Introduction
The seriousness of hepatitis C virus (HCV)
infections lies in their elevation of risks pertaining to various
life-threatening liver conditions such as liver cirrhosis (LC),
chronic hepatitis (CH) as well as hepatocellular carcinoma (HCC).
For instance, the estimated risk of hepatocellular carcinoma has
been reported to be often 15 to 20 times higher in subjects
infected with HCV (1). Statistics
also suggest that 75–85% of people infected will become chronic
carriers, and 10–15% of HCV infection cases will advance to
cirrhosis within the first 20 years (2), along with increased risks of
developing HCC. In Japan, ~80% of patients suffering from HCC is
caused by prior or concurrent HCV (3), suggesting potential links between
possible genetic predisposition and infection incidence. While
genetic aberrations can direct and drive carcinogenesis, exact
details, especially with the ethnic Japanese population, linking
HCV virulence to HCC susceptibility (and to a lesser extent LC and
CH) remain opaque. Preventative treatment options for HCV are also
lacking in a similar regard, although a meta-analysis by Colombo
and Iavarone had suggested the possibility of reducing HCC risks in
a small fraction of cases with interferon administration (4). To-date, while various studies have
sought to characterize the effect of genotypic alterations in liver
diseases, the role of SNPs in liver diseases still remain poorly
understood (5–11). Additionally, while genes such as
IFNγ and IL-28B have been reported to harbor potential associations
to HCV infections leading to conditions such as fibrosis and
jaundice (12–14), no clinical screening and
characterization studies have been conducted between HCV and severe
liver conditions such as CH, LC and HCC.
With an estimated 143 million people infected with
hepatitis C worldwide (15), early
genetic screening can have a beneficial role in greatly improving
the quality of life for those afflicted with liver diseases. With
the establishment of the BioBank Japan Project that stores and
maintains a number of annotated liver disease cases, we decide to
explore the possibility, if any, that certain genetic risk factors
are associated with the prevalence of HCV infections to
hepatocellular carcinoma in the Japanese population by MassARRAY
genotyping, a MALDI-TOF mass spectrometry-based approach well
demonstrated in medium- to large-scale cohort studies (16) to analyze a set of specific
polymorphisms. We herein present one of the earliest studies
attempting to assess several clinically relevant marker gene and
variant candidates linking HCV to liver cancer, as well as CH and
LC for the purpose of comparison, in the Japanese population using
DNA extracted from blood specimens. For each candidate, we then
assessed its significance by comparing genotypic frequencies
against records from the Japan Single Nucleotide Polymorphisms
databank (JSNP), NCBI dbSNP and the Tohoku Medical Megabank (TMM)
datasets (17–20) to elucidate their potential roles as
risk factors in HCV-induced liver diseases and their pertinence in
other ethnic groups.
Material and methods
SNP selection
Among published SNPs and polymorphisms in genes
relevant to cancer in recent literature, we generated a preliminary
list of SNP candidates for screening. To broaden the scope, this
list was later expanded to include a small number of SNPs that had
few reports of clinical significance to-date, but were
well-conserved, disease-associated and believed to impact protein
structure by SNPs3D searches (21). In all, 131 SNPs over 4 primer sets
were found to meet the MassARRAY prerequisites and selected for
analysis (tabulated data of SNPs and corresponding primers
available upon request).
Specimen collection and subject
demographics
Blood DNA samples from a collection of CH (200), LC
(80) and HCC (200) subjects were obtained from the BioBank Japan
Project (22). Hepatitis subjects
had a gender makeup of 103/97 male/female, with 102 over the age of
50 (mean 54±15). Samples were confirmed to be HCV-negative and
cancer-free. The Cirrhosis test group consisted of 45 males and 35
females, with 71 people over the age of 50 (mean 65±11) and
similarly confirmed to be HCV-negative and cancer-free. Among HCC
patients of 103 men and 97 women, 194 were over 50 (mean 69±8) and
all cases were confirmed to be HCV-positive. No other medical
history (including history of hepatitis virus B infection) or
personal information was obtained. For the purpose of statistical
assessment, a simulated control ('healthy') set based on JSNP (last
updated May, 2014) and NCBI data (build 142, October, 2014) was
used. In cases where multiple genotypes exist from both JSNP and
NCBI, prevailing JSNP results were used for the control set. The
TMM 2KJPN database (release June, 2016) was also consulted for
comparative analysis of variant allele frequencies for the Japanese
population.
