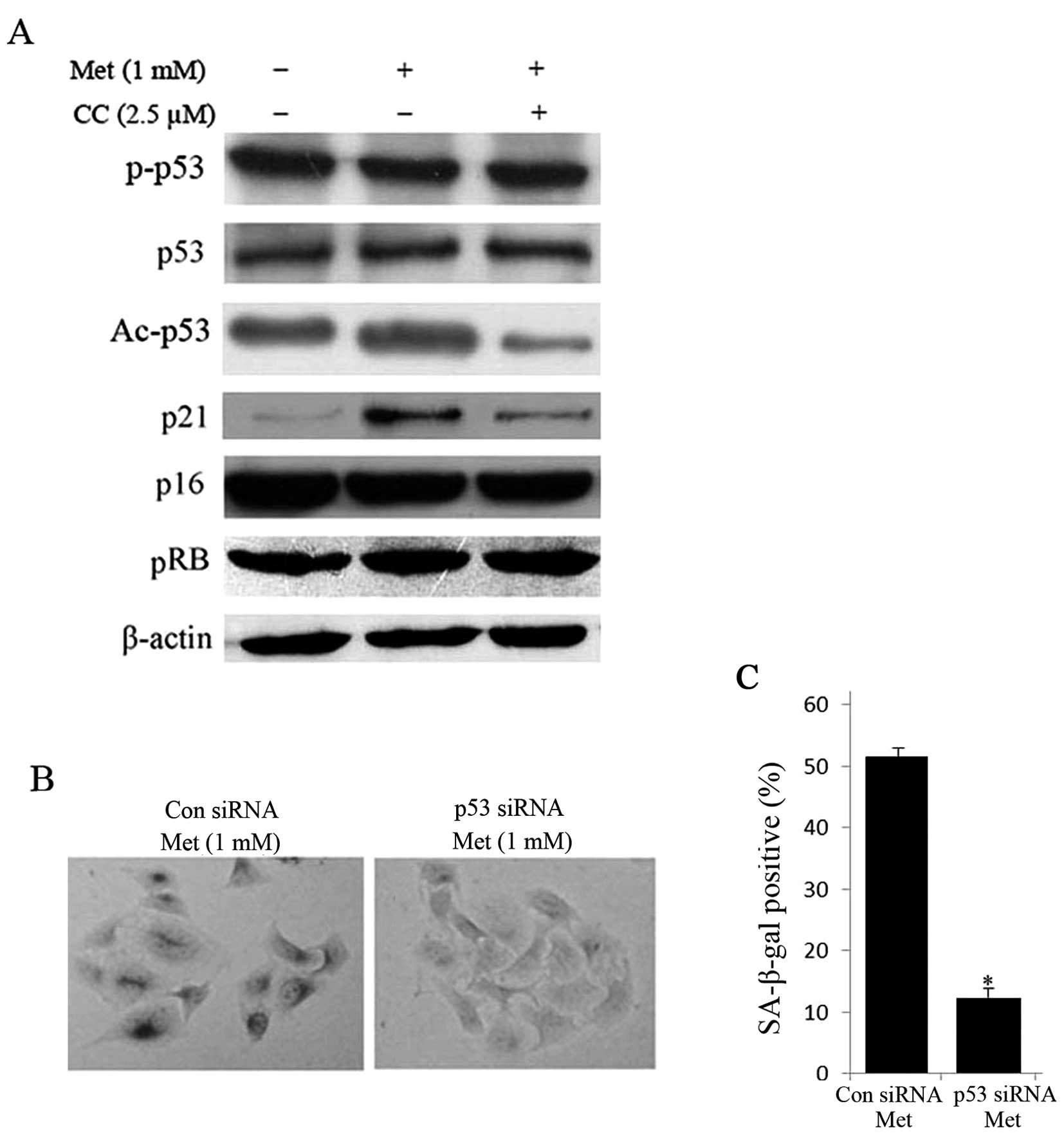
It is well known that senescence is mainly
manipulated through canonical p53/p21 and p16/pRB signaling pathway
(19). Thus, we sought to
determine whether activation of AMPK pathway depends on these two
pathways. Interestingly, the acetylation of p53 (Ac-p53) and p21
were increased in metformin-treated HepG2 cells, while the protein
expression of phosphorylation of p53 at Serine 15, p53, p16 and pRB
was unaltered (Fig. 5A). On the
other hand, treatment with compound C for 24 h obviously reduced
protein expression of Ac-p53 and p21 compared with untreated cells
(Fig. 5A). Furthermore,
siRNA-mediated knockdown of p53 diminished metformin-induced
SA-β-gal positive cells (Fig. 5B and
C). These data imply that the activation of AMPK pathway is
dependent on the p53.
