1. Introduction
The crucial role of transforming growth factor β
(TGFβ) in tumor progression, metastasis and treatment has been well
recognized and has become the topic of extensive research. Among
the effects, TGFβ can regulate cancer cell proliferation,
contribute to epithelial-to-mesenchymal transition (EMT), suppress
the function of immune cells compromising immune response,
contribute to the conversion of fibroblasts to myofibroblasts and
cause overproduction of extracellular matrix (ECM) in the tumor.
While it has been known for over two decades that anti-cancer drugs
cannot penetrate deep into collagen-rich tumors (e.g., pancreatic
cancers) and, more significantly, that depletion of collagen fibers
can improve drug delivery, only recently TGFβ has become a target
to reduce tumor fibrosis and thus, increase intratumoral drug
concentration and treatment efficacy. Preclinical data of this new
strategy are promising and it has already reached clinical trials.
In this review, we first present a brief description of TGFβ
synthesis and activation along with its signaling pathways.
Following, we discuss the effects of TGFβ on tumor progression, its
pathway alterations in cancer as well as its effects on EMT, immune
cells function, fibroblasts behavior and ECM remodeling. Finally,
based on the above, we review the barriers to the effective
delivery of drugs caused by TGFβ and how regulation of TGFβ
signaling can be employed to optimize delivery of therapeutic
agents and overall survival (1–3).
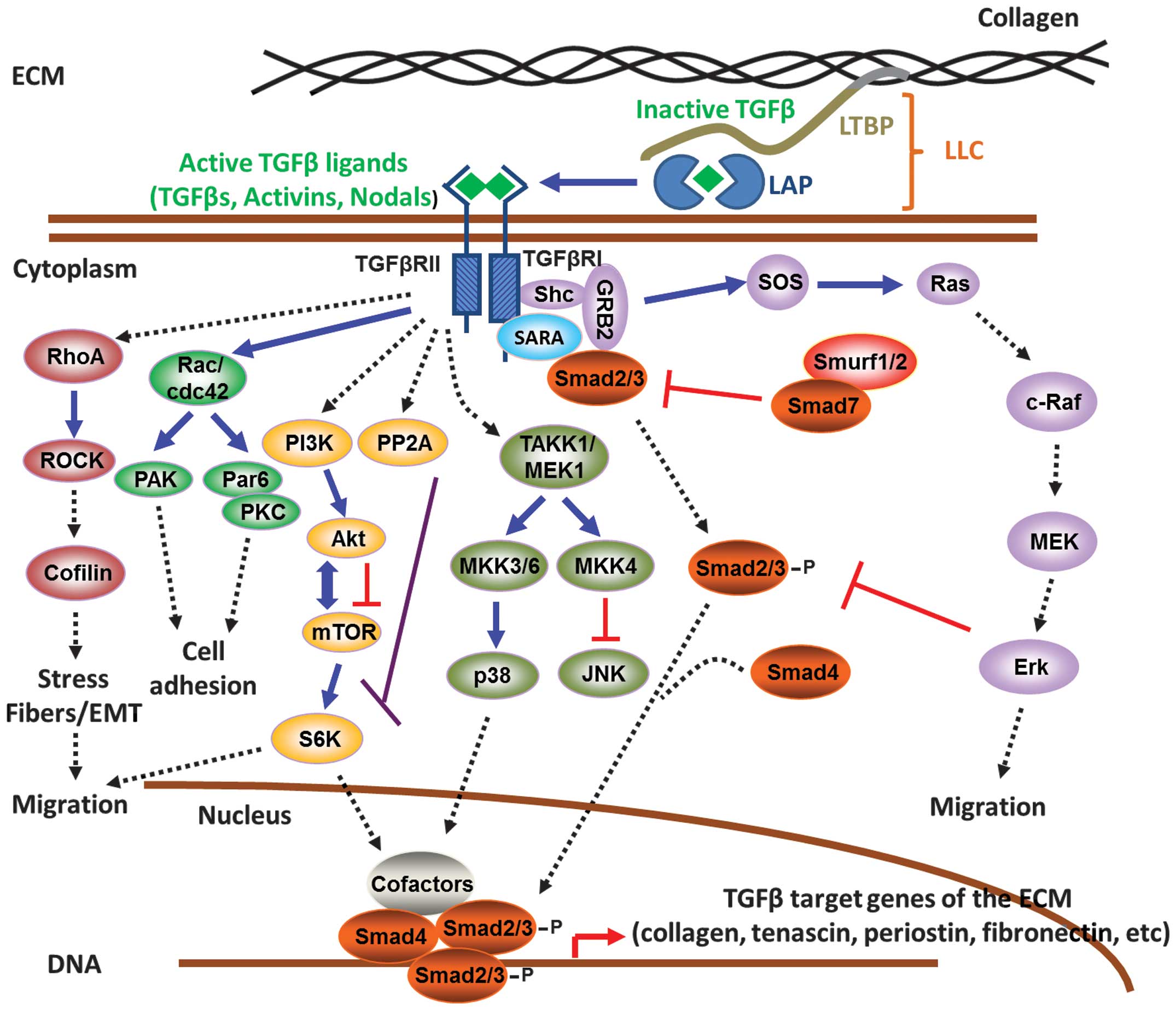
2. TGFβ synthesis and activation
The TGFβ superfamily encompasses around 40 secreted
cytokines, including TGFβ, bone morphogenetic proteins (BMPs),
activins, nodal, lefty, myostatin, anti-Müllerian hormone (AMH) and
growth differentiation factors (GDFs). These cytokines regulate a
plethora of biological functions such as cell proliferation and
apoptosis, embryonic patterning, stem cell maintenance, cell
differentiation, migration and immune surveillance. Importantly,
the effects of these factors are characterized as cell-type
specific as well as context dependent (1–3). The
TGFβ isoforms, with most common being TGFβ1, 2 and 3, are initially
synthesized as 75 kDa inactive homodimers, known as pro-TGFβ, which
consist of TGFβ associated with latency-associated proteins (LAPs)
at the N-terminal part of the pro-peptide. This is part of the TGFβ
large latent complex (LLC), comprised of the LAPs and the latency
TGFβ-binding proteins (LTBPs) (4–7), and
is covalently associated to the ECM via the N-terminal region of
LTBPs (8,9) (Fig.
1). While TGFβ is part of the LLC complex, it remains in an
inactive form since the high affinity association of LAPs with TGFβ
prevents the interaction with its receptors (10). During TGFβ activation, LAPs undergo
conformational changes induced by thrombospondin-1 (TSP-1)
(11,12) followed by cleavage mediated by
furin convertase, plasmin or matrix metalloproteinases MMP-2/9
resulting in the release of the mature 24 kDa TGFβ dimer (13–15).
The active ligand is then able to bind and activate TGFβ receptors
(TGFβRs) to propagate downstream intracellular signaling events.
Therefore, the processing of pro-TGFβ into the active TGFβ ligand
is a critical regulatory step which determines its
bioavailability.
3. TGFβ signaling pathways
The TGFβ and TGFβ-like cytokines mediate downstream
intracellular signaling via the Smad family of proteins, which
consists of eight human structurally related members (16–20)
(Fig. 1). Smads can be
functionally classified into three groups: the receptor activated
Smads (R-Smads), which include Smad1, 2, 3, 5, 8; the common
mediator Smad (Co-Smad), Smad4; and the inhibitory Smads (I-Smads),
Smad6 and 7 (17,21). Three types of TGFβRs are
responsible for initiating signaling; TGFβRI, II and III. There are
seven TGFβRI, five TGFβRII and two TGFβRIII known so far. TGFβRIs
include activin receptor-like kinases 1–7 (ALK1–7), TGFβRIIs
include the TGFβRII, bone morphogenetic protein receptor II
(BMPRII), activin receptor II (ACTRII), ACTRIIB, anti-Müllerian
hormone receptor II (AMHRII), while beta-gycan and endoglin belong
to the TGFβRIIIs (22) and mostly
function as co-receptors to enhance activin signaling (23). In most tissues, TGFβ ligands
function through heteromeric complex formation between two TGFβRI
and two TGFβRII molecules. While both receptors possess Ser/Thr
kinase activity, TGFβRIIs function as the ‘activator’ and TGFβRIs
as the ‘signal propagating’ component (24). The TGFβRII-ALK5 complex transduces
the signal from all three TGFβ isoforms in multiple cell types,
whereas association of TGFβRII with ALK1 is involved in endothelial
cells and with ALK2 in cardiovascular tissues (25). ALK5 activates Smad2 and 3 via the
canonical TGFβ signaling pathway whereas ALK2, 3 and 6 can activate
Smad1, 5 and 8, which are transducers of the BMP signaling pathway
(26,27). The TGFβ signaling pathways can be
classified in two major categories; the canonical or Smad-dependent
and the non-canonical or Smad-independent pathways.
