Introduction
Acute myeloid leukemia (AML) is a clonal
hematopoietic disorder characterized by multiple genetic
abnormalities in normal hematopoietic stem cells, which results in
accumulation of immature cells - the blasts (1). AML is a highly heterogeneous disease
and is categorized into eight FAB (French-American-British)
subtypes, based on morphological, cytochemical, and
immunophenotypic characteristics of leukemic blasts (2). In addition, genetic and molecular
alterations can stratify specific groups and determine their
prognosis, since some entities exhibit quite different clinical
outcomes (3). AML treatment is
mainly based on anthracyclines and cytarabine (4) and has changed little in the past
three decades, except for the acute promyelocytic leukemia subtype,
in which combination therapy with both anthracycline and
transretinoic acid has resulted in a high complete remission rate
(5). However, for the other AML
subtypes, complete remission rates seldom exceed 70% in younger
patients and 50% in older patients (4). For adults, 5-year overall survival is
between 30 and 50%, with even more disappointing results in terms
of disease-free survival, as most patients relapse within 3–5 years
from diagnosis (6,7). Taking into account that AML patients
have a poor clinical outcome, which is mainly associated with
chemoresistance or relapse after previous response (8), and that there has not been any
significant advance in treatment it is crucial to develop
innovative therapeutic approaches to increase response rates and
survival for AML patients.
Pterocarpanquinones are hybrid molecules originated
from the molecular hybridization of pterocarpans and naphtoquinones
(9), which have been shown to have
antitumoral activities (9,10). Among them, LQB-118 emerged as one
of the most promising compound, because it was able to induce high
apoptotic indexes in cells from chronic myeloid leukemia (CML)
patients with the multidrug resistance (MDR) phenotype (11), while displaying little toxicity
toward peripheral blood mononuclear cells (PBMC) derived from
healthy individuals (12). This
effect involves not only changes in mitochondrial membrane
potential and regulation of the endoplasmic reticulum stress
pathway (13), but also inhibition
of both the Survivin and XIAP antiapoptotic proteins and the drug
efflux transporter P-glycoprotein (Pgp) (11), proteins known to impact prognosis
and confer resistance to chemotherapy in multiple leukemia models,
including myeloid leukemia (14–20).
In AML cells, we have described that the LQB-118 compound triggers
apoptosis and suppresses Survivin and XIAP expression in cells from
AML patients displaying variable levels of Pgp activity and
expression (21). Moreover, there
was no nuclear translocation of the NF-κB oncogenic transcription
factor, differently from the effect observed after treatment of the
AML Kasumi cell line with idarubicin, an anthracyclin commonly used
in AML treatment (21).
Nevertheless, the mechanisms of action of the LQB-118 compound and
pathways involved in its antitumoral effect in AML cells are not
fully elucidated.
Forkhead box (Fox) transcription factors are crucial
regulators of a wide range of key biological processes and some are
also deregulated in cancer (22).
FoxO3a is a member of the Fox class ‘O’ subfamily frequently found
in AML leukemic blasts (23). It
is known to orchestrate signaling pathways involved in apoptosis,
cell cycle arrest, metabolism, DNA repair and response to stress,
through modulating transcription of direct targets, such as Bim,
p27kip1, glucose-6-phosphatase,
GADD45a, and manganese superoxide dismutase (MnSod),
respectively (24,25). Conversely, the Fox protein M1
(FoxM1) acts like a typical oncogene and transcriptionally
activates genes associated with cell cycle progression (i.e.,
cyclin B1, Aurora-B kinase, centromere protein A and B, cdc25, and
Survivin) (26,27). In AML, aberrant FoxM1
expression was found to contribute to proliferation of leukemic
blasts (28). The modulation of
FoxO3a and FoxM1 transcription factors has been shown to mediate
the cytotoxic and cytostatic effects of multiple chemotherapeutic
agents, playing a pivotal role in determining drug sensitivity and
resistance (29).
In this study, we investigated the ability of the
LQB-118 compound to sensitize AML cell lines and target FoxO3a and
FoxM1 transcription factors. Our data show that LQB-118 can induce
high apoptotic indexes in AML cells of distinct molecular subtypes.
LQB-118 promotes FoxO3a nuclear translocation and Bim upregulation
in HL60 cells. However, FoxO3a is relocated to the cytoplasm and
Bim expression is suppressed in U937 cells, in which
chemosensitivity correlates with FoxM1 and Survivin inhibition,
suggesting that the LQB-118 compound is able to differentially
target Fox transcription factors to sensitize AML cells of distinct
molecular subtypes. Studying different AML molecular subtypes is
important for preclinical drug development, as they display
different outcomes and drug resistance patterns in clinical
scenario. In addition, LQB-118 was administered to Swiss mice
without detectable toxicity to normal bone marrow-derived cells,
which indicates that it has selectivity for tumor cells.
Materials and methods
Materials
The pterocarpanquinone LQB-118 was synthesized in
the Laboratory of Bioorganic Chemistry, at the Federal University
of Rio de Janeiro (UFRJ) as described (10). The compound was dissolved in
dimethyl sulfoxide (DMSO), and serial dilutions in culture medium
were performed before use. DMSO in the concentration paired to the
highest concentration of the LQB-118 compound was used as a vector
control in all experiments. The LQB-118 compound was used at
concentrations of 1.5, 3, 6 and 9 μM, which were demonstrated to
cause no toxicity to PBMC isolated from healthy individuals
(12). All materials are described
in detail in each section.
Animals
Swiss mice were bred in the animal facilities at the
Leopoldo de Meis Biochemistry Institute from UFRJ (IBqM-UFRJ), and
housed under standard laboratory conditions (20–25°C, 12-h light
regimen) with free access to water and standard chow (ad
libitum). Two-month female mice were used in the experiments.
Procedures were approved by the Center of Health Sciences Ethics
Committee for Animal Use (CEUA-CCS, UFRJ) under the protocol
IBQM082.
Isolation of bone marrow cells
Mice were anaesthetized with ethyl ether (Reagen,
Rio de Janeiro, Brazil) and euthanized by cervical dislocation.
Bone marrow was aspirated from femoral bones with a syringe
containing RPMI-1640 medium (Sigma-Aldrich Corp. St. Louis, MO,
USA). Bone marrow cells were homogenized in cold RPMI-1640 medium,
centrifuged at 200 g for 10 min and then resuspended in 3 ml of
cold RPMI medium supplemented with 5% fetal bovine serum (FBS;
Gibco, Grand Island, NY, USA). The cell suspension was incubated
with 0.08% Trypan blue staining (Sigma-Aldrich) and counted in an
optical microscope.
LQB-118 in vivo administration
The in vivo concentration of LQB-118 used was
based upon its in vitro EC50 obtained from
previous work. Calculations were made considering a medium of 30 g
weight and 4 ml of bodily fluids, the final concentration of 3.8
mg/kg is representative of ~15X EC50. Mice were
intraperitoneally injected with 3.8 mg/kg LQB-118 diluted in
phosphate buffered saline (PBS; Sigma-Aldrich) and DMSO
(Sigma-Aldrich). Control group received only the vehicle
(diluent).
Ex vivo analysis of bone marrow
subpopulations
Cells from the bone marrow of femoral bones were
washed with PBS supplemented with 5% FBS (Gibco), resuspended in
300 μl of RPMI-1640 medium for the final concentration of
106 cells/ml. Subsequently, cells were analyzed by flow
cytometry in a FACSCalibur (Becton-Dickinson, San Jose, CA, USA).
Ten thousand cells were acquired based on forward and side scatter
parameters, representative of cell size and granulosity. Analysis
was performed on Summit v4.3 software (Dako Colorado, Inc., Fort
Collins, CO, USA).