SNP detection by single-base
extension
DNA specimens (5 µg) were diluted to the
stock concentration of 10 ng/µl prior to PCR using the iPLEX
Gold reagent kit (Sequenom, San Diego, CA, USA). Each specimen (1
µl) was mixed with the primer mix (500 nM) prior to SNP
isolation, and 6/96 samples were then randomly selected to confirm
the extent of PCR by gel electrophoresis. Four primer mixes, each
containing 33 SNPs, were prepared to the same final composition per
Sequenom's instructions. PCR products underwent shrimp alkaline
phosphatase (0.5 unit) treatment to dephosphorylate unincorporated
deoxynucleotides at 37°C for 40 min followed by 85°C for 5 min.
Extension reactions were performed following the iPLEX-Extend
protocol by preparing each sample in a solution of 0.62 µl
water, 0.2 µl 10X iPLEX buffer plus, 0.2 µl iPLEX
termination mix, 0.94 µl extend primer mix, 0.04 µl
iPLEX enzyme for reaction as follows: 94°C (step 1, 30 sec), 94°C
(step 2, 5 sec), 52°C (step 3, 5 sec), 80°C (step 4, 5 sec), in
which steps III and IV were repeated for 5 cycles followed by 40
cycles of steps II–IV before the final step at 72°C for 3 min.
Following instructions from the SpectroChip Chip and Resin kit
(Sequenom), samples were characterized by mass spectrometric
analysis on a MassARRAY compact MALDI-TOF mass spectrometric
analyzer. Oligonucleotides were binned with replicates per
manufacturer's recommendations to minimize calling failures. Data
were acquired and processed using EpiTYPER 1.0 in the Sequenom
MassARRAY Workstation suite. For illustrative purposes, some data
and cluster plot/spectrum snapshots were exported to XML and
tab-delimited text files, respectively, for replotting and
annotation with R 3.2 (available R-project.org).
Statistical analysis
Statistical analyses were performed using SPSS 15.0
(IBM Corporation, Armonk, NY, USA) and R. Allele and genotypic
differences between healthy (see Specimen collection) and diseased
subjects as well as ethnicity were evaluated by the χ2
test of significance. Ancestral/reference and alternative allele
were reordered for presentation according to dbSNP conventions; in
certain cases where the ancestral allele was ambiguous, e.g.
PMS2 (rs1805321), the statistically dominant allele in dbSNP
was selected as the ancestral allele (e.g. C for rs1805321).
Comparisons to the TMM dataset were determined in R, but not used
to determine significance as only allele frequencies were
available. A predefined α-level of 0.05 was used for statistical
assessment. Per the exploratory and targeted nature of this study,
P-values were not corrected for multiple comparisons; instead, the
authors advised readers to use corrected significance levels, for
instance the Dunn-Šidák corrected α′=0.00039 (m=131) when
cross-examining results here with other non-targeted genotyping
association studies to account for potential consequences of
multiple hypothesis testing.
Changes to local protein structure
As specimens obtained for this study came from a
Japanese population, we consulted annotated records from TMM to
determine potential changes to local protein structures. For
non-synonymous SNPs meeting the significance level, such changes
were predicted by evaluating the impact of amino acid substitutions
by a composite probability from HumVar PolyPhen-2 (2.2.2r398) and
Grantham scores (23,24). PolyPhen2 FPR thresholds were
appraised at 10/20%. For the purpose of calculation, the Grantham
value of all non-identical amino acid replacements were transformed
to a kernel density function in R and re-evaluated as cumulative
probabilities. Scores were averaged by the geometric mean (square
root of the product of two probabilities).
Interaction and gene enrichment
analysis
Potential interactions among the list of significant
candidate genes were analyzed by GeneMANIA to identify the top 25
related genes. Interactions were selected to allow at most 25 genes
with 25 attributes, and filtered by the presence of possible
associations via co-expression, co-localization, genetic and
physical interactions, pathway information as well as predicted
interactions. Networks were analyzed and visualized in Cytoscape.
Statistically enriched Reactome pathways (version 58) were
identified by the Panther overrepresentation test with a predefined
α-level of 0.05 to assess individual pathway significance.
Linkage disequilibrium analysis
Potential linkage disequilibria were examined using
LDproxy in the LDlink suite (25)
to possible linkages among the significant SNPs, using the Japanese
population genotype data from the Phase 3 of the 1000 Genomes
Project as input. Proxy locus pairs with correlation coefficients
(R2) >0.80, and RegulomeDB scores of 1-5
(including subgroups of 1–3) were retained for further analysis.