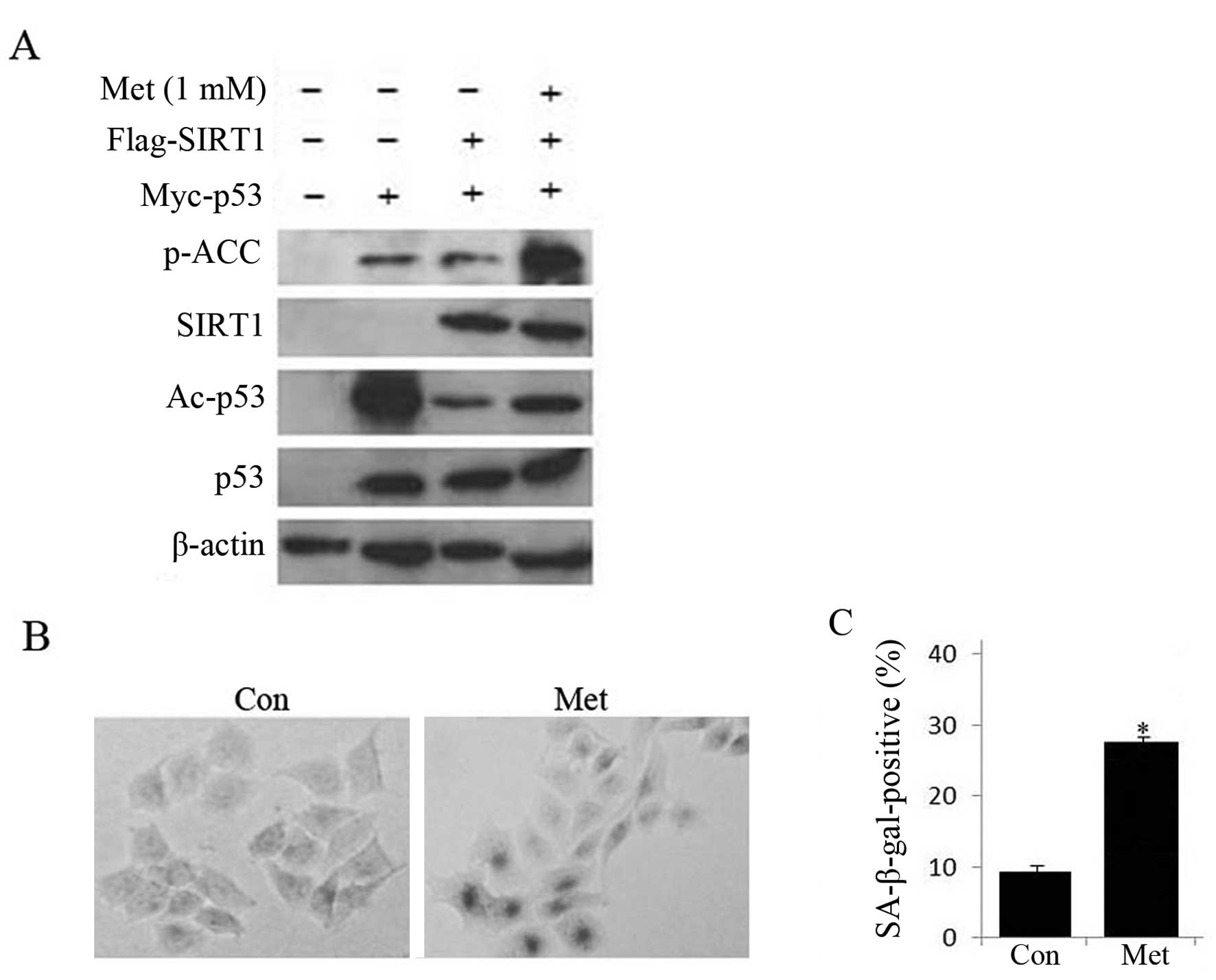
As AMPK activation promotes acetylation of p53, we
investigated whether SIRT1, a NAD-dependent protein deacetylase
(20), was involved in the
regulation of p53 transcriptional activity. Toward this goal, HepG2
cells were co-transfected with Myc-p53 and Flag-SIRT1 plasmid and
then treated with metformin. As shown in Fig. 6, co-expression of SIRT1 and p53
significantly suppressed the level of acetylated p53, but 1 mM
metformin treatment partially rescued the effect of SIRT1 on p53
acetylation and increased SA-β-gal staining, suggesting that
activating AMPK by metformin treatment might inhibit the SIRT1
deacetylase activity on p53, resulting in promotion of HepG2 cell
senescence.