Cell lines
The human AML-derived cell lines HL60 and U937 were
obtained originally from American Type Culture Collection (ATCC;
VA, USA) and cultured in RPMI-1640 medium (Gibco; BRL, UK)
supplemented with 10% heat-inactivated FBS and glutamine (2 mM) and
maintained in a humidified atmosphere at 37°C and 5%
CO2. HL60 cells are derived from promyelocytic leukemia
(M3 FAB subtype) (30,31) and U937 cells, from monocytic type
(M4/M5 subtypes) (32). The cell
lines were genotyped for confirmation of authenticity by the Sonda
Laboratory at UFRJ and monitored for mycoplasm contamination. Cells
were exposed to LQB-118 for 24 h at a density of
2×105/ml.
Assessment of DNA content
After LQB-118 treatment, 2×106 cells were
washed twice in PBS and incubated with 500 μl of propidium iodide
(PI) staining solution (PI 50 μg/ml diluted in citrate buffer 4 mM
and Triton X-100 0.3%) and 500 μl ribonuclease A (RNAse 100 μg/ml
diluted in citrate buffer 40 mM) for 15 min at room temperature.
DNA content was determined on a flow cytometer (FACSCalibur; BD
Biosciences, San Jose, CA, USA) and a total of ten thousand events
were acquired per sample. Analysis was performed using CellQuest
Pro software. This assay was performed to analyze the cell cycle
profile and assess DNA fragmentation, which was quantified by the
percentage of cells in sub-G0/G1 phase.
Apoptosis assay
For detection of apoptosis, the Annexin V assay
(Genzyme Diagnostics, Cambridge, MA, USA) was performed. Cells were
stained with FITC-labeled Annexin V as previously described
(11). The compound-induced
apoptotic index was analyzed using flow cytometry, and results were
calculated as a percentage of Annexin V -positive cells in the
presence of the drug subtracting the positivity in the absence of
the drug.
Detection of caspase-3 expression using
flow cytometry
After exposure to LQB-118 for 24 h, a total of
2×106 cells were washed with PBS and centrifuged. Then,
cells were submitted to a caspase-3 staining protocol as previously
described (20). Anti-active
caspase-3 antibody was purchased from BD Biosciences. For each
condition, a negative control without antibody was also analyzed.
The results were expressed as the percentage of cell with caspase-3
positivity.
Immunofluorescence
After LQB-118 treatment for 24 h, an
immunofluorescence assay using an anti-FoxO3a monoclonal antibody
was performed. Cytospins were prepared with HL60 and U937
105 cells treated or not with 1.5 μM of LQB118 and DMSO.
Cytospins were fixed with 4% paraformaldehyde for 20 min, and
permeabilized with 1% PBS (pH 8.0)-Triton X-100 for 10 min. Cells
were blocked with PBS (pH 8.0) containing 3% BSA for 1 h and then,
stained with a monoclonal antibody against FoxO3a (1:200 dilution,
Cell Signaling, Danvers, MA, USA) diluted in PBS (pH 8.0) 1% bovine
serum albumin (BSA) overnight. The antibody-antigen complexes were