Variant pair associations, as a function of the correlation
coefficient between loci pairs, were visually examined in Cytoscape
as network diagrams to identify common functionally active proxy
variants.
Cohort allele frequency dilution
VCF files containing individual variant data
information from the Phase 3 1000 Genomes Project (26) were acquired via the Data Slicer
(GRCh37 release 89, last accessed August, 2017) for each of the 12
candidate polymorphisms. To test the effect on changes in candidate
allele frequencies if samples from this population were mixed with
the cohort. n=0–480 randomly selected genotypes from G1000 were
mixed with the cohort, also randomly selected to make up a final
sample size of 480, in order to examine the resultant reference (A)
and alternative (B) allele counts as well as the mean allele
frequencies (%B) as a function of n. In a separate validation, both
the cohort and G1000 populations were randomly sampled (100
individuals/set) to evaluate the likelihood that both populations
contained different mean allele frequencies. Hundred random trials
were performed for both validations in R, with statistical
significance by two-sample t-tests.
Results
We found 12 out of the 131 SNPs investigated
(Table I) to be potentially
associated; among those, 10 SNPs, 1, [α-methylacyl-CoA
racemase (AMACR) rs34677], 2 (ARHGAP8,
rs2071762), 3 (KDR, rs2305948), 4
(SELE, rs5361), 5 (XPC, rs2228001), 6
(FANCA, rs2239359), 8 (BRCA2, rs766173),
9 (PCDHGB7, rs17208397), 11 (PMS2,
rs1805321) and 12 (ADAMTS16, rs16875054) were
non-synonymous; SNP 7 (LRRN3, rs17439799) was
intronic and 10 (ERCC4, rs2020955) was synonymous. A
full table containing genotypes, alleles and call rates for the 131
selected SNPs are available upon request and at github.com/cccrijlin/marray_si. Discrepancies did
exist in allele distribution between database annotations, e.g.
WRN (rs1346044) contained T/C alleles in NCBI but A/G in
JSNP.
 | Table IGenotypes, alleles and impact on
local protein structure of statistically significant SNPs and
corresponding JSNP and dbSNP entries. |
Table I
Genotypes, alleles and impact on
local protein structure of statistically significant SNPs and
corresponding JSNP and dbSNP entries.
| ID | Gene (dbSNP
rsID) | A/B | DP | AA | AB | BB | Sum | A | B | ST | dbSNP
χ2 | dbSNP P-value | dbSNP ss# | JSNP
χ2 | JSNP P-value | TMM A/B | TMM
χ2 | TMM P-value | Mut | PV | GV | PG | GMI |
|---|
| 1 | AMACR (34677) | T/G | HCC | 4 | 22 | 149 | 175 | 30 | 320 | GT | 41.7253 | 0.0000 | 68931918 | 8.1811 | 0.0167 | C/A (3717/379) | 0.1793 | 0.6720 | Q>H | 0.320 | 24 | 0.047 | 0.123 |