Canonical pathway (Smad-dependent)
Even though TGFβ isoforms may elicit diverse
cellular responses, they all activate signaling via a similar
sequence of events. Binding of the active TGFβ1 ligand to the
Ser/Thr kinase TGFβRII followed by recruitment of the ALK5 (TGFβRI)
on the cell surface initiates intracellular signaling. Within the
heterotetrameric receptor-ligand complex formed, TGFβRII
phosphorylates TGFβRI allowing it to interact with the R-Smads
(Smad2/3) which, in turn, become phosphorylated at the conserved
SSXS C-terminal motif (28,29).
Recruitment of R-Smads to the activated TGFβRI is facilitated by
Smad anchor for receptor activation (SARA) protein (30). Subsequently, this triggers the
formation of a heterotrimeric complex between phosphorylated
R-Smads (Smad2/3) and Co-Smad (Smad4), which can translocate into
the nucleus to regulate gene expression (3) (Fig.
1). Smads can differentially modulate gene expression by acting
as transcription factors in co-operation with co-activators, such
as p300/CREB-binding protein (CBP), p300/CBP-associated factor
(PCAF), Smad4-interacting factor (SMIF), forkhead transcription
factors 1, 3, 4 (FoxO1/3/4), specificity protein 1 (Sp1),
c-Jun/c-Fos, Sertad1, or co-repressors, such as E2F4/5-p107,
activating transcription factor 3 (ATF3), TGFβ-induced factor
(TGIF), Ski, SnoN, forkhead transcription factor G1 (FoxG1),
ecotropic viral integration site 1 protein (EVI1) and C-terminal
binding protein (CTBP) (28,31–47).
In addition, Smads are able to epigenetically regulate gene
expression either by inducing chromatin remodeling (48,49)
or by maintaining DNA methylation and silencing of selected genes
(50). Importantly, the I-Smad,
Smad7, is a key target gene induced by TGFβ signaling and
acts as negative feedback regulator of the pathway (51). In the absence of TGFβ stimulation,
Smad7 resides in the cell nucleus and translocates to the plasma
membrane upon TGFβ-mediated receptor activation (52). Smad7 is then able to interfere and
block interactions between the R-Smads and the activated receptors
to inhibit downstream signaling events (53). In addition, Smad7 can target the
TGFβRs for proteasomal degradation via the E3-ubiquitin ligases
Smurf1 and 2 (54,55). Finally, Smad7 antagonizes the
formation of a functional Smad-DNA complex by directly binding to
DNA via its MH2 domain and therefore blocks TGFβ-mediated
transcriptional responses (56).
Non-canonical pathways
(Smad-independent)
It is also well established that TGFβ-mediated
effects can also be exerted through non-canonical Smad-independent
pathways (57). TGFβ has been
shown to induce activation of Erk signaling in various tissues
including epithelial and endothelial cells, fibroblasts, breast and
colorectal cancer cells in order to promote disassembly of adherens
junctions and cell migration (58–64).
TGFβRI phosphorylation can recruit and activate ShcA, thus
promoting the formation of a ShcA/Grb2/Sos complex. In turn, this
complex is able to activate Ras on the plasma membrane followed by
sequential activation of c-Raf, MEK and Erk (65).
Moreover, TGFβ can mediate the activation of the
c-Jun N-terminal kinase (JNK) and p38/mitogen-activated protein
kinase (MAPK) pathways, which are responsible for promoting
apoptosis or cell migration depending on cellular context (66–68),
via the mitogen-activated protein kinase kinase (MKK)4 and 3/6,
respectively (69,70). Further upstream, MKKs are
phosphorylated by the TGFβ-activated kinase 1 (TAK1) (71,72)
which is recruited to the TGFβRs via the scaffold protein TNF
receptor-associated factor 6 (TRAF6) (73,74).
Besides TAK1, two other mitogen-activated protein kinase kinase
kinases (MAPKKKs), namely MEKK1 and mixed lineage kinase 3 (MLK3),
were also shown to mediate TGFβ-induced activation of JNK and
p38-MAPK by MKK4 and 3/6 (75,76).
The Rho-like small GTPases, predominantly RhoA, Rac
and cell division cycle 42 (cdc42), are additional molecules that
mediate important TGFβ cellular functions, such as cytoskeletal
organization, cell polarity, cell migration and gene expression
(77). TGFβ is able to rapidly
activate the RhoA and cdc42/Rac1 pathways, in a Smad2/3-independent
manner, to promote actin polymerization, formation of stress fibers
and EMT (78,79). TGFβ may also downregulate RhoA
protein levels by recruitment of Par6 at the TGFβRI–II complex.
Phosphorylation of Par6 by TGFβRII triggers binding of the E3
ligase Smurf1 to the complex followed by ubiquitination and
degradation of RhoA at sites of cellular protrusions. Subsequently,
this leads to the dissolution of tight junctions, rearrangement of
actin cytoskeleton and EMT (80).
Some of the effects exerted by TGFβ could also be
mediated by activation of the phosphatidylinositol-4,5-bisphosphate
3-kinase/Akt (PI3K/Akt) pathway. This is evident from studies
showing that TGFβ can rapidly induce PI3K activation followed by
phosphorylation of its effector Akt to promote EMT, cell migration
and survival (81,82). One of the most important effector
molecules downstream of PI3K/Akt pathway appears to be the
mammalian target of rapamycin (mTOR), a key regulator of protein
synthesis, which can subsequently phosphorylate S6 kinase (S6K) and
eukaryotic initiation factor 4E-binding protein 1 (4EBP1) (83). Activation of the mTOR pathway by
TGFβ is thought to be important for regulating cell size, EMT and
invasion (84) (Fig. 1).
4. TGFβ signaling in cancer initiation and
tumor progression
It is well established that the multipotent actions
of TGFβ are highly context dependent. The complexity of these
functions is increased due to the fact that TGFβ exerts distinct
effects depending on the tissue type as well as the genetic and
epigenetic background of cells (85). It is clearly evident that TGFβ
plays dual roles during carcinogenesis. In early stages TGFβ
promotes growth inhibition and apoptosis of normal epithelial and
lymphoid cells as well as pre-malignant tumors, whereas during late
stages TGFβ acquires pro-oncogenic and pro-metastatic roles, which
are associated with a progressive increase in the locally secreted
TGFβ levels (86–88). Therefore, one of the hallmarks of
cancer is that the vast majority of cases exhibits insensitivity to
TGFβ-mediated growth inhibition.
Regulation of cell proliferation
It has long been noted that TGFβ has a cytostatic
effect on normal epithelial (89),
endothelial (90,91) and neuronal cells (92) as well as certain cells of the
immune system, such as T cells (93). These functions of TGFβ are
extremely important for physiological tissue homeostasis in order
to restrain cell proliferation and prevent the generation of
hyperproliferative disorders, like cancer. These anti-proliferative
effects primarily control the G1/S phase transition events
(94) and are mediated via
induction of the cyclin-dependent kinase inhibitors CDKN2B
(encoding p15/INK4B) (95),
CDKN1A (encoding p21/Cip/Waf1) (96) and p27/Kip1 (97) by TGFβ. Cell cycle arrest can also
be achieved by repression of the proliferation-inducing
transcription factors c-Myc (98)
and the family of inhibitor of DNA-binding proteins ID1, 2 and 3
(36,99). On the other hand, the effects of
TGFβ in proliferation can be opposing, depending on the tissue
type. It is also well recognized that TGFβ enhances proliferation
of fibroblasts (89) and it is
often mediated indirectly by TGFβ-induced connective tissue growth
factor (CTGF) secretion, which is responsible for stimulating
fibroblast proliferation and ECM synthesis (100). It is now unambiguously accepted
that cancer-associated fibroblasts (CAFs) play critically important
roles in the tumor microenvironment and cancer progression and
their functions are further discussed below.
Pathway alterations in human cancers
Numerous human studies have identified that
components of the TGFβ pathway become genetically or epigenetically
altered in various tumor types thus explaining, at least in part,
the escape from TGFβ-mediated growth control. Loss of function or
truncating mutations in TGFβRI and TGFβRII as well as
in Smad2 and Smad4 have been detected in colorectal,
pancreatic, gastric and prostate cancers (18,101–105). In addition, loss of the 18q21
chromosome region, harboring the Smad4 gene, is commonly
observed in ~60% of pancreatic and 30% of colorectal cancers
(106–109) has been shown to promote
angiogenesis and tumor growth by inducing vascular endothelial
growth factor (VEGF) expression (60,110). However, in other tumor types like
breast, the frequency of Smad gene mutations is rare
(18,104,105) suggesting that alternative
mechanisms for acquiring resistance to growth inhibition by TGFβ
exist. These include activation of the Ras oncogene which leads to
Erk-mediated Smad2/3 phosphorylation and suppression of functional
Smad complex formation (111–113). Furthermore, overexpression of the
dominant-negative CCAAT/enhancer-binding protein β (C/EBPβ) isoform
LIP in breast cancer patients was found to suppress TGFβ-mediated
growth inhibition (114).
Finally, another mechanism which TGFβ may exploit in order to
switch from a tumor suppressor to a metastasis-promoting factor is
through differential regulation of the ID1 gene. While ID1
expression is suppressed by TGFβ in normal tissues, it was found to
be induced in patient-derived metastatic breast cancer cells
(115).