detected by incubation for 1 h with secondary anti-rabbit Alexa
Fluor 488 or 594 antibodies (1:300 dilution, Life Technologies, NY,
USA) in PBS (pH 8.0) 1% BSA. Cells were washed and treated with
DAPI (1:1,000, 4′,6-diamidino-2-phenylindole) for 10 min for
nuclear staining. Coverslips were mounted with N-propylgallate and
the cells were subsequently analyzed with Nikon fluorescence
microscope using the Nikon software (Leica Microsystem).
Western blot analysis
For detection of Bim, FoxM1, Survivin, Beclin1 and
β-actin expression, pellets of 2×106 were harvested and
submitted to protein extraction and immunoblotting as previously
described (17). Primary
antibodies comprised Bim (1:1,000 dilution; Stressgen, Brussels,
Belgium), FoxM1 (1:500 dilution; Cell Signaling), Survivin (1:1,000
dilution; R&D Systems, Minneapolis, MN, USA), Beclin1 (1:1,000;
BD Biosciences), and β-actin (1:5,000 dilution; Sigma-Aldrich).
Secondary anti-mouse (1:500 dilution) and anti-rabbit (1:1,000
dilution) were purchased from GE Healthcare (Buckinghamshire, UK).
Antibody complexes were visualized using the ECL detection system
(GE Healthcare) and band intensities were quantified using the
VisionWorks software. Protein expression was normalized with
respect to β-actin.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 4.0 software (San Diego, CA, USA). Statistical significance
was calculated by non-parametric and unpaired Student’s t-test
since experiments compared different groups of animals. Data
presented normal distribution according to normality test.
Results
The LQB-118 compound induces apoptosis in
AML cell lines derived from different FAB subtypes
Recently, we demonstrated that the novel compound
LQB-118 is capable of sensitizing the Kasumi AML cell line, derived
from the M2 FAB subtype, to undergo apoptosis (21). Since AML is a highly heterogeneous
myeloproliferative disease (1,33),
the first step of this study was to investigate if LQB-118
cytotoxic effects extend to cell lines derived from other AML FAB
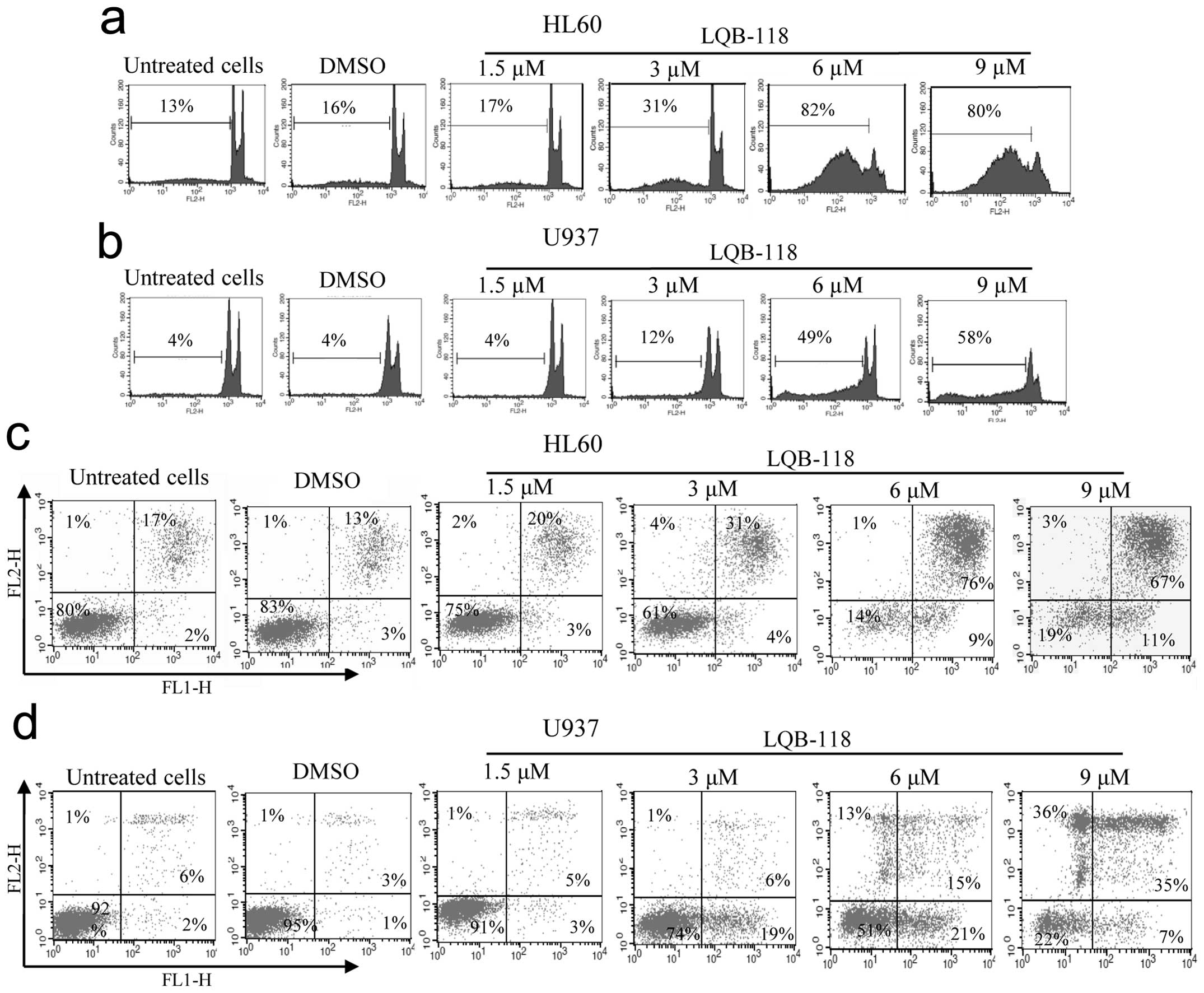
subtypes. As observed in Fig. 1a and
b, exposure to LQB-118 compound for 24 h resulted in high
percentages of DNA fragmentation, mainly at concentrations of 6 and
9 μM. We also found that both cell lines had an increase in Annexin
V staining after LQB-118 treatment in a dose-dependent manner
(Fig. 1c and d), suggesting that
the compound might exert its effects on AML cells by triggering
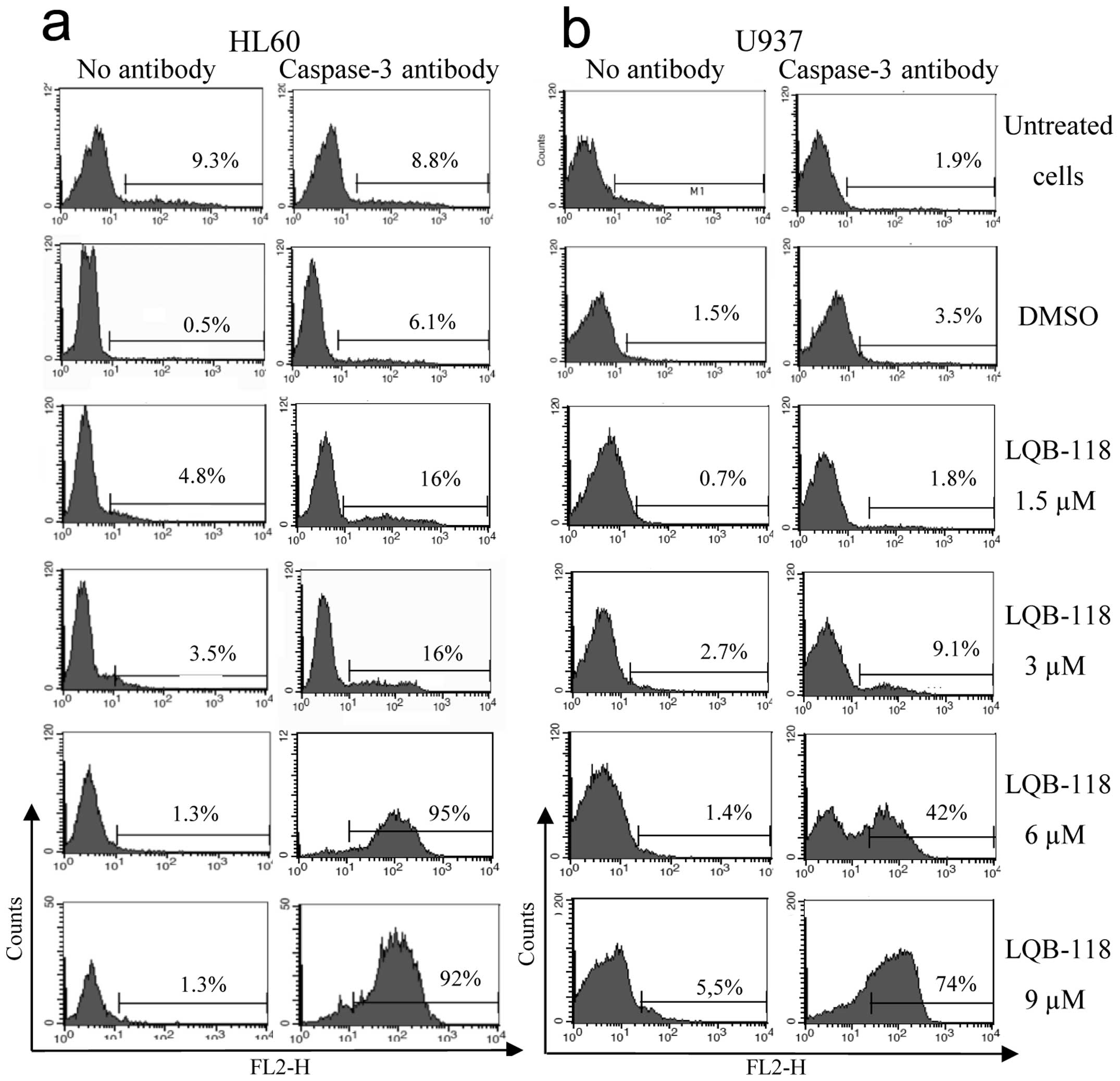
apoptosis. To confirm these data, we assessed caspase-3 levels and
observed that LQB-118 induced caspase-3 activation in both cell
lines (Fig. 2). It is important to
highlight that HL60 cells were slightly more sensitive than U937
cells in all cytotoxicity assays performed. We also questioned if
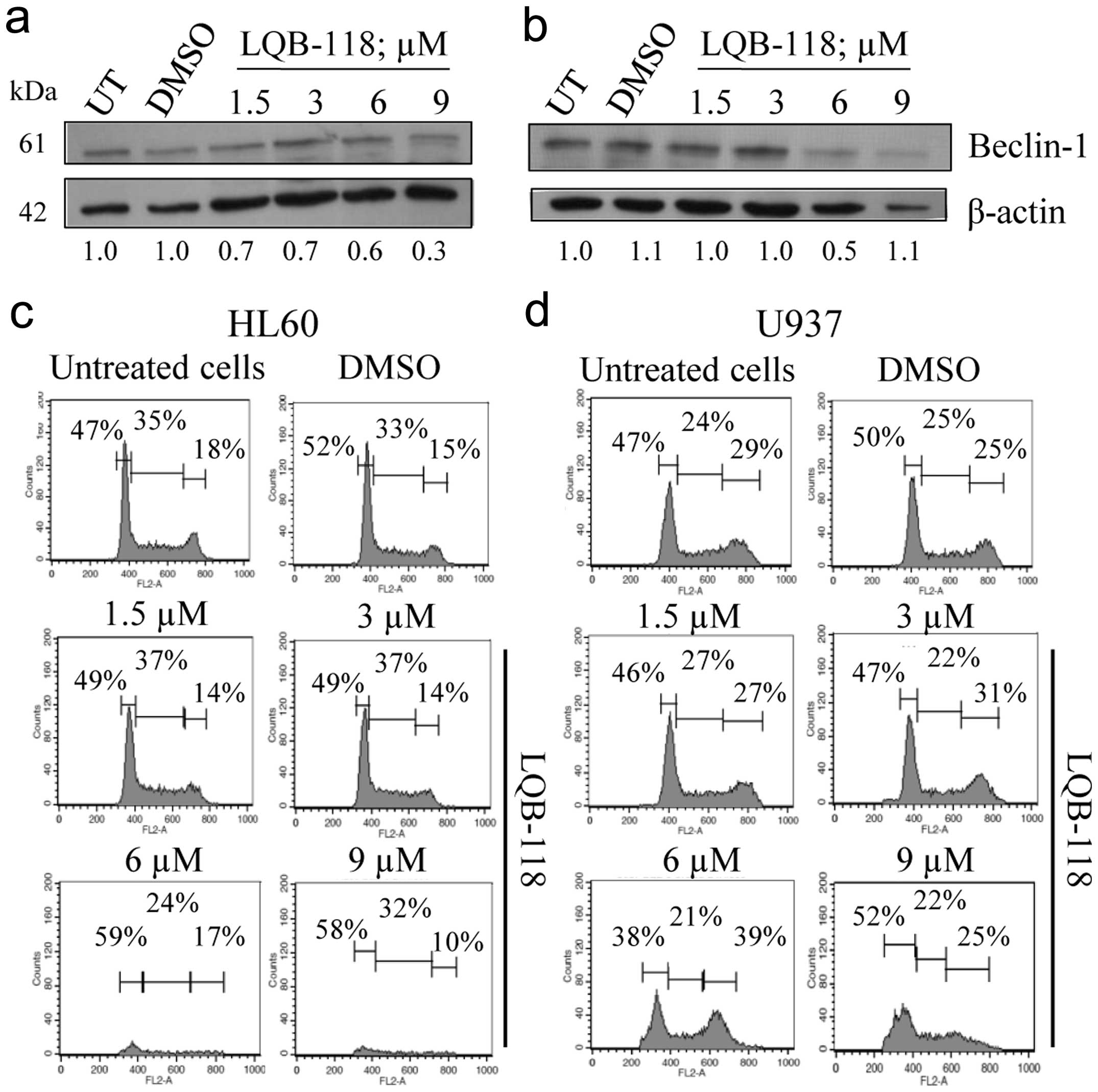
the LQB-118 compound would somehow modulate the autophagic process.