| | | LC | 3 | 6 | 56 | 65 | 12 | 118 | GT | 18.0383 | 0.0001 | | 12.0369 | 0.0024 | | 0.0001 | 0.9932 | | | | | |
| | | CH | 1 | 17 | 158 | 176 | 19 | 333 | GT | 49.3707 | 0.0000 | | 11.4293 | 0.0033 | | 13.2155 | 0.0003 | | | | | |
| 2 | ARHGAP8
(2071762) | T/C | HCC | 38 | 73 | 78 | 189 | 149 | 229 | N/S | | | 69271108 | | | C/T
(2456/1580) | | | P>L | 0.018 | 98 | 0.521 | 0.097 |
| | | LC | 13 | 29 | 36 | 78 | 55 | 101 | N/S | | | | | | | | | | | | | |
| | | CH | 20 | 94 | 81 | 195 | 134 | 256 | GT | 8.5177 | 0.0141 | | 7.0856 | 0.0289 | | 3.4369 | 0.0638 | | | | | |
| 3 | KDR (2305948) | T/C | HCC | 0 | 25 | 158 | 183 | 25 | 341 | N/S | | | | 6.2351 | 0.0443 | C/T (3662/436) | 5.2632 | 0.0218 | V>I | 0.999 | 29 | 0.079 | 0.281 |
| | | LC | 0 | 9 | 57 | 66 | 9 | 123 | N/S | | | | | | | | | | | | | |
| | | CH | 2 | 27 | 148 | 177 | 31 | 323 | N/S | | | | | | | | | | | | | |
| 4 | SELE (5361) | A/C | HCC | 180 | 2 | 0 | 182 | 362 | 2 | AL | | | | 8.0262 | 0.0046 | T/G (3953/145) | 9.3738 | 0.0022 | S>R | 0.944 | 110 | 0.637 | 0.775 |
| | | LC | 74 | 2 | 0 | 76 | 150 | 2 | N/S | | | | | | | | | | | | | |
| | | CH | 180 | 2 | 0 | 182 | 362 | 2 | AL | | | | 8.0262 | 0.0046 | | 9.3738 | 0.0022 | | | | | |
| 5 | XPC (2228001) | A/C | HCC | 90 | 80 | 15 | 185 | 260 | 110 | GT | | | | 13.1123 | 0.0014 | G/T
(1694/2398) | 19.1813 | 0.0000 | Q>K | 0.000 | 53 | 0.179 | 0.000 |
| | | LC | 25 | 35 | 28 | 78 | 85 | 91 | N/S | | | | | | | | | | | | | |
| | | CH | 63 | 89 | 21 | 173 | 215 | 131 | N/S | | | | | | | | | | | | | |
| 6 | FANCA
(2239359) | T/C | HCC | 105 | 49 | 8 | 162 | 259 | 65 | GT | | | | 7.0776 | 0.0290 | C/T (652/3396) | 3.4226 | 0.0643 | G>S | 0.000 | 56 | 0.200 | 0.000 |
| | | LC | 51 | 51 | 0 | 69 | 153 | 51 | N/S | | | | | | | | | | | | | |
| | | CH | 138 | 40 | 4 | 182 | 316 | 48 | N/S | | | | | | | | | | | | | |
| 7 | LRRN3
(17439799) | T/C | HCC | 120 | 55 | 10 | 185 | 295 | 75 | GT | | | | 6.6997 | 0.0351 | T/C (3113/981) | 2.5606 | 0.1096 | Int | | | | |
| | | LC | 27 | 1 | 12 | 40 | 55 | 25 | GT | | | | 65.0216 | 0.0000 | | 2.2784 | 0.1312 | | | | | |
| | | CH | 114 | 43 | 13 | 170 | 271 | 69 | GT | | | | 14.8948 | 0.0006 | | 2.3367 | 0.1264 | | | | | |
| 8 | BRCA2 (766173) | T/G | HCC | 140 | 47 | 1 | 188 | 327 | 49 | GT | 10.5751 | 0.0051 | 66862596
69130493 | | | A/C (3569/521) | 0.0266 | 0.8703 | N>H | 0.034 | 68 | 0.279 | 0.097 |
| | | LC | 47 | 24 | 0 | 71 | 118 | 24 | AL | 15.4693 | 0.0001 | | | | | 2.1199 | 0.1454 | | | | | |