EMT and cancer metastasis
EMT is an integral process during embryonic
development which can be abnormally reactivated in adult tissues
under pathological conditions, such as cancer and fibrosis
(116). It involves the
activation of a coordinated reversible transcriptional program
whereby epithelial cells undergo dissolution of cell junctions,
lose their polarity and epithelial characteristics concomitantly
with acquisition of mesenchymal features and dramatic remodeling of
their cytoskeleton. During this process, the expression of
epithelial genes, such as E-cadherin, γ- and
β-catenin, zonula occludens (ZO), and
claudins is suppressed with concurrent expression of
mesenchymal components, such as N-cadherin, vimentin, fibronectin
and α-smooth muscle actin (α-SMA) (50,117,118). This program can be initiated by
several pleiotropically acting transcription factors regulated by
signaling pathways such as TGFβ, Wnt and receptor tyrosine kinases
(RTKs). Some of the better characterized examples include Snail
(119), Slug (120), zinc-finger E-box binding homeobox
1 (ZEB1/δEF1) (121), zinc-finger
E-box binding homeobox 2/Smad interacting protein 1 (ZEB2/SIP1)
(122), Twist (117), high mobility group AT-hook 2
(HMGA2) (123) and forkhead box
protein C2 (FOXC2) (124). In
addition, recent studies indicate that overactive TGFβ-TGFβR-Smad2
signaling axis could further contribute to the establishment of an
EMT phenotype by maintaining the epigenetic silencing of epithelial
genes during this process (50).
Besides Smads, other signaling pathways have also been implicated
in TGFβ-induced EMT, including Erk, PI3K/Akt, RhoA, p38-MAPK and
cofilin (125–127). Induction of EMT is one of the
major mechanisms by which TGFβ has been shown to promote cell
motility, invasiveness and metastasis of cancer cells (128). EMT significantly enhances
intravasation of carcinoma in situ cells through the
basement membrane, survival in the circulation, extravasation at
the distal tissues and formation of micrometastases in secondary
organs (116,117,129).
5. The effects of TGFβ on the tumor
microenvironment
Under physiological conditions, the sustained local
release of basal TGFβ levels is sufficient to maintain normal
tissue homeostasis. However, under conditions of tissue injury, the
local TGFβ secretion from stromal cells and blood platelets is
rapidly increased to facilitate wound repair as well as to prevent
uncontrolled regenerative cell proliferation and inflammation
(130,131). A similar situation is commonly
observed in pre-malignant tumors where TGFβ is secreted in the
microenvironment initially to control proliferation and cancer
progression, but it is ultimately utilized by cancer cells to
promote their malignant properties. Local TGFβ release produces a
tumor microenvironment which is conducive to tumor growth, invasion
and metastasis (132). Secretion
of TGFβ can be derived from epithelial cancer cells thus regulating
their own properties within the tumor mass in an autocrine or
paracrine fashion (125).
Moreover, infiltrating stromal cells, including fibroblasts,
leukocytes, macrophages, bone-marrow derived endothelial,
mesenchymal and myeloid precursor cells, is another major source of
this cytokine (133). Finally,
TGFβ can be stored in the ECM of the bone and can be activated
during development of osteolytic metastatic lesions (134). In the following paragraphs, we
summarize the effects of TGFβ on the main and better characterized
components of the tumor microenvironment and particularly on
fibroblasts, immune cells and the ECM.
Effect of TGFβ on immune cells
TGFβ exhibits immunosuppressive effects on all arms
of the immune system because it functions as antagonist of several
functions of the immune cells (132,135). As a result, the anti-tumor immune
response is compromised, reducing cancer cell recognition and
clearance. Specifically, TGFβ affects the function of natural
killer cells, CD4+ and 8+ T cells,
macrophages, neutrophils, dendritic, mast and B cells (136–138). Specifically, a TGFβ-rich tumor
microenvironment is a suppressor of T-cell proliferation, reduces
their effector function and inhibits the maturation of T helper
cells (137,139,140). It also induces macrophage M2
polarization from a type I to a type II phenotype, which hinders
the suppression of monocyte-mediated cell death, reduces effector
function and increases chemotaxis (141,142). Additionally, TGFβ induces an N2
neutrophil phenotype which, as with the macrophages, reduces
effector function and increases secretion of inflammatory cytokines
(143). Finally, high levels of
TGFβ can cause apoptosis of B cells, inhibit the maturation of
dendritic and natural killer cells and induce chemotaxis of mast
cells (144–146). The combined immuno-suppressive
effects of TGFβ compromise the ability of the host to resist tumor
progression and thus consist a barrier to immunotherapy.
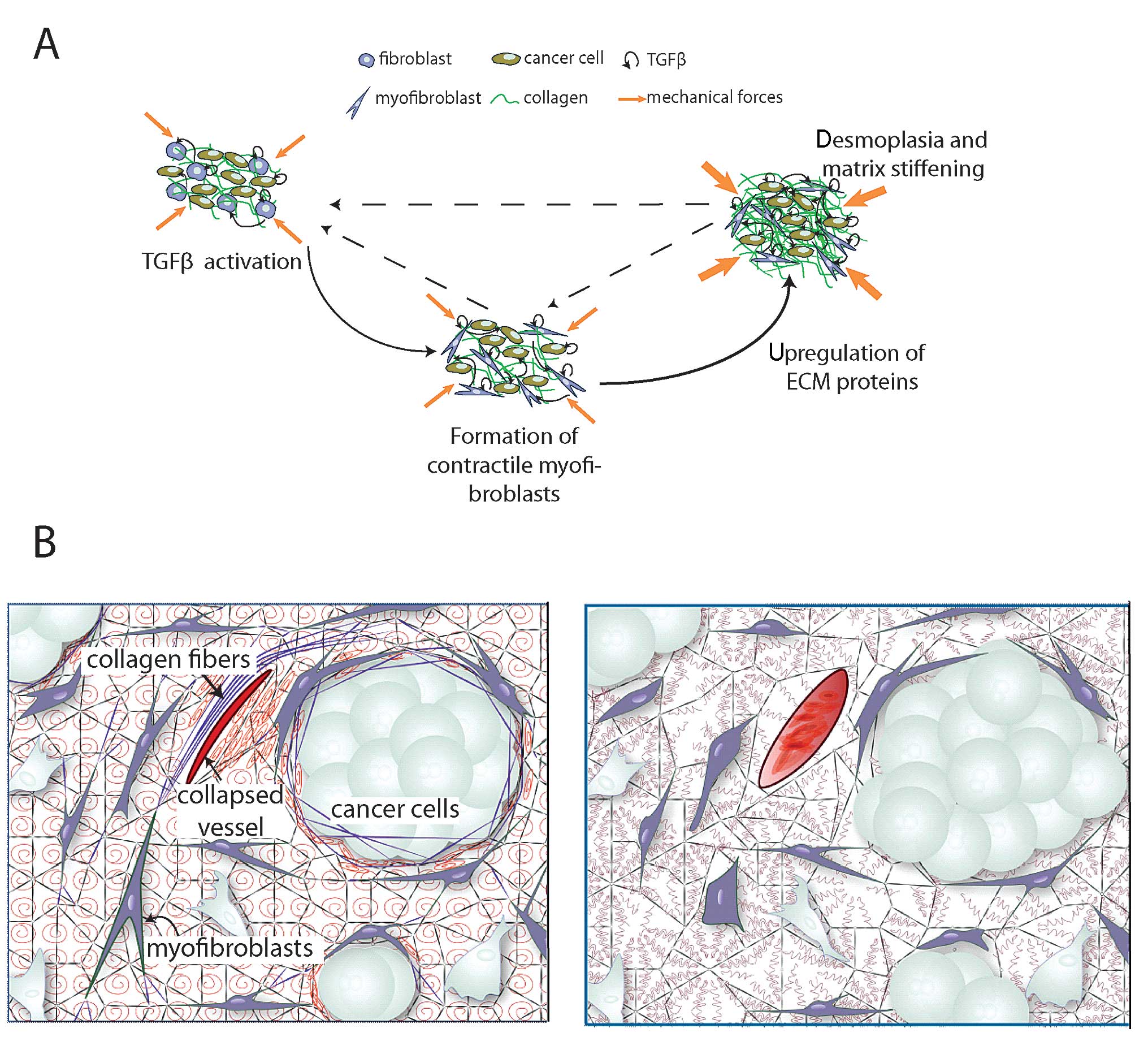
Effect of TGFβ on fibroblasts
A primary role of TGFβ in modulating the tumor
microenvironment is its contribution to the conversion of
fibroblasts to myofibroblasts, also known as CAFs (147,148). Specifically, the compressive
forces developed inside a tumor, due to its growth in the confined
space of the host tissue, can facilitate the conversion of
fibroblasts to proto-myofibroblasts. Subsequently, TGFβ increases
the levels of collagens I and III and fibronectin, which promote
cellular adhesion to extracellular fibers, and thus, enhances the
communication of mechanical signals between the ECM of the tumor
and the fibroblasts (149,150).