Our results show that the levels of Beclin1, which is an important
regulator of autophagy (34,35),
were not induced upon LQB-118 exposure (Fig. 3a and b). This suggests that the
compound does not induce Beclin1-mediated autophagy. Our data show
that the LQB-118 compound is highly effective in inducing apoptotic
cell death in AML cell lines derived from different FAB
subtypes.
LQB-118-induced apoptosis occurs
independently of changes in the cell cycle
Since many chemotherapeutic agents exert their
cytotoxic effects through the modulation of the phases of cell
cycle, we investigate the ability of LQB-118 compound to interfere
with the cell cycle. The histograms shown in Fig. 3c and 3d indicate that the treatment
of both cell lines with the compound did not result in significant
changes in cell cycle distribution. This indicates that apoptosis
induced by LQB-118 occurs independently of alterations in the cell
cycle profile.
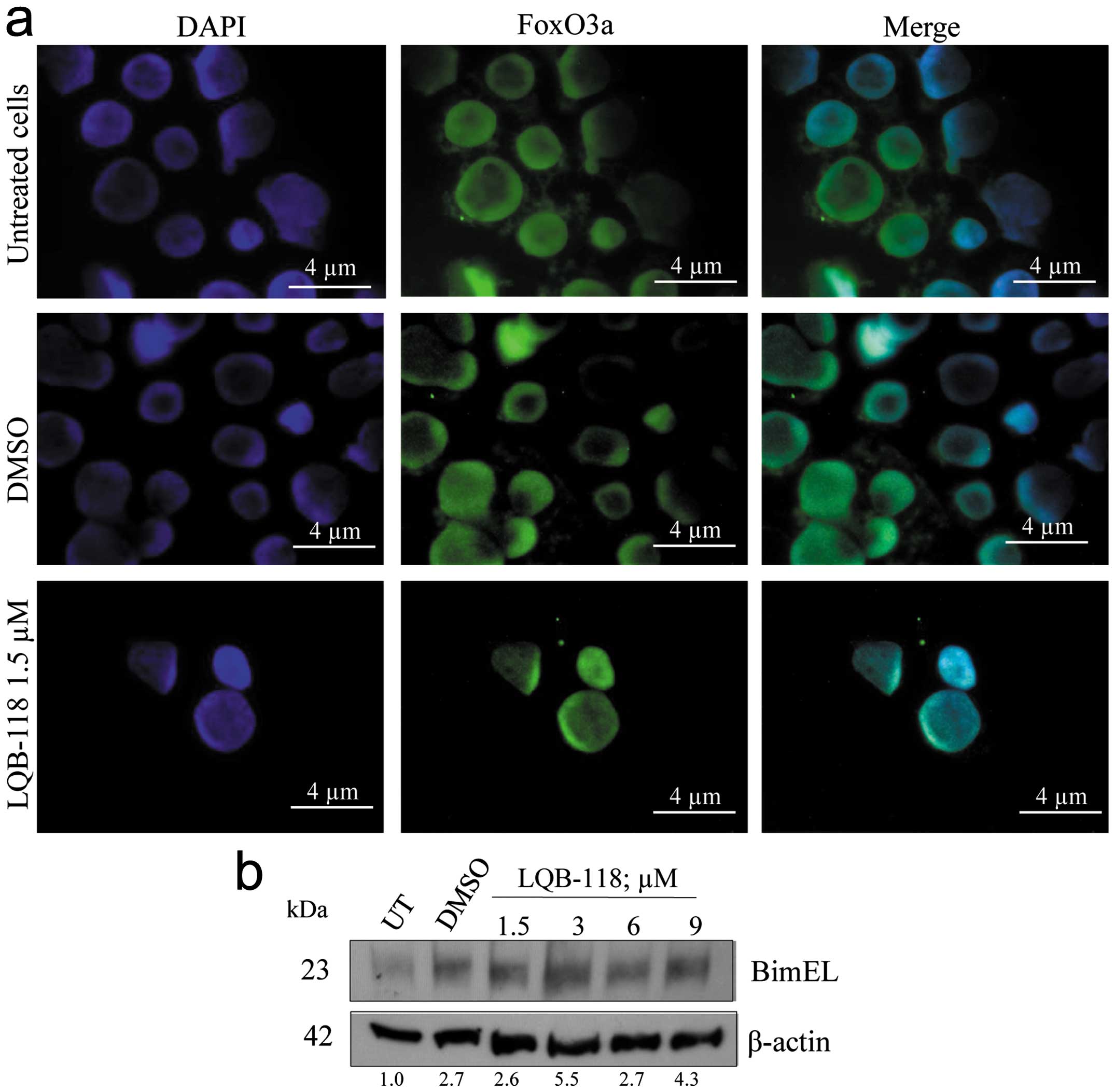
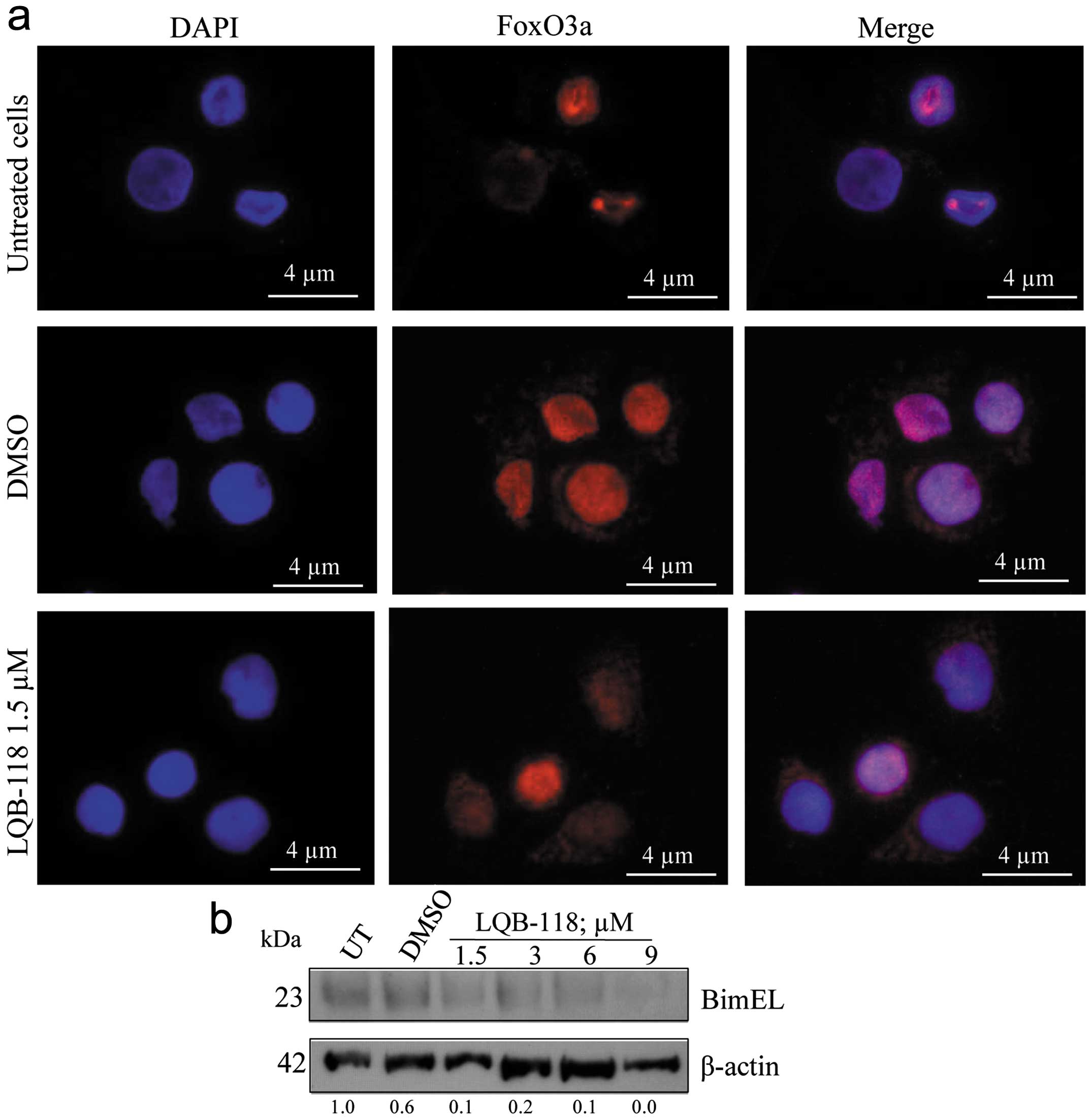
LQB-118 treatment results in FoxO3a
transcription factor relocation and Bim induction in HL60 cells,
but not in U937 cells
Next, we investigated if LQB-118 compound would be
able to target FoxO3a, as it is known that the transcription factor
can mediate the cytotoxic effects of several drugs in cancer cells
(24). We could observe that both
cell lines displayed a diffuse, predominantly nuclear pattern of
FoxO3a staining (Fig. 4a and
5a). Upon LQB-118 treatment at 1.5
μM, we observed nuclear translocation of FoxO3a in HL60 cells
(Fig. 4a). We also analyzed Bim
expression, which is a direct transcriptional target for FoxO3a,