Discussion
The role of senescence in suppressing tumor
progression and as a determinant of the outcome of conventional
anticancer therapies have indicated that pro-senescence therapies
could be an alternative strategy for treating cancer (21,22).
In contrast to apoptosis, driving cells to undergo senescence have
fewer side effects related to cytotoxicity (23) and senescent tumor cells are
efficiently eliminated by immune cells, resulting in efficient
tumor progression (7), which might
provide a more realistic approach for the chronic management of
some cancers (2). Epidemiological
and laboratory studies have shown that metformin has unanticipated
cancer prevention and treatment activity in patients with HCC
(24–26). However, the role of low doses of
metformin action in HCC remains unclear. Hence, the aim of this
study was to elucidate the effect of low concentration of metformin
on hepatoma cell proliferation and a more detailed understanding of
the mechanisms as well as to ascertain the feasibility of metformin
being used as a promising adjuvant agent for treatment or
chemoprevention in HCC.
Many studies suggest a role of AMPK signaling
pathway in the senescence process. For instance, senescence is
accompanied by a marked elevation of AMPK activity in senescent
fibroblasts (27). Activation of
AMPK contributes to premature aging phenotype of
Zmpste24−/− mice (28).
In addition, AMPK inhibition resulted in significant
anti-senescence effect to vascular smooth muscle cells (29). However, the role of AMPK in cancer
cell senescence was largely unknown. Our present study revealed
that AMPK activation is involved in promoting senescence in
hepatoma cells exposed to low doses of metformin. Inhibition of
AMPK activity by compound C led to profoundly decreased senescence
and rescued proliferation of hepatoma cells. These results
indicated that AMPK plays an important role in regulating
senescence in human hepatoma cells.
Recently, it has been shown that activation of AMPK
by AICAR (5-amino-1-β-Dffff-ribofuranosyl-imidazole-4-carboxamide)
leads to cell cycle arrest in a human HCC cell line via activating
p53-p21 pathway (30).
Additionally, Jones et al (31) found AMPK activation promotes p53
activity through phosphorylation at Serine 15 and this
phosphorylation event is essential for mediating the effects of
AMPK on p53-dependent mouse embryonic fibroblast cell cycle arrest.
However, persistent activation of AMPK results in accelerated
p53-dependent cellular senescence. By contrast, in this study we
demonstrated although the levels of phosphorylation of p53 at
Serine 15 were not changed in metformin-treated senescent hepatoma
cells, the levels of p53 acetylation at Lys382 were significantly
increased, suggesting that AMPK activation is capable of inducing
cellular senescence by stabilizing p53 via promoting p53
acetylation.
Previously, Luo and colleagues (32) found that SIRT1 can directly
deacetylate p53 both in vitro and in vivo and
promotes cell survival under stress. Furthermore, another
independent study (33) reported
that p53 is among the first non-histone substrates identified to be
functionally involved in the anti-senescence activity of SIRT1.