As a result, the mechanical forces are more actively transmitted in
the interior of the cell and contribute to the conversion of
proto-myofibroblasts to differentiated myofibroblasts.
Myofibroblasts are characterized by more extensively developed
stress fibers in the cytoskeleton compared to proto-myofibroblasts,
presumably to balance the extracellular forces, and by the de
novo expression of α-SMA. The contraction of myofibroblasts is
sustained by α-SMA stress fibers and it is regulated by Rho/ROCK
signaling activation. The produced contractile forces remodel the
ECM due to the ability of fibroblasts to stretch collagen fibers
and produce ECM molecules (151,152). Additionally, these forces can be
transmitted to the LLC via integrins. LLC is also bound to
extracellular fibers (Fig. 1),
which resists the pulling of the LLC by myofibroblasts and gives
rise to a mechanically-induced liberation of TGFβ (147). The stiffer the ECM, the stronger
the interactions among myofibroblasts, LLC and extracellular fibers
and thus, the release of TGFβ becomes more pronounced. Therefore,
myofibroblast contraction within a collagen-rich, and thus, stiff
microenvironment further stimulates the release of active TGFβ from
its latent form.
Effect of TGFβ on ECM
TGFβ upregulates the expression and synthesis of
many matrix proteins, primarily through the recruitment of
myofibroblast. Proteins upregulated by TGFβ include collagens I–V,
basement membrane proteins (laminin, entactin, perlecan) and ECM
proteins (fibronectin, osteopontin, thrombospontin, tenascin,
osteonectin/SPARC, elastin, biglycan, decorin, and hyaluronan)
(153). Additionally, in the
early stages of carcinogenesis, TGFβ stimulates myofibroblasts and
other stromal cells to enhance the synthesis of collagen
crosslinking enzymes, particularly lysyl oxidase, which increases
the rigidity of the collagen network (154). On the contrary, TGFβ
downregulates the synthesis of matrix-depleting proteins, such as
matrix metalloproteinases (MMP-1, -8, -13). As a result, the
increase in matrix protein synthesis and decrease in matrix
proteinase activity, owing to the TGFβ activity, contributes to the
remodeling of the tumor ECM and can result in a fibrotic response,
known as desmoplasia, which is commonly observed in many types of
tumors and particularly in pancreatic, colon and breast cancers as
well as in various sarcomas (155,156).
Tumor fibrotic response stiffens the tumor tissue,
and as a result, it increases the compressive physical forces in
the interior of the tumor (157).
Compression of cancer cells alters their gene expression profile to
enhance their invasive and metastatic phenotype (158,159). Furthermore, as mentioned
previously, matrix stiffening along with the high contractile
forces of myofibroblasts, cause further liberation of TGFβ from the
LLC. These events suggest a positive feedback loop between TGFβ
activation, myofibroblast contraction and ECM remodeling and
production (Fig. 2A) (148). Finally, compression of
intratumoral blood vessels reduces tumor perfusion, and thus, the
delivery of oxygen (160).
Hypo-perfusion and hypoxia, in turn contribute to immune-evasion,
promote malignant progression and metastasis, and reduce the
efficacy of a number of therapies including radiation treatment and
systemic administration of chemo- and nanotherapy (161–163).
6. TGFβ, tumor desmoplasia and barriers to
drug delivery
The desmoplastic reaction of solid tumors hinders
all three transport steps of the systemic delivery of drugs, namely
vascular, transvascular and interstitial transport (156,163). As mentioned above, increased
levels of collagen in the ECM, result in intratumoral blood vessel
compression and hypo-perfusion. Hypo-perfusion, in turn, reduces
the concentration of the drug that can reach the tumor site. Apart
from compromised drug delivery, hypo-perfusion also decreases the
supply of oxygen rendering the tumor hypoxic, which in turn reduces
the efficacy of radiation therapy. Additionally, desmoplasia
reduces the hydraulic conductivity of the tumor interstitial space,
i.e., the ease with which the interstitial fluid percolates through
the interstitial space of a tissue. High hydraulic conductivity
allows fluid to rapidly flow in the interstitial space and be
drained by peripheral lymphatic vessels. The accumulation of
collagen and other ECM proteins in tumors decrease the available
spaces for interstitial fluid flow and because the fluid cannot
freely move, the interstitial fluid pressure (IFP) increases.
Interstitial hypertension is a hallmark of tumor pathophysiology.
IFP reaches and even exceeds micro-vascular fluid pressure, which
eliminates pressure gradients across the tumor vessel wall and
thus, the transvascular transport of drugs (164). Therefore, the only mechanism of
transport is through diffusion (i.e., due to a concentration
difference), which is inversely proportional to the size of the
therapeutic agent. Chemotherapeutic agents, with a size <1 nm,
are able to diffuse fast and exit the tumor vasculature.
Nanoparticles, however, with sizes >60 nm cannot effectively
extravasate into the tumor interstitial space (165).
Furthermore, the dense interstitial matrix of
desmoplastic tumors hinders the homogeneous distribution of large
nanoparticles. As with transvascular transport, nanoparticles with
a size >60 nm often cannot penetrate deep into the tumor because
their size is comparable to the size of the pores of the
interstitial collagen network and they often get trapped (166). Therefore, even if large
nanoparticles extravassate from the leaky vessels of the tumor,
they will not be able to effectively diffuse into the tissue but
they will concentrate in the perivascular regions, causing only
local effects. Apart from these steric interactions between the
interstitial matrix and nanoparticles, the increased levels of
collagen and hyaluronan give rise to electrostatic interactions.
Indeed, hyaluronan has a highly negative charge, while collagen
fibers carry a slight positive charge. Nanoparticles of a
non-neutral surface charge density can be attracted
electrostatically and bind to these proteins, which further
inhibits their uniform delivery inside the tumor (167).
7. Therapeutic applications of TGFβ
targeting
Pharmacological inhibition of TGFβ has been used in
preclinical and clinical studies as a therapeutic strategy to
either hinder tumor progression directly or modify the tumor
micro-environment in order to improve perfusion and drug delivery
and thus, increase indirectly the efficacy of the treatment. There
is a large number of TGFβ inhibitory drugs employed in these
studies (137). Particularly,
targeting with TGFβ agents (e.g., 1D11, AP12009, SD-208) as well as
non-specific targeting with other TGFβ inhibitory drugs (e.g.,
tranilast) have shown to reduce tumor progression and metastasis
in vivo, mainly owing to augmentation of the immune response
and inhibition of EMT (132,168–171). However, there are also studies
that relate inhibition of TGFβ with promotion of tumor progression
owing to an increase in inflammatory cell infiltration (172). Particularly, it has been shown
that inflammatory infiltrates mediate the pro-tumorigenic functions
of fibroblasts that lack TGFβ signalling. Clinical trials for the
use of TGFβ inhibitory drugs have been in progress
(ClinicalTrials.gov identifiers: NCT00368082, NCT01582269 and
NCT00844064), but their results are not conclusive yet, presumably
owing to differences in the degree of desmoplasia among tumor types
or even among tumors of the same type, but also owing to the
various effects of TGFβ on tumor biology.
Targeting of TGFβ to reduce desmoplasia has the
ability to alleviate physical forces in tumors, decompress tumor
blood vessels and improve perfusion (Fig. 2B) (160). Restoration of tumor perfusion,
however, can increase nutrients supply to the tumor, and thus,
increase its growth rate. Also, the decompressed vessels could
allow more metastatic cells to leave the primary tumor. Indeed, in
some cases, inhibition of TGFβ has been shown to facilitate tumor
progression and metastases in mouse tumor models (173,174), whereas other studies, not related
to TGFβ, have shown a correlation between improved perfusion and
increased metastases (175,176). Therefore, based on this
rationale, judicious doses of TGFβ inhibitory drugs should be used
to alleviate physical forces, decompress blood vessels and improve
perfusion when these agents are combined with cytotoxic treatments,
such as chemo-, nano-, immuno- and radiotherapy. In these combined
treatments the role of the anti-TGFβ drug is to enhance the
delivery of the cytotoxic agent and thus, optimize its efficacy.
This therapeutic strategy is known as stress alleviation treatment
(156,163,165).
Detailed in vivo studies have shown that
re-purposing the anti-hypertensive, angiotensin receptor blocker
(ARB) drug losartan reduced expression of TGFβ1 and decreased
stromal collagen and hyaluronan production, in doses that did not
affect blood pressure. Reduction of collagen and hyaluronan, in
turn, reduced stress levels in the tumor decompressing intratumoral
blood vessels and improving perfusion. Furthermore, reduction of
the ECM components improved the interstitial fluid flow and thus,
reduced levels of IFP. Improved perfusion and reduced IFP enhanced
the delivery and efficacy of chemotherapy in orthotopic breast and
pancreatic murine tumor models (160). Also, in another study combined
treatment of mice bearing tumors with losartan and nanomedicine
(Doxil) increased the distribution of the drug and the overall
survival of the mice (177).