and LQB-118 was able to induce Bim expression (Fig. 4b) (36). Interestingly, LQB-118 induced
nuclear exclusion of FoxO3a in U937 cells (Fig. 5a). Corroborating these data, there
was no Bim upregulation, and its expression was inhibited in a
dose-dependent manner (Fig. 5b).
It is relevant to emphasize that U937 cells displayed a
resistant-phenotype at lower concentrations (e.g., 1.5 μM).
Notably, immunofluorescence experiments were not performed after
treatment with 3, 6 and 9 μM of LQB-118 because cells were not
suitable for morphologic analysis. Together, these data indicate
that LQB-118 can promote a differential modulation of FoxO3a
pathway in AML cells with distinct molecular characteristics.
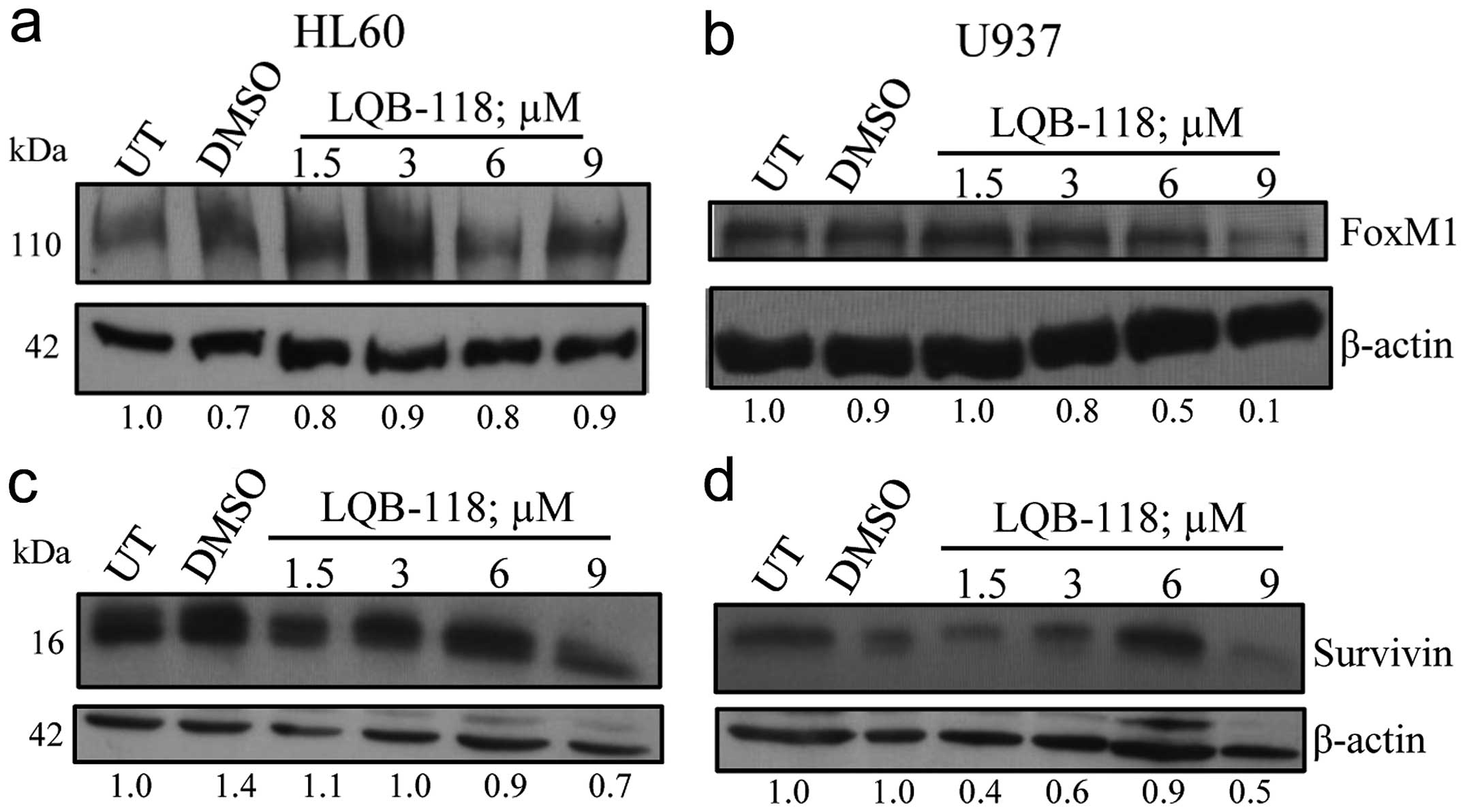
The LQB-118 compound promotes repression
of FoxM1 transcription factor in U937 cells
Because FoxM1 has been shown to induce cell
proliferation in AML cells (28)
its suppression would be a viable approach for these leukemic
subtypes. As a consequence, we questioned if FoxM1 would be a
target for the LQB-118 compound. Fig.
6b shows that FoxM1 expression is abolished in U937 cells upon
treatment with 9 μM of LQB-118. This effect was not so pronounced
in HL60 cells, in which FoxM1 levels remained almost unaltered
(Fig. 6a). Survivin expression was
evaluated because it can be positively regulated by FoxM1 and it
has a known role in drug resistance and as a therapeutic target
(28,37). After LQB-118 treatment, we observed
that U937 cells displayed a decrease in Survivin levels, which was
less pronounced in HL60 cells (Fig. 6c
and d). These data show that the LQB-118 compound targets the
FoxM1 pathway to induce apoptosis in U937, but not in HL60
cells.