SIRT1 antagonizes p53-induced cellular senescence in primary mouse
embryonic fibroblasts by interacting with and acetylating p53.
Consistently, we present evidence that co-expression of SIRT1 and
p53 remarkably impaired p53 acetylation, while low doses of
metformin in part suppressed SIRT1-mediated deacetylation and
enchanced the SA-β-gal staining, implying that SIRT1 is a negative
regulator of p53 and is associated with the metformin-mediated
senescence in hepatoma cells. As is known, AMPK and SIRT1 are the
two key energy sensor systems regulating cell survival,
proliferation and senescence (34). AMPK and SIRT1 are vital links in an
orchestrated network regulating cellular senescence. For example,
SIRT1 was able to prevent endothelial senescence triggered by
LKB1-dependent AMPK activation, implying that anti-senescence
effect of SIRT1 is achieved at least in part by regulating the
LKB1-AMPK pathway (35). However,
whether AMPK counter-regulates SIRT1 in mammalian cellular
senescence is still unknown. Intriguingly, in our preliminary
study, interaction between AMPK and SIRT1 was clearly demonstrated
using co-immunoprecipitation in cell-free systems (data not shown).
Further studies are required to determine whether a possible
regulation of SIRT1 by AMPK is involved in metformin-stimulated
senescence in hepatoma cells.
In conclusion, the data provide evidence, for the
first time, that exposure of hepatoma cells to low doses of
metformin results in the induction of senescence instead of
apoptosis and this correlates with activation of the
senescence-promoting AMPK pathway in a p53-dependent manner. These
findings imply that metformin-induced senescence might be a viable
and relatively safe option for developing HCC adjuvant
therapeutics.
Acknowledgements
We would like to thank Dr Kai Luo, Min
Deng, Min Hou and Min Liang for technical assistance and
suggestions. This study was supported by grant A20110231 from Star
of Science and Technology of Guangzhou and 20121A011163 from Health
Bureau of Guangzhou.
References
|
1.
|
Collado M, Blasco MA and Serrano M:
Cellular senescence in cancer and aging. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Collado M and Serrano M: Senescence in
tumors: evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kang TW, Yevsa T, Woller N, et al:
Senescence surveillance of pre-malignant hepatocytes limits live
cancer development. Nature. 479:547–551. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Dimri GP, Lee X, Basile G, et al: A
biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Debacq-Chainiaux F, Erusalimsky JD,
Campisi J and Toussaint O: Protocols to detect
senescence-associated beta-galactosidase (SA-βgal) activity, a
biomarker of senescent cells in culture and in vivo. Nat Protoc.
4:1798–1806. 2009.PubMed/NCBI
|
|
7.
|
Xue W, Zender L, Miething C, et al:
Senescence and tumour clearance is triggered by p53 restoration in
murine liver carcinomas. Nature. 445:656–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
9.
|
Pollak M and Gonzalez-Angulo AM: Metformin
and hepatic carcinogenesis. Cancer Prev Res. 5:500–502. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Qu Z, Zhang Y, Liao M, Chen Y, Zhao J and
Pan Y: In vitro and in vivo antitumoral action of metformin on
hepatocellular carcinoma. Hepatol Res. 42:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chen HP, Shieh JJ, Chang CC, et al:
Metformin decreases hepatocellular carcinoma risk in a
dose-dependent manner: population-based and in vitro studies. Gut.
62:606–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Liu J, Hou M, Yuan T, et al: Enhanced
cytotoxic effect of low doses of metformin combined with ionizing
radiation on hepatoma cells via ATP deprivation and inhibition of
DNA repair. Oncol Rep. 28:1406–1412. 2012.PubMed/NCBI
|
|
13.
|
Zhou G, Myers R, Li Y, Chen Y, et al: Role
of AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ben Sahra I, Regazzetti C, Robert G, et
al: Metformin, independent of AMPK, induces mTOR inhibition and
cell-cycle arrest through REDD1. Cancer Res. 71:4366–4372.
2011.PubMed/NCBI
|
|
15.
|
Del Barco S, Vazquez-Martin A, Cufí S, et
al: Metformin: multi-faceted protection against cancer. Oncotarget.