Furthermore, retrospective analyses of clinical data have shown
increased survival in patients with lung or renal cancers treated
with ARBs (178,179). Similar retrospective analysis has
shown that patients with pancreatic ductal adenocarcinomas (PDACs)
receiving ARBs survived ~6 months longer than those who did not
(180). These preclinical and
clinical data have led to a phase II clinical trial with losartan
and FOLFIRINOX in PDAC patients (ClinicalTrials.gov identifier
NCT01821729). Apart from the use of ARBs, the TGFβ neutralizing
antibody 1D11 improved the distribution and efficacy of
therapeutics in breast carcinomas by reducing the tumor stroma
(181). Additionally,
re-purposing the drug pirfenidone, a TGFβ inhibitor clinically
approved for the treatment of idiopathic pulmonary fibrosis, was
shown to suppress desmoplasia in mice bearing pancreatic tumors and
improve the efficacy of chemotherapy (182). Apart from chemotherapy, radiation
therapy has been also improved after treatment with TGFβ
inhibitors. Efficacy of radiotherapy depends on the oxygenation of
the tissue, which is regulated by tumor perfusion (183,184).
8. Conclusions and future perspectives
Owing to the pleiotropic effects of TGFβ on tumor
microenvironment and progression, targeting TGFβ signaling to
directly treat tumor growth remains controversial. Recent studies
have suggested an alternative therapeutic strategy, which involves
the use of anti-TGFβ agents in a stress alleviation treatment. The
scope of this strategy is to hinder but not completely inhibit the
activation of TGFβ ultimately aiming to reduce tumor desmoplasia
and particularly the levels of collagen. As described in this
review, reduced collagen levels can lead to improved delivery of
both chemo- and nano-therapeutics by alleviating mechanical forces
and decompressing intratumoral blood vessels. Thus, blocking of
TGFβ can improve indirectly the efficacy of conventional
treatments. It is promising that many anti-TGFβ agents exist that
are already clinically approved for other diseases (e.g., ARBs for
hypertension). Re-purposing of these drugs can lead to more
effective anti-cancer therapies. Therefore, we need to identify
safe and well-tolerated pharmaceutical agents that may complement
the treatment regimen of cancer patients. Anti-TGFβ agents are not
the only drugs that have the ability to modify the tumor
microenvironment. In principle, any clinically approved agent that
has the ability to reduce collagen levels could be employed as an
alternative strategy. Also, collagen is not the only target for the
stress alleviation treatment. Reduction of stromal cells or
hyaluronan has also the potential to enhance drug delivery through
the same mechanism (157).
Acknowledgements
We thank Dr Christiana Polydorou for useful
discussions. This study received funding from the European Research
Council under the European Union’s Seventh Framework Programme
(FP7/2007-2013)/ERC grant agreement no.
336839-ReEngineeringCancer.
References
|
1
|
Derynck R and Akhurst RJ: Differentiation
plasticity regulated by TGF-beta family proteins in development and
disease. Nat Cell Biol. 9:1000–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wakefield LM and Hill CS: Beyond TGFβ:
roles of other TGFβ superfamily members in cancer. Nat Rev Cancer.
13:328–341. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Massagué J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Annes JP, Munger JS and Rifkin DB: Making
sense of latent TGFbeta activation. J Cell Sci. 116:217–224. 2003.
View Article : Google Scholar
|
|
5
|
Gleizes PE, Beavis RC, Mazzieri R, Shen B
and Rifkin DB: Identification and characterization of an
eight-cysteine repeat of the latent transforming growth factor-beta
binding protein-1 that mediates bonding to the latent transforming
growth factor-beta1. J Biol Chem. 271:29891–29896. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyazono K, Olofsson A, Colosetti P and
Heldin CH: A role of the latent TGF-beta 1-binding protein in the
assembly and secretion of TGF-beta 1. EMBO J. 10:1091–1101.
1991.PubMed/NCBI
|
|
7
|
Saharinen J, Taipale J and Keski-Oja J:
Association of the small latent transforming growth factor-beta
with an eight cysteine repeat of its binding protein LTBP-1. EMBO
J. 15:245–253. 1996.PubMed/NCBI
|
|
8
|
Unsöld C, Hyytiäinen M, Bruckner-Tuderman
L and Keski-Oja J: Latent TGF-beta binding protein LTBP-1 contains
three potential extracellular matrix interacting domains. J Cell
Sci. 114:187–197. 2001.
|
|
9
|
Nunes I, Gleizes PE, Metz CN and Rifkin
DB: Latent transforming growth factor-beta binding protein domains
involved in activation and transglutaminase-dependent cross-linking
of latent transforming growth factor-beta. J Cell Biol.
136:1151–1163. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lawrence DA, Pircher R, Krycève-Martinerie
C and Jullien P: Normal embryo fibroblasts release transforming
growth factors in a latent form. J Cell Physiol. 121:184–188. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crawford SE, Stellmach V, Murphy-Ullrich
JE, et al: Thrombospondin-1 is a major activator of TGF-beta1 in
vivo. Cell. 93:1159–1170. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ribeiro SM, Poczatek M, Schultz-Cherry S,
Villain M and Murphy-Ullrich JE: The activation sequence of
thrombos-pondin-1 interacts with the latency-associated peptide to
regulate activation of latent transforming growth factor-beta. J
Biol Chem. 274:13586–13593. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dubois CM, Laprise MH, Blanchette F,
Gentry LE and Leduc R: Processing of transforming growth factor
beta 1 precursor by human furin convertase. J Biol Chem.
270:10618–10624. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato Y and Rifkin DB: Inhibition of
endothelial cell movement by pericytes and smooth muscle cells:
activation of a latent transforming growth factor-beta 1-like
molecule by plasmin during co-culture. J Cell Biol. 109:309–315.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu Q and Stamenkovic I: Cell
surface-localized matrix metalloproteinase-9 proteolytically
activates TGF-beta and promotes tumor invasion and angiogenesis.
Genes Dev. 14:163–176. 2000.PubMed/NCBI
|
|
16
|
Derynck R, Zhang Y and Feng XH: Smads:
transcriptional activators of TGF-beta responses. Cell. 95:737–740.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Massagué J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Riggins GJ, Thiagalingam S, Rozenblum E,
et al: Mad-related genes in the human. Nat Genet. 13:347–349. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lagna G, Hata A, Hemmati-Brivanlou A and
Massagué J: Partnership between DPC4 and SMAD proteins in TGF-beta
signalling pathways. Nature. 383:832–836. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakao A, Imamura T, Souchelnytskyi S, et
al: TGF-beta receptor-mediated signalling through Smad2, Smad3 and
Smad4. EMBO J. 16:5353–5362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bierie B and Moses HL: Tumour
microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.
Nat Rev Cancer. 6:506–520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewis KA, Gray PC, Blount AL, et al:
Betaglycan binds inhibin and can mediate functional antagonism of
activin signalling. Nature. 404:411–414. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wrana JL, Attisano L, Wieser R, Ventura F
and Massagué J: Mechanism of activation of the TGF-beta receptor.
Nature. 370:341–347. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miyazono K, Maeda S and Imamura T: BMP
receptor signaling: transcriptional targets, regulation of signals,
and signaling cross-talk. Cytokine Growth Factor Rev. 16:251–263.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abdollah S, Macías-Silva M, Tsukazaki T,
Hayashi H, Attisano L and Wrana JL: TbetaRI phosphorylation of
Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex
formation and signaling. J Biol Chem. 272:27678–27685. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Feng X, We R and Derynck R:
Receptor-associated Mad homologues synergize as effectors of the
TGF-beta response. Nature. 383:168–172. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsukazaki T, Chiang TA, Davison AF,
Attisano L and Wrana JL: SARA, a FYVE domain protein that recruits
Smad2 to the TGFbeta receptor. Cell. 95:779–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng XH, Zhang Y, Wu RY and Derynck R: The
tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300
are coactivators for smad3 in TGF-beta-induced transcriptional
activation. Genes Dev. 12:2153–2163. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Janknecht R, Wells NJ and Hunter T:
TGF-beta-stimulated cooperation of smad proteins with the
coactivators CBP/p300. Genes Dev. 12:2114–2119. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Itoh S, Ericsson J, Nishikawa J, Heldin CH
and ten Dijke P: The transcriptional co-activator P/CAF potentiates
TGF-beta/Smad signaling. Nucleic Acids Res. 28:4291–4298. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bai RY, Koester C, Ouyang T, et al: SMIF,
a Smad4-interacting protein that functions as a co-activator in
TGFbeta signalling. Nat Cell Biol. 4:181–190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CR, Kang Y, Siegel PM and Massagué J:
E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to
c-myc repression. Cell. 110:19–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang Y, Chen CR and Massagué J: A
self-enabling TGFbeta response coupled to stress signaling: Smad
engages stress response factor ATF3 for Id1 repression in
epithelial cells. Mol Cell. 11:915–926. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wotton D, Knoepfler PS, Laherty CD,
Eisenman RN and Massagué J: The Smad transcriptional corepressor
TGIF recruits mSin3. Cell Growth Differ. 12:457–463.