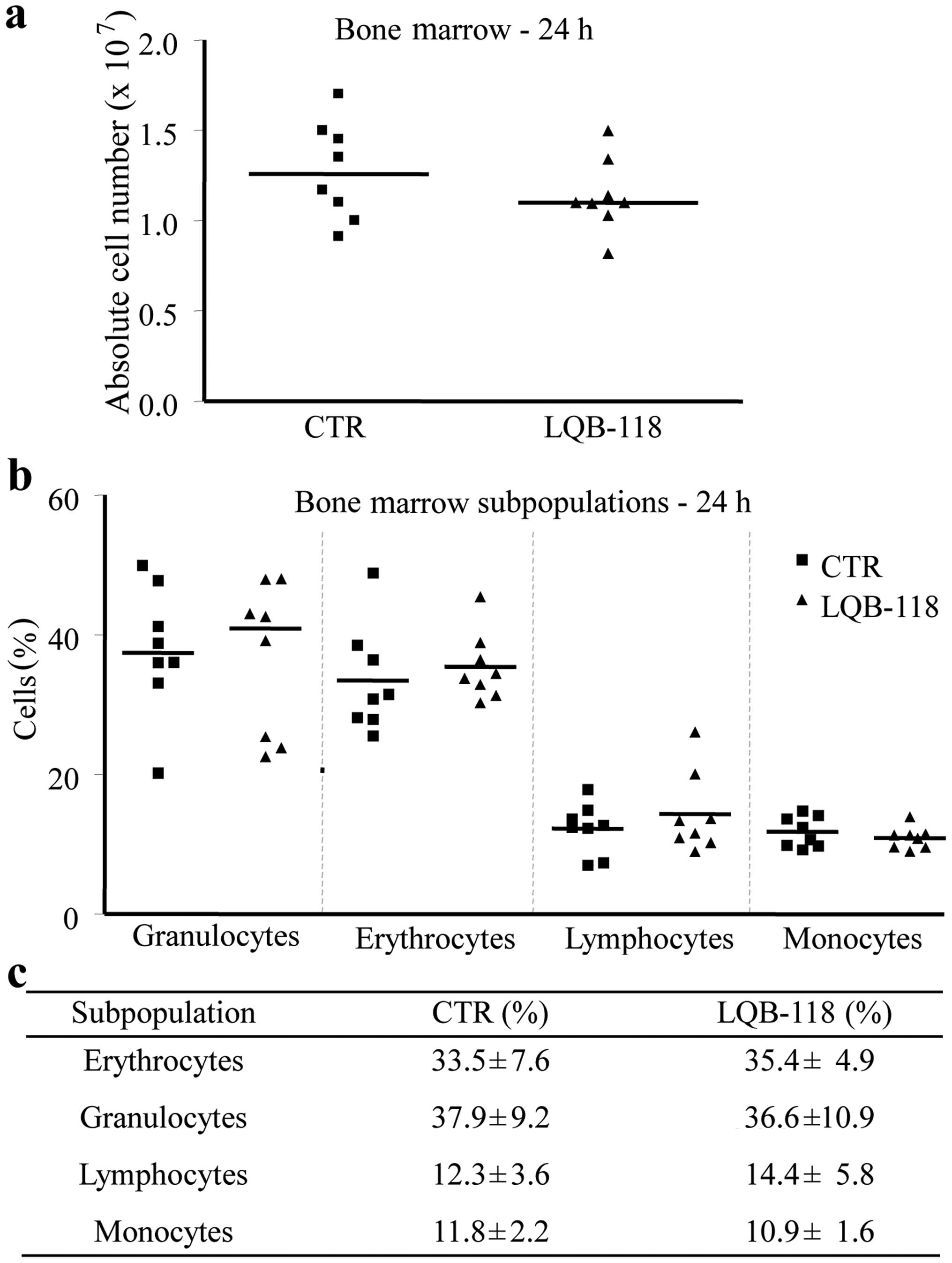
LQB-118 compound is not cytotoxic to
normal bone-marrow derived cells
Next, we asked if the LQB-118 compound would have a
further cytotoxic effect on normal bone marrow cells. To
investigate this conjecture, we intraperitoneally injected healthy
Swiss mice with LQB-118, and collected cells from their bone
marrow. As shown in Fig. 7a, there
was no statistical significance between the absolute cell count
from LQB-118-treated mice and those injected with the diluents. We
also analyzed bone marrow cellular populations and found that the
LQB-118 compound did not alter the distribution of the
subpopulations when compared to control group (Fig. 7b and c). These results indicate
that LQB-118 has a selective antitumoral activity, since it does
not affect normal cells from the bone marrow.
Discussion
AML is a highly heterogeneous disease, characterized
by a diverse spectrum of genetic abnormalities and the accumulation
of blastic cells (1,2). Despite effort having been made, no
significant advances were observed in AML treatment, complete
remission, and survival rates, except for the APL subtype (4,5).
Statistics from the Surveillance Epidemiology and End Results
(SEER) show that the overall 5-year relative survival is 24.2%,
among AML population in general. Poor outcomes in AML patients are
closely related to chemoresistance and/or disease relapse (8). Considering that ~15,000 people were
diagnosed with AML in the USA in 2013 (38), there is an urgent need for research
on treatment and development of novel therapeutic strategies. AML
cell lines represent in vitro models for studying
preclinical drug development. In recent studies, the novel
pterocarpanquinone LQB-118 demonstrated a satisfactory cytotoxic
effect against diverse neoplastic cell lines, with little toxicity
towards PBMC (10,12), indicating that it might be
selective in killing tumor cells. Interestingly, the LQB-118
compound induced apoptosis in cells from AML and CML patients
regardless of the MDR phenotype (11,21),
which strongly suggests that it may bypass mechanisms of resistance
while sensitizing tumor cells. Consistent with this, LQB-118
inhibited the expression of Survivin and XIAP antiapoptotic
proteins and Pgp efflux transporter in AML and CML cell lines
(11,21) and also triggered cascades involving
the endoplasmic reticulum stress pathway and depolarization of
mitochondrial membrane potential (13). Although these previous studies
provided some insights into LQB-118 mechanisms of action, they also
prompted us to investigate whether its effect extends to other AML
subtypes and which signaling pathways are regulated during
chemosensitivity.
In the present study, we evaluated the effects of
LQB-118 compound in HL60 (M3 subtype) and U937 (M5 subtype) AML
cell lines and observed that it induced apoptosis-associated DNA
fragmentation in both cell lines. Additionally, there was a great
increase in the population of cells positive for Annexin V, when
compared with the control of untreated cells. Confirming these
data, we found that ~70 and 90% of U937 and HL60 cells,
respectively, exhibited activated caspase-3 levels when exposed to
LQB-118, suggesting that the compound is able to induce high
apoptotic indexes in AML cells. Although HL60 cells were shown to
be more sensitive to the compound even at the lowest
concentrations, high LQB-118 concentrations could trigger U937
cells to undergo apoptosis. The LQB-118-mediated cytotoxic effects
previously mentioned occurred independently of changes in cell
cycle distribution and had no participation of Beclin1, a critical
regulator of the autophagic process (34,35).
Reinforcing these data, we have recently demonstrated that LQB-118
displays anticancer activities in the Kasumi cell line (M2 subtype)
as well as cells from AML patients (21). Altogether, these results show that
the LQB-118 compound is very effective in inducing apoptosis in
cells derived from distinct AML molecular subtypes. It is important
to emphasize that we observed no toxicity to normal bone marrow
cells isolated from LQB-118-treated mice when compared to control.
Thus, the low toxicity against these cells is indicative that this
pterocarpanquinone has a selective activity against leukemic cells.