| | | CH | 144 | 47 | 1 | 192 | 335 | 49 | GT | 10.0898 | 0.0064 | | | | | 0.0002 | 0.9901 | | | | | |
| 9 | PCDHGB7
(17208397) | G/C | HCC | 152 | 6 | 0 | 158 | 310 | 6 | AL | 5.0817 | 0.0242 | 24521861
68951534 | | | G/C (3955/129) | 1.5656 | 0.2109 | V>L | 0.261 | 32 | 0.089 | 0.153 |
| | | LC | 70 | 2 | 0 | 72 | 142 | 2 | AL | 4.0500 | 0.0442 | | | | | 1.4511 | 0.2284 | | | | | |
| | | CH | 107 | 1 | 0 | 108 | 215 | 1 | AL | 9.7059 | 0.0018 | | | | | 5.0848 | 0.0241 | | | | | |
| 10 | ERCC4
(1799801) | T/C | HCC | 120 | 61 | 11 | 192 | 301 | 83 | GT | 7.2055 | 0.0272 | 52072800
66861998 | | | T/C
(3060/1032) | 2.4396 | 0.1183 | Syn | | | | |
| | | LC | 44 | 32 | 3 | 79 | 120 | 38 | N/S | | | | | | | | | | | | | |
| | | CH | 116 | 72 | 9 | 197 | 304 | 90 | N/S | | | | | | | | | | | | | |
| 11 | PMS2 (1805321) | T/C | HCC | 13 | 50 | 74 | 137 | 76 | 198 | GT | 56.2044 | 0.0000 | 5259684 | | | N/A | | | P>S | 0.001 | 74 | 0.295 | 0.017 |
| | | LC | 3 | 42 | 28 | 73 | 48 | 98 | GT | 75.2905 | 0.0000 | | | | | | | | | | | |
| | | CH | 8 | 82 | 81 | 171 | 98 | 244 | GT | 70.9810 | 0.0000 | | | | | | | | | | | |
| 12 | ADAMTS16
(16875054) | A/G | HCC | 9 | 11 | 105 | 125 | 29 | 221 | GT | 16.3429 | 0.0003 | 48414945
66315014 | | | G/A (3559/537) | 0.5531 | 0.4570 | A>T | 0.590 | 58 | 0.211 | 0.352 |
| | | LC | 2 | 10 | 60 | 72 | 14 | 130 | N/S | | | 68925406 | | | | | | | | | | |
| | | CH | 2 | 22 | 141 | 165 | 26 | 304 | GT | 9.8724 | 0.0072 | | | | | 9.1853 | 0.0024 | | | | | |
We also evaluated the likelihood that these variants
differed from corresponding records in the TMM databank by their
maximum χ2 statistics between the JSNP and dbSNP
datasets as a potential screen to confirm the ethnic specificity of
those variants to the Japanese population, but did not assign
significance based on these statistics as the TMM databank
contained considerably less publicly available information than
JSNP or dbSNP at the time of analysis. Most of the variants
appeared to have allele frequencies that varied across different
phenotypes, some with only one subgroup that strongly deviated from
the other two, e.g. 3 in only HCC, and others in multiple
subgroups, e.g. 4 and 8. Generally, the HCC subgroup
frequently exhibited allele frequencies that deviated from the
other two phenotypes, for example candidates 5 and 6.
For SNPs with significant differences in genotype and allele
frequency, all but 1 (AMACR) expressed differential
compositions across different ethnic groups (Table II), suggesting that they may be
specific to the Japanese population.