2001.PubMed/NCBI
|
|
38
|
Akiyoshi S, Inoue H, Hanai J, et al: c-Ski
acts as a transcriptional co-repressor in transforming growth
factor-beta signaling through interaction with smads. J Biol Chem.
274:35269–35277. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luo K, Stroschein SL, Wang W, et al: The
Ski oncoprotein interacts with the Smad proteins to repress TGFbeta
signaling. Genes Dev. 13:2196–2206. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stroschein SL, Wang W, Zhou S, Zhou Q and
Luo K: Negative feedback regulation of TGF-beta signaling by the
SnoN onco-protein. Science. 286:771–774. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun Y, Liu X, Eaton EN, Lane WS, Lodish HF
and Weinberg RA: Interaction of the Ski oncoprotein with Smad3
regulates TGF-beta signaling. Mol Cell. 4:499–509. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Seoane J, Le HV, Shen L, Anderson SA and
Massagué J: Integration of Smad and forkhead pathways in the
control of neuroepithelial and glioblastoma cell proliferation.
Cell. 117:211–223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pardali K, Kurisaki A, Morén A, ten Dijke
P, Kardassis D and Moustakas A: Role of Smad proteins and
transcription factor Sp1 in p21(Waf1/Cip1) regulation by
transforming growth factor-beta. J Biol Chem. 275:29244–29256.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Feng XH and Derynck R: Smad3 and
Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced
transcription. Nature. 394:909–913. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin X, Liang YY, Sun B, et al: Smad6
recruits transcription corepressor CtBP to repress bone
morphogenetic protein-induced transcription. Mol Cell Biol.
23:9081–9093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Peng Y, Zhao S, Song L, Wang M and Jiao K:
Sertad1 encodes a novel transcriptional co-activator of SMAD1 in
mouse embryonic hearts. Biochem Biophys Res Commun. 441:751–756.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Izutsu K, Kurokawa M, Imai Y, Maki K,
Mitani K and Hirai H: The corepressor CtBP interacts with Evi-1 to
repress transforming growth factor beta signaling. Blood.
97:2815–2822. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xi Q, Wang Z, Zaromytidou AI, et al: A
poised chromatin platform for TGF-β access to master regulators.
Cell. 147:1511–1524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ross S, Cheung E, Petrakis TG, Howell M,
Kraus WL and Hill CS: Smads orchestrate specific histone
modifications and chromatin remodeling to activate transcription.
EMBO J. 25:4490–4502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Papageorgis P, Lambert AW, Ozturk S, et
al: Smad signaling is required to maintain epigenetic silencing
during breast cancer progression. Cancer Res. 70:968–978. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nakao A, Afrakhte M, Morén A, et al:
Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta
signalling. Nature. 389:631–635. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
52
|
Itóh S, Landström M, Hermansson A, et al:
Transforming growth factor beta1 induces nuclear export of
inhibitory Smad7. J Biol Chem. 273:29195–29201. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hayashi H, Abdollah S, Qiu Y, et al: The
MAD-related protein Smad7 associates with the TGFbeta receptor and
functions as an antagonist of TGFbeta signaling. Cell.
89:1165–1173. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ebisawa T, Fukuchi M, Murakami G, et al:
Smurf1 interacts with transforming growth factor-beta type I
receptor through Smad7 and induces receptor degradation. J Biol
Chem. 276:12477–12480. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kavsak P, Rasmussen RK, Causing CG, et al:
Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets
the TGF beta receptor for degradation. Mol Cell. 6:1365–1375. 2000.
View Article : Google Scholar
|
|
56
|
Zhang S, Fei T, Zhang L, et al: Smad7
antagonizes transforming growth factor beta signaling in the
nucleus by interfering with functional Smad-DNA complex formation.
Mol Cell Biol. 27:4488–4499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar :
|
|
58
|
Hartsough MT and Mulder KM: Transforming
growth factor beta activation of p44mapk in proliferating cultures
of epithelial cells. J Biol Chem. 270:7117–7124. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Frey RS and Mulder KM: TGFbeta regulation
of mitogen-activated protein kinases in human breast cancer cells.
Cancer Lett. 117:41–50. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Papageorgis P, Cheng K, Ozturk S, et al:
Smad4 inactivation promotes malignancy and drug resistance of colon
cancer. Cancer Res. 71:998–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Finlay GA, Thannickal VJ, Fanburg BL and
Paulson KE: Transforming growth factor-beta 1-induced activation of
the ERK pathway/activator protein-1 in human lung fibroblasts
requires the autocrine induction of basic fibroblast growth factor.
J Biol Chem. 275:27650–27656. 2000.PubMed/NCBI
|
|
62
|
Vinals F and Pouysségur J: Transforming
growth factor beta1 (TGF-beta1) promotes endothelial cell survival
during in vitro angiogenesis via an autocrine mechanism implicating
TGF-alpha signaling. Mol Cell Biol. 21:7218–7230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ellenrieder V, Hendler SF, Boeck W, et al:
Transforming growth factor beta1 treatment leads to an
epithelial-mesenchymal transdifferentiation of pancreatic cancer
cells requiring extra-cellular signal-regulated kinase 2
activation. Cancer Res. 61:4222–4228. 2001.PubMed/NCBI
|
|
64
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lee MK, Pardoux C, Hall MC, et al:
TGF-beta activates Erk MAP kinase signalling through direct
phosphorylation of ShcA. EMBO J. 26:3957–3967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liao JH, Chen JS, Chai MQ, Zhao S and Song
JG: The involvement of p38 MAPK in transforming growth factor
beta1-induced apoptosis in murine hepatocytes. Cell Res. 11:89–94.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kimura N, Matsuo R, Shibuya H, Nakashima K
and Taga T: BMP2-induced apoptosis is mediated by activation of the
TAK1-p38 kinase pathway that is negatively regulated by Smad6. J
Biol Chem. 275:17647–17652. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFbeta-mediated fibroblastic transdifferentiation and cell
migration. J Cell Sci. 115:3193–3206. 2002.PubMed/NCBI
|
|
69
|
Hocevar BA, Brown TL and Howe PH: TGF-beta
induces fibronectin synthesis through a c-Jun N-terminal
kinase-dependent, Smad4-independent pathway. EMBO J. 18:1345–1356.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yu L, Hébert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent
TGF-beta responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yamaguchi K, Shirakabe K, Shibuya H, et
al: Identification of a member of the MAPKKK family as a potential
mediator of TGF-beta signal transduction. Science. 270:2008–2011.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shim JH, Xiao C, Paschal AE, et al: TAK1,
but not TAB1 or TAB2, plays an essential role in multiple signaling
pathways in vivo. Genes Dev. 19:2668–2681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sorrentino A, Thakur N, Grimsby S, et al:
The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a
receptor kinase-independent manner. Nat Cell Biol. 10:1199–1207.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yamashita M, Fatyol K, Jin C, Wang X, Liu
Z and Zhang YE: TRAF6 mediates Smad-independent activation of JNK
and p38 by TGF-beta. Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang L, Wang W, Hayashi Y, et al: A role
for MEK kinase 1 in TGF-beta/activin-induced epithelium movement
and embryonic eyelid closure. EMBO J. 22:4443–4454. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kim KY, Kim BC, Xu Z and Kim SJ: Mixed
lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates
transforming growth factor-beta-induced apoptosis in hepatoma
cells. J Biol Chem. 279:29478–29484. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jaffe AB and Hall A: Rho GTPases:
biochemistry and biology. Annu Rev Cell Dev Biol. 21:247–269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bhowmick NA, Ghiassi M, Bakin A, et al:
Transforming growth factor-beta1 mediates epithelial to mesenchymal
transdifferentiation through a RhoA-dependent mechanism. Mol Biol
Cell. 12:27–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Edlund S, Landström M, Heldin CH and
Aspenström P: Transforming growth factor-beta-induced mobilization
of actin cytoskeleton requires signaling by small GTPases Cdc42 and
RhoA. Mol Biol Cell. 13:902–914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ozdamar B, Bose R, Barrios-Rodiles M, Wang
HR, Zhang Y and Wrana JL: Regulation of the polarity protein Par6
by TGFbeta receptors controls epithelial cell plasticity. Science.
307:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor beta-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shin I, Bakin AV, Rodeck U, Brunet A and
Arteaga CL: Transforming growth factor beta enhances epithelial
cell survival via Akt-dependent regulation of FKHRL1. Mol Biol
Cell. 12:3328–3339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hidalgo M and Rowinsky EK: The
rapamycin-sensitive signal transduction pathway as a target for
cancer therapy. Oncogene. 19:6680–6686. 2000. View Article : Google Scholar
|
|
84
|
Lamouille S and Derynck R: Cell size and
invasion in TGF-beta-induced epithelial to mesenchymal transition
is regulated by activation of the mTOR pathway. J Cell Biol.