In 2011, da Cunha-Júnior et al described an anti-leishmanial
effect of LQB-118 in BALB/c mice infected with Leishmania
amazonensis (39). The
compound was administered intraleasionally, orally and
intraperitoneally, the latter being effective to control both
lesion and parasite growth in a concentration of 4.5 mg/kg/day,
comparable to our study. Furthermore, this concentration did not
induce liver damage, since it did not alter serological markers of
toxicity such as the enzymes aspartate aminotransferase (AST),
alanine aminotransferase (ALT) and creatinine. However, it does not
exclude the possibility of the molecule undergoing liver metabolism
first before reaching the bone marrow. However, since it
selectively induced oxidative stress and apoptosis against L.
Amazonensis (40), this
possibility seems less likely.
We also explored the signaling pathways regulated by
LQB-118 in AML cells. First, we assessed FoxO3a localization,
because its transcriptional activity is regulated by
nuclear-cytoplasmic shuttling (41) and observed that LQB-118 treatment
of HL60 cells resulted in FoxO3a nuclear translocation.
Accordingly, the expression of its direct target Bim was
upregulated upon LQB-118 exposure. In contrast, LQB-118 treatment
of U937 cells led to FoxO3a nuclear exclusion and Bim
downregulation. It is important to note that the immunofluorescence
was performed with cells exposed to the lowest LQB-118
concentration, which is cytotoxic to HL60, but not to U937 cells.
This result suggests that LQB-118 promotes differential modulation
of FoxO3a localization in different molecular subtypes and that
FoxO3a cytoplasmic localization, and consequent prevention from its
transcriptional activity, correlates with LQB-118 resistance in our
model. Although some reports propose the idea that FoxO3a might
have an oncogenic role in AML (42,43)
most studies point FoxO3a as a tumor suppressor transcription
factor. It was demonstrated that the FLT3 tyrosine kinase promotes
phosphorylation-mediated FoxO3a inactivation, repression of Bim and
p27kip1 gene expression and apoptosis inhibition in
FLT3-expressing AML cells (44).
Also, high phosphorylated FoxO3a levels were found to be an
independent predictor of shorter remission duration and drug
resistance (44). Corroborating
these data, FoxO3a activation has been shown to be an essential
event in mediating the effects of transretinoic acid (45), hypomethylating agents (46), Aurora A kinase inhibitors (47) and FLT3 inhibitors (48) in AML cells, through positive
regulation of its targets TRAIL, Bim, Puma, and p27kip1.
These studies further emphasize FoxO3a usefulness as a target for
therapeutic approaches aiming to overcome resistance in AML.
Based on the fact that FoxO3a and FoxM1
transcription factors have opposing functions (49), our next step was to investigate
FoxM1 modulation upon LQB-118 treatment. Our results show that
FoxM1 expression was progressively decreased in LQB-118-treated
U937 cells. Survivin, an antiapoptotic protein known to be
transcriptionally regulated by FoxM1, was also downregulated after
exposure to the compound. The modulation in the FoxM1/Survivin axis
was less pronounced in HL60 cells, mainly at the concentration of 9
μM, indicating that this signaling pathway undergoes differential
modulation by the LQB-118 compound, depending on the molecular
subtype. FoxM1 is a master regulator of cell cycle progression
(27), whose overexpression was
found in a wide variety of tumors (reviewed in ref. 50). Although the role of FoxM1 in AML
has not been fully elucidated, FoxM1 was found to be aberrantly
expressed in leukemic blast cells (28,51).
Consistently, FoxM1 depletion inhibited colony formation in cells
derived from AML patients, indicating its relevance for cell cycle
progression, and also resulted in Survivin suppression in a panel
of cell lines (28). Survivin
expression has already been shown to correlate with poor prognosis
in AML and chemoresistance, as Survivin knockdown sensitized AML
cells to LQB-118 and idarubicin-induced apoptosis (14,21).
This group of data suggests that the LQB-118 compound negatively
regulates the oncogenic FoxM1/Survivin axis while sensitizing AML
cells.
In conclusion, this study demonstrated that the
LQB-118 compound can regulate multiple pathways to induce apoptosis
and target FoxO3a and FoxM1 transcription factors in AML cells,
with no toxicity to normal bone marrow cells. LQB-118 effects were
different in cell lines derived from distinct molecular subtypes,
which suggests that its mechanisms of action may vary according to
the subtype under investigation. In addition, measuring FoxO3a and
FoxM1 expression might possibly be a valuable tool to identify
sensitive and resistant patients, to stratify groups with different
clinical outcomes. We are currently conducting further in
vivo experiments in order to confirm the efficacy of LQB-118 in
tumor-bearing mice. Taken together, our findings propose the novel
pterocarpanquinone LQB-118 compound as a potential future
therapeutics for treatment of AML.
Acknowledgements
We thank Professor Vívian M. Rumjanek for valuable
comments on this manuscript. This study was supported by grants
from Instituto Nacional de Ciência e Tecnologia (INCT), Conselho
Nacional de Desenvolvimento Científico e Tecnológico (CNPq),
Fundação de Amparo à Pesquisa do Rio de Janeiro (FAPERJ), Programa
de Oncobiologia and Coordenação de Aperfeiçoamento de Pessoal de
Nível Superior (CAPES).
References
|
1
|
Kumar CC: Genetic abnormalities and
challenges in the treatment of acute myeloid leukemia. Genes
Cancer. 2:95–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bennett JM, Catovsky D, Daniel MT, et al:
Proposed revised criteria for the classification of acute myeloid
leukemia. A report of the French-American-British Cooperative
Group. Ann Intern Med. 103:620–625. 1985. View Article : Google Scholar
|
|
3
|
Harris NL, Jaffe ES, Diebold J, et al:
World Health Organization classification of neoplastic diseases of
the hematopoietic and lymphoid tissues: report of the Clinical
Advisory Committee meeting - Airlie House, Virginia, November 1997.
J Clin Oncol. 17:3835–3849. 1999.
|
|
4
|
O’Donnell MR, Abboud CN, Altman J, et al:
Acute myeloid leukemia. J Natl Compr Cancer Netw. 10:984–1021.
2012.
|
|
5
|
Tallman MS, Nabhan C, Feusner JH, et al:
Acute promyelocytic leukemia: evolving therapeutic strategies.
Blood. 99:759–767. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith ML, Hills RK and Grimwade D:
Independent prognostic variables in acute myeloid leukaemia. Blood
Rev. 25:39–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wood WA and Lee SJ: Malignant hematologic
diseases in adolescents and young adults. Blood. 117:5803–5815.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaffer BC, Gillet JP, Patel C, et al:
Drug resistance: still a daunting challenge to the successful
treatment of AML. Drug Resist Updat. 15:62–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Netto CD, Santos ES, Castro CP, et al:
(+/−)-3,4-Dihydroxy-8,9-methylenedioxypterocarpan and derivatives:
cytotoxic effect on human leukemia cell lines. Eur J Med Chem.
44:920–925. 2009.
|
|
10
|
Netto CD, da Silva AJ, Salustiano EJ, et
al: New pterocarpanquinones: synthesis, antineoplasic activity on
cultured human malignant cell lines and TNF-alpha modulation in
human PBMC cells. Bioorg Med Chem. 18:1610–1616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maia RC, Vasconcelos FC, de Sá Bacelar T,
et al: LQB-118, a pterocarpanquinone structurally related to
lapachol [2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone]: a
novel class of agent with high apoptotic effect in chronic myeloid
leukemia cells. Invest New Drugs. 29:1143–1155. 2011.
|
|
12
|
Salustiano EJ, Netto CD, Fernandes RF, et
al: Comparison of the cytotoxic effect of lapachol, alpha-lapachone
and pentacyclic 1,4-naphthoquinones on human leukemic cells. Invest
New Drugs. 28:139–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Sá Bacelar T, da Silva AJ, Costa PR, et
al: The pterocarpanquinone LQB 118 induces apoptosis in tumor cells
through the intrinsic pathway and the endoplasmic reticulum stress
pathway. Anticancer Drugs. 24:73–83. 2013.PubMed/NCBI
|
|
14
|
Adida C, Recher C, Raffoux E, et al:
Expression and prognostic significance of survivin in de novo acute
myeloid leukaemia. Br J Haematol. 111:196–203. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tamm I, Kornblau SM, Segall H, et al:
Expression and prognostic significance of IAP-family genes in human
cancers and myeloid leukemias. Clin Cancer Res. 6:1796–1803.