2:896–917. 2011.PubMed/NCBI
|
|
16.
|
Wang W, Yang X, López de Silanes I,
Carling D and Gorospe M: Increased AMP:ATP ratio and AMP-activated
protein kinase activity during cellular senescence linked to
reduced HuR function. J Biol Chem. 278:27016–2723. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Cairney CJ, Bilsland AE, Evans T, et al:
Cancer cell senescence: a new frontier in drug development. Drug
Discov Today. 17:269–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Qian Y, Zhang J, Yan B and Chen X: DEC1, a
basic helix-loop-helix transcription factor and a novel target gene
of the p53 family, mediates p53-dependent premature senescence. J
Biol Chem. 283:2896–2905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Mallette FA, Goumard S, Gaumont-Leclerc
MF, Moiseeva O and Ferbeyre G: Human fibroblasts require the Rb
family of tumor suppressors, but not p53, for PML-induced
senescence. Oncogene. 23:91–99. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Guarente L: Sir2 links chromatin
silencing, metabolism and aging. Genes Dev. 14:1021–1026.
2000.PubMed/NCBI
|
|
21.
|
Nardella C, Clohessy JG, Alimonti A and
Pandolfi PP: Pro-senescence therapy for cancer treatment. Nat Rev
Cancer. 11:503–511. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Acosta JC and Gil J: Senescence: a new
weapon for cancer therapy. Trends Cell Biol. 22:211–219. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Aljada A and Mousa SA: Metformin and
neoplasia: implications and indications. Pharmacol Ther.
133:108–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lee MS, Hsu CC, Wahlqvist ML, Tsai HN,
Chang YH and Huang YC: Type 2 diabetes increases and metformin
reduces total, colorectal, liver and pancreatic cancer incidence in
taiwanese a representative population prospective cohort study of
800,000 individuals. BMC Cancer. 11:202011. View Article : Google Scholar
|
|
26.
|
Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q
and Kip KE: Metformin for liver cancer prevention in patients with
type 2 diabetes: a systematic review and meta-analysis. J Clin
Endocrinol Metab. 97:2347–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Zwerschke W, Mazurek S, Stöckl P, Hütter
E, Eigenbrodt E and Jansen-Dürr P: Metabolic analysis of senescent
human fibroblasts reveals a role for AMP in cellular senescence.
Biochem J. 376:403–411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Mariño G, Ugalde AP, Salvador-Montoliu N,
et al: Premature aging in mice activates a systemic metabolic
response involving autophagy induction. Hum Mol Genet.
17:2196–2211. 2008.PubMed/NCBI
|
|
29.
|
Sung JY, Woo CH, Kang YJ, Lee KY and Choi
HC: AMPK induces vascular smooth muscle cell senescence via LKB1
dependent pathway. Biochem Biophys Res Commun. 413:143–148. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Imamura K, Ogura T, Kishimoto A, Kaminishi
M and Esumi H: Cell cycle regulation via p53 phosphorylation by a
5′-AMP activated protein kinase activator,
5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human
hepatocellular carcinoma cell line. Biochem Biophys Res Commun.
287:562–567. 2001.
|
|
31.
|
Jones RG, Plas DR, Kubek S, et al:
AMP-activated protein kinase induces a p53-dependent metabolic
checkpoint. Mol Cell. 18:283–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Luo J, Nikolaev AY, Imai S, et al:
Negative control of p53 by Sir2alpha promotes cell survival under
stress. Cell. 107:137–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Langley E, Pearson M, Faretta M, et al:
Human SIR2 deacetylates p53 and antagonizes PML/p53-induced
cellular senescence. EMBO J. 21:2383–2396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Wang Y, Liang Y and Vanhoutte PM: SIRT1
and AMPK in regulating mammalian senescence: a critical review and
a working model. FEBS Lett. 585:986–994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Zu Y, Liu L, Lee MY, et al: SIRT1 promotes
proliferation and prevents senescence through targeting LKB1 in
primary porcine aortic endothelial cells. Circ Res. 106:1384–1393.
2010. View Article : Google Scholar : PubMed/NCBI
|