178:437–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Roberts AB and Wakefield LM: The two faces
of transforming growth factor beta in carcinogenesis. Proc Natl
Acad Sci USA. 100:8621–8623. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tang B, Vu M, Booker T, et al: TGF-beta
switches from tumor suppressor to prometastatic factor in a model
of breast cancer progression. J Clin Invest. 112:1116–1124. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wakefield LM and Roberts AB: TGF-beta
signaling: positive and negative effects on tumorigenesis. Curr
Opin Genet Dev. 12:22–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Siegel PM, Shu W, Cardiff RD, Muller WJ
and Massagué J: Transforming growth factor beta signaling impairs
Neu-induced mammary tumorigenesis while promoting pulmonary
metastasis. Proc Natl Acad Sci USA. 100:8430–8435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Siegel PM and Massagué J: Cytostatic and
apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Choi ME and Ballermann BJ: Inhibition of
capillary morphogenesis and associated apoptosis by dominant
negative mutant transforming growth factor-beta receptors. J Biol
Chem. 270:21144–21150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hyman KM, Seghezzi G, Pintucci G, et al:
Transforming growth factor-beta1 induces apoptosis in vascular
endothelial cells by activation of mitogen-activated protein
kinase. Surgery. 132:173–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Rich JN, Zhang M, Datto MB, Bigner DD and
Wang XF: Transforming growth factor-beta-mediated p15(INK4B)
induction and growth inhibition in astrocytes is SMAD3-dependent
and a pathway prominently altered in human glioma cell lines. J
Biol Chem. 274:35053–35058. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yang X, Letterio JJ, Lechleider RJ, et al:
Targeted disruption of SMAD3 results in impaired mucosal immunity
and diminished T cell responsiveness to TGF-beta. EMBO J.
18:1280–1291. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Laiho M, DeCaprio JA, Ludlow JW,
Livingston DM and Massagué J: Growth inhibition by TGF-beta linked
to suppression of retinoblastoma protein phosphorylation. Cell.
62:175–185. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Hannon GJ and Beach D: p15INK4B is a
potential effector of TGF-beta-induced cell cycle arrest. Nature.
371:257–261. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y
and Wang XF: Transforming growth factor beta induces the
cyclin-dependent kinase inhibitor p21 through a p53-independent
mechanism. Proc Natl Acad Sci USA. 92:5545–5549. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Polyak K, Kato JY, Solomon MJ, et al:
p27Kip1, a cyclin-Cdk inhibitor, links transforming growth
factor-beta and contact inhibition to cell cycle arrest. Genes Dev.
8:9–22. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pietenpol JA, Stein RW, Moran E, et al:
TGF-beta 1 inhibition of c-myc transcription and growth in
keratinocytes is abrogated by viral transforming proteins with pRB
binding domains. Cell. 61:777–785. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
100
|
Grotendorst GR: Connective tissue growth
factor: a mediator of TGF-beta action on fibroblasts. Cytokine
Growth Factor Rev. 8:171–179. 1997. View Article : Google Scholar
|
|
101
|
Park K, Kim SJ, Bang YJ, et al: Genetic
changes in the transforming growth factor beta (TGF-beta) type II
receptor gene in human gastric cancer cells: correlation with
sensitivity to growth inhibition by TGF-beta. Proc Natl Acad Sci
USA. 91:8772–8776. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kim IY, Ahn HJ, Zelner DJ, et al: Genetic
change in transforming growth factor beta (TGF-beta) receptor type
I gene correlates with insensitivity to TGF-beta 1 in human
prostate cancer cells. Cancer Res. 56:44–48. 1996.PubMed/NCBI
|
|
103
|
Markowitz S, Wang J, Myeroff L, et al:
Inactivation of the type II TGF-beta receptor in colon cancer cells
with microsatellite instability. Science. 268:1336–1338. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Riggins GJ, Kinzler KW, Vogelstein B and
Thiagalingam S: Frequency of Smad gene mutations in human cancers.
Cancer Res. 57:2578–2580. 1997.PubMed/NCBI
|
|
105
|
Schutte M, Hruban RH, Hedrick L, et al:
DPC4 gene in various tumor types. Cancer Res. 56:2527–2530.
1996.PubMed/NCBI
|
|
106
|
Eppert K, Scherer SW, Ozcelik H, et al:
MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related
protein that is functionally mutated in colorectal carcinoma. Cell.
86:543–552. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Hahn SA, Hoque AT, Moskaluk CA, et al:
Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer
Res. 56:490–494. 1996.PubMed/NCBI
|
|
108
|
Hahn SA, Schutte M, Hoque AT, et al: DPC4,
a candidate tumor suppressor gene at human chromosome 18q21.1.
Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Thiagalingam S, Lengauer C, Leach FS, et
al: Evaluation of candidate tumour suppressor genes on chromosome
18 in colorectal cancers. Nat Genet. 13:343–346. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Schwarte-Waldhoff I, Volpert OV, Bouck NP,
et al: Smad4/DPC4-mediated tumor suppression through suppression of
angiogenesis. Proc Natl Acad Sci USA. 97:9624–9629. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kretzschmar M, Doody J, Timokhina I and
Massagué J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kretzschmar M, Doody J and Massagué J:
Opposing BMP and EGF signalling pathways converge on the TGF-beta
family mediator Smad1. Nature. 389:618–622. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Massagué J: Integration of Smad and MAPK
pathways: a link and a linker revisited. Genes Dev. 17:2993–2997.
2003. View Article : Google Scholar
|
|
114
|
Gomis RR, Alarcón C, Nadal C, Van Poznak C
and Massagué J: C/EBPbeta at the core of the TGFbeta cytostatic
response and its evasion in metastatic breast cancer cells. Cancer
Cell. 10:203–214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Padua D, Zhang XH, Wang Q, et al: TGFbeta
primes breast tumors for lung metastasis seeding through
angiopoietin-like 4. Cell. 133:66–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yang J, Mani SA, Donaher JL, et al: Twist,
a master regulator of morphogenesis, plays an essential role in
tumor metastasis. Cell. 117:927–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Cano A, Pérez-Moreno MA, Rodrigo I, et al:
The transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Savagner P, Yamada KM and Thiery JP: The
zinc-finger protein slug causes desmosome dissociation, an initial
and necessary step for growth factor-induced epithelial-mesenchymal
transition. J Cell Biol. 137:1403–1419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Eger A, Aigner K, Sonderegger S, et al:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Comijn J, Berx G, Vermassen P, et al: The
two-handed E box binding zinc finger protein SIP1 downregulates
E-cadherin and induces invasion. Mol Cell. 7:1267–1278. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Thuault S, Valcourt U, Petersen M,
Manfioletti G, Heldin CH and Moustakas A: Transforming growth
factor-beta employs HMGA2 to elicit epithelial-mesenchymal
transition. J Cell Biol. 174:175–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Mani SA, Yang J, Brooks M, et al:
Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is
associated with aggressive basal-like breast cancers. Proc Natl
Acad Sci USA. 104:10069–10074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhu B, Fukada K, Zhu H and Kyprianou N:
Prohibitin and cofilin are intracellular effectors of transforming
growth factor beta signaling in human prostate cancer cells. Cancer
Res. 66:8640–8647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Deckers M, van Dinther M, Buijs J, et al:
The tumor suppressor Smad4 is required for transforming growth
factor beta-induced epithelial to mesenchymal transition and bone
metastasis of breast cancer cells. Cancer Res. 66:2202–2209. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Grande JP: Role of transforming growth
factor-beta in tissue injury and repair. Proc Soc Exp Biol Med.
214:27–40. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Singer AJ and Clark RA: Cutaneous wound
healing. N Engl J Med. 341:738–746. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Pickup M, Novitskiy S and Moses HL: The
roles of TGFβ in the tumour microenvironment. Nat Rev Cancer.
13:788–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Dalal BI, Keown PA and Greenberg AH:
Immunocytochemical localization of secreted transforming growth
factor-beta 1 to the advancing edges of primary tumors and to lymph
node metastases of human mammary carcinoma. Am J Pathol.
143:381–389. 1993.PubMed/NCBI
|
|
134
|
Kingsley LA, Fournier PG, Chirgwin JM and
Guise TA: Molecular biology of bone metastasis. Mol Cancer Ther.
6:2609–2617. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Prud’homme GJ: Pathobiology of
transforming growth factor beta in cancer, fibrosis and immunologic
disease, and therapeutic considerations. Lab Invest. 87:1077–1091.
2007. View Article : Google Scholar
|
|
136
|
Wrzesinski SH, Wan YY and Flavell RA:
Transforming growth factor-beta and the immune response:
implications for anti-cancer therapy. Clin Cancer Res.
13:5262–5270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Flavell RA, Sanjabi S, Wrzesinski SH and
Licona-Limón P: The polarization of immune cells in the tumour
environment by TGFbeta. Nat Rev Immunol. 10:554–567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Laouar Y, Sutterwala FS, Gorelik L and
Flavell RA: Transforming growth factor-beta controls T helper type
1 cell development through regulation of natural killer cell
interferon-gamma. Nat Immunol. 6:600–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Rubtsov YP and Rudensky AY: TGFbeta
signalling in control of T-cell-mediated self-reactivity. Nat Rev
Immunol. 7:443–453. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Gong D, Shi W, Yi SJ, Chen H, Groffen J
and Heisterkamp N: TGFβ signaling plays a critical role in
promoting alternative macrophage activation. BMC Immunol.