2000.PubMed/NCBI
|
|
16
|
Wuchter C, Leonid K, Ruppert V, et al:
Clinical significance of P-glycoprotein expression and function for
response to induction chemotherapy, relapse rate and overall
survival in acute leukemia. Leukemia. 14:1018–1024. 2000.
|
|
17
|
Nestal de Moraes G, Silva KL, Vasconcelos
FC, et al: Survivin overexpression correlates with an
apoptosis-resistant phenotype in chronic myeloid leukemia cells.
Oncol Rep. 25:1613–1619. 2011.PubMed/NCBI
|
|
18
|
Nestal de Moraes G, Souza PS, Costas FC,
et al: The interface between BCR-ABL-dependent and -independent
resistance signaling pathways in chronic myeloid leukemia. Leuk Res
Treatment. 2012:6717022012.PubMed/NCBI
|
|
19
|
Silva KL, de Souza PS, Nestal de Moraes G,
et al: XIAP and P-glycoprotein co-expression is related to imatinib
resistance in chronic myeloid leukemia cells. Leuk Res.
37:1350–1358. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Souza PS, Vasconcelos FC, De Souza Reis
FR, et al: P-glycoprotein and survivin simultaneously regulate
vincristine-induced apoptosis in chronic myeloid leukemia cells.
Int J Oncol. 39:925–933. 2011.
|
|
21
|
de Souza Reis FR, de Faria FC, Castro CP,
et al: The therapeutical potential of a novel pterocarpanquinone
LQB-118 to target inhibitor of apoptosis proteins in acute myeloid
leukemia cells. Anticancer Agents Med Chem. 13:341–351.
2013.PubMed/NCBI
|
|
22
|
Myatt SS and Lam EW: The emerging roles of
forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chapuis N, Park S, Leotoing L, et al: IκB
kinase overcomes PI3K/Akt and ERK/MAPK to control FoxO3a activity
in acute myeloid leukemia. Blood. 116:4240–4250. 2010.
|
|
24
|
Yang JY and Hung MC: A new fork for
clinical application: targeting forkhead transcription factors in
cancer. Clin Cancer Res. 15:752–757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Tang N, Hadden TJ, et al: Akt,
FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leung TW, Lin SS, Tsang AC, et al:
Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS
Lett. 507:59–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang IC, Chen YJ and Hughes D: Forkhead
box M1 regulates the transcriptional network of genes essential for
mitotic progression and genes encoding the SCF (Skp2-Cks1)
ubiquitin ligase. Mol Cell Biol. 25:10875–10894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakamura S, Hirano I, Okinaka K, et al:
The FOXM1 transcriptional factor promotes the proliferation of
leukemia cells through modulation of cell cycle progression in
acute myeloid leukemia. Carcinogenesis. 31:2012–2021. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wilson MS, Brosens JJ, Schwenen HD, et al:
FOXO and FOXM1 in cancer: the FOXO-FOXM1 axis shapes the outcome of
cancer chemotherapy. Curr Drug Targets. 12:1256–1266. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gallagher R, Collins S, Trujillo J, et al:
Characterization of the continuous, differentiating myeloid cell
line (HL-60) from a patient with acute promyelocytic leukemia.
Blood. 54:713–733. 1979.PubMed/NCBI
|
|
31
|
Lee KH, Chang MY, Ahn JI, et al:
Differential gene expression in retinoic acid-induced
differentiation of acute promyelocytic leukemia cells, NB4 and
HL-60 cells. Biochem Biophys Res Commun. 296:1125–1133. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ralph P, Harris PE, Punjabi CJ, et al:
Lymphokine inducing ‘terminal differentiation’ of the human
monoblast leukemia line U937: a role for gamma interferon. Blood.
62:1169–1175. 1983.
|
|
33
|
Cáceres-Cortés JR: Blastic leukaemias
(AML): a biologist’s view. Cell Biochem Biophys. 66:13–22.
2013.PubMed/NCBI
|
|
34
|
Lorin S, Hamaï A, Mehrpour M, et al:
Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wrighton KH: Autophagy: kinase crosstalk
through beclin 1. Nat Rev Mol Cell Biol. 14:402–403. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sunters A, Fernández de Mattos S, Stahl M,
et al: FoxO3a transcriptional regulation of Bim controls apoptosis
in paclitaxel-treated breast cancer cell lines. J Biol Chem.
278:49795–49805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kelly RJ, Lopez-Chavez A, Citrin D, et al:
Inhibitors of apoptosis proteins (IAPs) as potential molecular
targets for therapy of hematological malignancies. Curr Mol Med.
11:633–649. 2011. View Article : Google Scholar
|
|
38
|
Howlader N, Noone AM, Krapcho M, et al:
SEER Cancer Statistics Review, 1975–2010. National Cancer
Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2010/,
based on November 2012 SEER data submission, posted to the SEER web
site. 2013
|
|
39
|
da Cunha EF Júnior, Pacienza-Lima W,
Ribeiro GA, et al: Effectiveness of the local or oral delivery of
the novel naphthop-terocarpanquinone LQB-118 against cutaneous
leishmaniasis. J Antimicrob Chemother. 66:1555–1559. 2011.
|
|
40
|
Ribeiro GA, Cunha-Júnior EF, Pinheiro RO,
et al: LQB-118, an orally active pterocarpanquinone, induces
selective oxidative stress and apoptosis in Leishmania amazonensis.
J Antimicrob Chemother. 68:789–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Van Der Heide LP, Hoekman MF and Smidt MP:
The ins and outs of FoxO shuttling: mechanisms of FoxO
translocation and transcriptional regulation. Biochem J.
380:297–309. 2004.PubMed/NCBI
|
|
42
|
Santamaría CM, Chillón MC, García-Sanz R,
et al: High FoxO3a expression is associated with a poorer prognosis
in AML with normal cytogenetics. Leuk Res. 33:1706–1709.
2009.PubMed/NCBI
|
|
43
|
Sykes SM, Lane SW, Bullinger L, et al:
AKT/FOXO signaling enforces reversible differentiation blockade in
myeloid leukemia. Cell. 146:697–708. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kornblau SM, Singh N, Qiu Y, et al: Highly
phosphorylated FOXO3A is an adverse prognostic factor in acute
myeloid leukemia. Clin Cancer Res. 16:1865–1874. 2010. View Article : Google Scholar
|
|
45
|
Sakoe Y, Sakoe K, Kirito K, et al: FOXO3A
as a key molecule for all-trans retinoic acid-induced granulocytic
differentiation and apoptosis in acutepromyelocytic leukemia.
Blood. 115:3787–3795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thépot S, Lainey E, Cluzeau T, et al:
Hypomethylating agents reactivate FOXO3A in acute myeloid leukemia.
Cell Cycle. 10:2323–2330. 2011.PubMed/NCBI
|
|
47
|
Kelly KR, Nawrocki ST, Espitia CM, et al:
Targeting Aurora A kinase activity with the investigational agent
alisertib increases the efficacy of cytarabine through a
FOXO-dependent mechanism. Int J Cancer. 131:2693–2703. 2012.
View Article : Google Scholar
|
|
48
|
Scheijen B, Ngo HT, Kang H, et al: FLT3
receptors with internal tandem duplications promote cell viability
and proliferation by signaling through Foxo proteins. Oncogene.
23:3338–3349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao F and Lam EW: Role of the forkhead
transcription factor FOXO-FOXM1 axis in cancer and drug resistance.
Front Med. 6:376–380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Koo CY, Muir KW and Lam EW: FOXM1: from
cancer initiation to progression and treatment. Biochim Biophys
Acta. 1819:28–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang X, Zeng J, Zhou M, et al: The tumor
suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid
leukemia. Mol Cancer. 11:562012. View Article : Google Scholar : PubMed/NCBI
|