13:312012. View Article : Google Scholar
|
|
143
|
Fridlender ZG, Sun J, Kim S, et al:
Polarization of tumor-associated neutrophil phenotype by TGF-beta:
“N1” versus “N2” TAN. Cancer Cell. 16:183–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yamaguchi Y, Tsumura H, Miwa M and Inaba
K: Contrasting effects of TGF-beta 1 and TNF-alpha on the
development of dendritic cells from progenitors in mouse bone
marrow. Stem Cells. 15:144–153. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ramesh S, Wildey GM and Howe PH:
Transforming growth factor beta (TGFbeta)-induced apoptosis: the
rise & fall of Bim. Cell Cycle. 8:11–17. 2009. View Article : Google Scholar
|
|
146
|
Marcoe JP, Lim JR, Schaubert KL, et al:
TGF-β is responsible for NK cell immaturity during ontogeny and
increased susceptibility to infection during mouse infancy. Nat
Immunol. 13:843–850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wipff PJ, Rifkin DB, Meister JJ and Hinz
B: Myofibroblast contraction activates latent TGF-beta1 from the
extracellular matrix. J Cell Biol. 179:1311–1323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Wipff PJ and Hinz B: Myofibroblasts work
best under stress. J Bodyw Mov Ther. 13:121–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier
C and Brown RA: Myofibroblasts and mechano-regulation of connective
tissue remodelling. Nat Rev Mol Cell Biol. 3:349–363. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Karagiannis GS, Poutahidis T, Erdman SE,
Kirsch R, Riddell RH and Diamandis EP: Cancer-associated
fibroblasts drive the progression of metastasis through both
paracrine and mechanical pressure on cancer tissue. Mol Cancer Res.
10:1403–1418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Paszek MJ, Zahir N, Johnson KR, et al:
Tensional homeostasis and the malignant phenotype. Cancer Cell.
8:241–254. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Samuel MS, Lopez JI, McGhee EJ, et al:
Actomyosin-mediated cellular tension drives increased tissue
stiffness and β-catenin activation to induce epidermal hyperplasia
and tumor growth. Cancer Cell. 19:776–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Branton MH and Kopp JB: TGF-beta and
fibrosis. Microbes Infect. 1:1349–1365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Egeblad M, Rasch MG and Weaver VM: Dynamic
interplay between the collagen scaffold and tumor evolution. Curr
Opin Cell Biol. 22:697–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Smith NR, Baker D, Farren M, et al: Tumor
stromal architecture can define the intrinsic tumor response to
VEGF-targeted therapy. Clin Cancer Res. 19:6943–6956. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Stylianopoulos T and Jain RK: Combining
two strategies to improve perfusion and drug delivery in solid
tumors. Proc Natl Acad Sci USA. 110:18632–18637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Stylianopoulos T, Martin JD, Chauhan VP,
et al: Causes, consequences, and remedies for growth-induced solid
stress in murine and human tumors. Proc Natl Acad Sci USA.
109:15101–15108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Demou ZN: Gene expression profiles in 3D
tumor analogs indicate compressive strain differentially enhances
metastatic potential. Ann Biomed Eng. 38:3509–3520. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Tse JM, Cheng G, Tyrrell JA, et al:
Mechanical compression drives cancer cells toward invasive
phenotype. Proc Natl Acad Sci USA. 109:911–916. 2012. View Article : Google Scholar :
|
|
160
|
Chauhan VP, Martin JD, Liu H, et al:
Angiotensin inhibition enhances drug delivery and potentiates
chemotherapy by decompressing tumor blood vessels. Nat Commun.
4:25162013. View Article : Google Scholar
|
|
161
|
Facciabene A, Peng X, Hagemann IS, et al:
Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and
T(reg) cells. Nature. 475:226–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Wilson WR and Hay MP: Targeting hypoxia in
cancer therapy. Nat Rev Cancer. 11:393–410. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Jain RK, Martin JD and Stylianopoulos T:
The role of mechanical forces in tumor growth and therapy. Annu Rev
Biomed Eng. 16:321–346. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Jain RK and Stylianopoulos T: Delivering
nanomedicine to solid tumors. Nat Rev Clin Oncol. 7:653–664. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Chauhan VP and Jain RK: Strategies for
advancing cancer nanomedicine. Nat Mater. 12:958–962. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Popovi Z, Liu W, Chauhan VP, et al: A
nanoparticle size series for in vivo fluorescence imaging. Angew
Chem Int Ed Engl. 49:8649–8652. 2010. View Article : Google Scholar
|
|
167
|
Stylianopoulos T, Poh MZ, Insin N, et al:
Diffusion of particles in the extracellular matrix: the effect of
repulsive electrostatic interactions. Biophys J. 99:1342–1349.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhong Z, Carroll KD, Policarpio D, et al:
Anti-transforming growth factor beta receptor II antibody has
therapeutic efficacy against primary tumor growth and metastasis
through multi-effects on cancer, stroma, and immune cells. Clin
Cancer Res. 16:1191–1205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Uhl M, Aulwurm S, Wischhusen J, et al:
SD-208, a novel transforming growth factor beta receptor I kinase
inhibitor, inhibits growth and invasiveness and enhances
immunogenicity of murine and human glioma cells in vitro and in
vivo. Cancer Res. 64:7954–7961. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Kim S, Buchlis G, Fridlender ZG, et al:
Systemic blockade of transforming growth factor-beta signaling
augments the efficacy of immunogene therapy. Cancer Res.
68:10247–10256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Chakrabarti R, Subramaniam V, Abdalla S,
Jothy S and Prud’homme GJ: Tranilast inhibits the growth and
metastasis of mammary carcinoma. Anticancer Drugs. 20:334–345.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Achyut BR, Bader DA, Robles AI, et al:
Inflammation-mediated genetic and epigenetic alterations drive
cancer development in the neighboring epithelium upon stromal
abrogation of TGF-β signaling. PLoS Genet. 9:e10032512013.
View Article : Google Scholar
|
|
173
|
Bragado P, Estrada Y, Parikh F, et al:
TGF-β2 dictates disseminated tumour cell fate in target organs
through TGF-β-RIII and p38α/β signalling. Nat Cell Biol.
15:1351–1361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Biswas T, Gu X, Yang J, Ellies LG and Sun
LZ: Attenuation of TGF-β signaling supports tumor progression of a
mesenchymal-like mammary tumor cell line in a syngeneic murine
model. Cancer Lett. 346:129–138. 2014. View Article : Google Scholar
|
|
175
|
Stockmann C, Doedens A, Weidemann A, et
al: Deletion of vascular endothelial growth factor in myeloid cells
accelerates tumorigenesis. Nature. 456:814–818. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Rhim AD, Mirek ET, Aiello NM, et al: EMT
and dissemination precede pancreatic tumor formation. Cell.
148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Diop-Frimpong B, Chauhan VP, Krane S,
Boucher Y and Jain RK: Losartan inhibits collagen I synthesis and
improves the distribution and efficacy of nanotherapeutics in
tumors. Proc Natl Acad Sci USA. 108:2909–2914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Wilop S, von Hobe S, Crysandt M, Esser A,
Osieka R and Jost E: Impact of angiotensin I converting enzyme
inhibitors and angiotensin II type 1 receptor blockers on survival
in patients with advanced non-small-cell lung cancer undergoing
first-line platinum-based chemotherapy. J Cancer Res Clin Oncol.
135:1429–1435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Keizman D, Huang P, Eisenberger MA, et al:
Angiotensin system inhibitors and outcome of sunitinib treatment in
patients with metastatic renal cell carcinoma: a retrospective
examination. Eur J Cancer. 47:1955–1961. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Nakai Y, Isayama H, Ijichi H, et al: Phase
I trial of gemcitabine and candesartan combination therapy in
normotensive patients with advanced pancreatic cancer: GECA1.
Cancer Sci. 103:1489–1492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Liu J, Liao S, Diop-Frimpong B, et al:
TGF-β blockade improves the distribution and efficacy of
therapeutics in breast carcinoma by normalizing the tumor stroma.
Proc Natl Acad Sci USA. 109:16618–16623. 2012. View Article : Google Scholar
|
|
182
|
Kozono S, Ohuchida K, Eguchi D, et al:
Pirfenidone inhibits pancreatic cancer desmoplasia by regulating
stellate cells. Cancer Res. 73:2345–2356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Bouquet F, Pal A, Pilones KA, et al: TGFβ1
inhibition increases the radiosensitivity of breast cancer cells in
vitro and promotes tumor control by radiation in vivo. Clin Cancer
Res. 17:6754–6765. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Zhang M, Kleber S, Röhrich M, et al:
Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor
LY2109761 enhances radiation response and prolongs survival in
glioblastoma. Cancer Res. 71:7155–7167. 2011. View Article : Google Scholar : PubMed/NCBI
|