| Table IIStratified comparisons of significant
SNPs across different ethnic groups. |
Table II
Stratified comparisons of significant
SNPs across different ethnic groups.
| Cytoband | Rs# Symbol | NCBI SSID | Population | Groups | P-value
|
|---|
| GT | AL |
|---|
| 5p13.2 | rs34677 | 12675253 | JPT, HCB, CEU,
YRI | G1 | 0.096 | 0.089 |
| AMACR | 68931918 | | | 0.070 | 0.029 |
| 98712196 | | | ** | ** |
| 22q13.31 | rs2071762 | 12527661 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| ARHGAP8 | 69271108 | | | ** | ** |
| 71647951 | | | ** | ** |
| 4q12 | rs2305948 | 48430022 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| KDR | 68899172 | | | ** | ** |
| 1q24.2 | rs5361 | 48428825 | JPT, HCB, CEU,
YRI | G1 | 0.001 | ** |
| SELE | 68784305 | | | ** | ** |
| 3p25.1 | rs2228001 | 44411054 | JPT, HCB, CEU,
YRI | G1 | 0.009 | 0.004 |
| XPC | 66856826 | JPT, HSP, CEU,
AAM, | G2 | 0.001 | 0.002 |
| | CHB, YRI GENO
PANEL | | | |
| 68853585 | JPT, HCB, CEU,
YRI | G1 | 0.009 | 0.007 |
| 16q24.3 | rs2239359 | 16702260 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| FANCA | 43872290 | AoD Japanese,
African | G3 | N/A | ** |
| | American,
Caucasian, Chinese | | | |
| 66860952 | JPT, HSP, CEU,
AAM, | G2 | ** | ** |
| | CHB, YRI GENO
PANEL | | | |
| 69355410 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| 7q31.1 | rs17439799 | 24455640 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| LRRN3 | | | | | |
| 13q13.1 | rs766173 | 5586314 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| BRCA2 | 66862596 | JPT, HSP, CEU,
AAM, | G2 | ** | ** |
| | CHB, YRI GENO
PANEL | | | |
| 69130493 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| 5q31.3 | rs17208397 | 24621861 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| PCDHGB7 | 68951534 | | | ** | ** |
| 16p31.12 | rs1799801 | 52072800 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| ERCC4 | 66861998 | JPT, HSP, CEU,
AAM, | G2 | ** | ** |
| | CHB, YRI GENO
PANEL | | | |
| 5p15.32 | rs16875054 | 48414945 | JPT, HCB, CEU,
YRI | G1 | ** | ** |
| ADAMTS16 | 66315014 | | | ** | ** |
| 68925406 | | | ** | ** |
We also explored whether potential linkage
disequilibria existed among candidates in Table I to ascertain the possibility of
non-random association with other alleles as means to confirm their
true phenotypic implications on the liver diseases. While some of
those SNPs did have proxy pairs that would suggest some extent of
linkage disequilibria (available upon request), we observed no
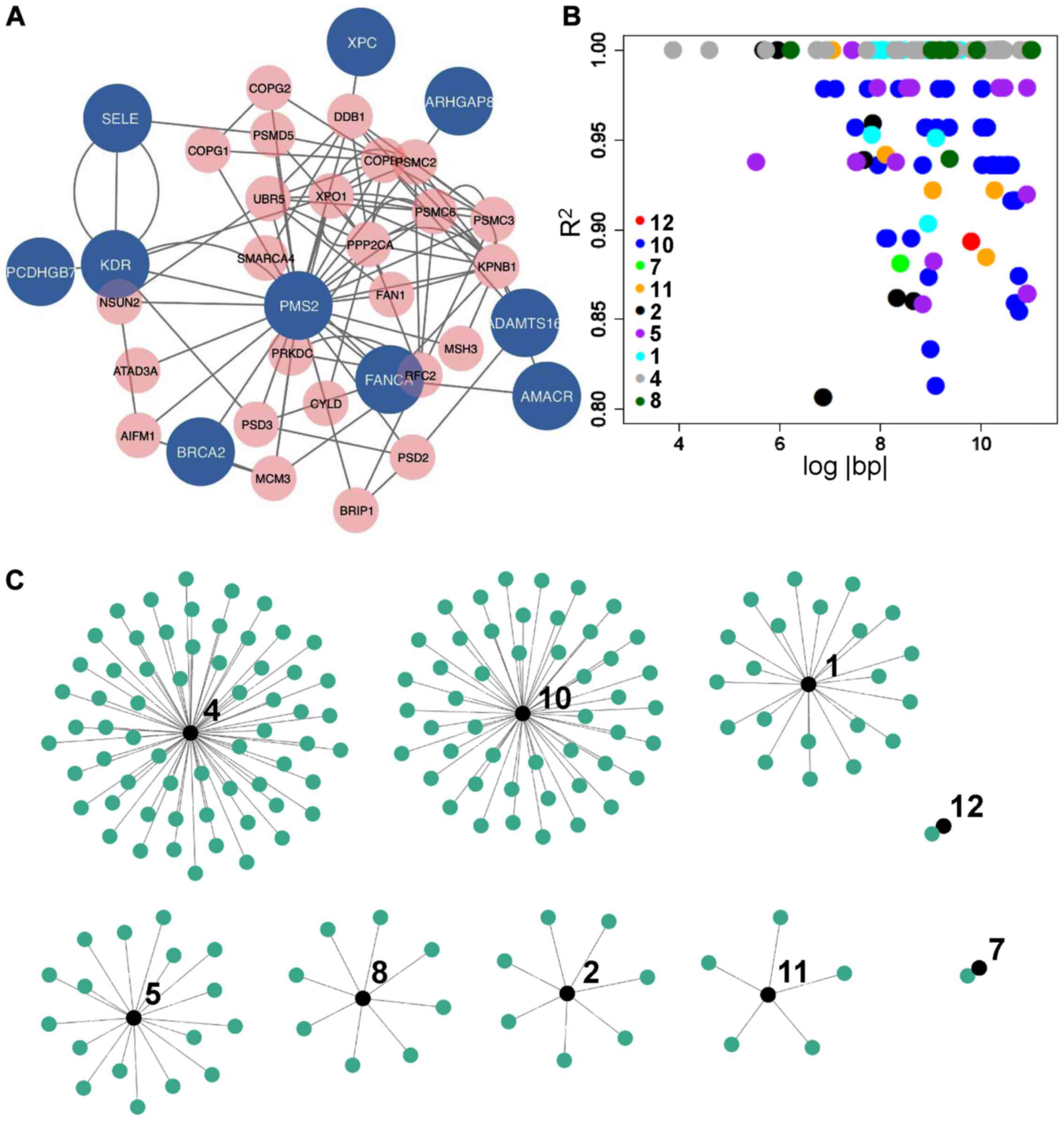
common proxy variants among them (Fig.
1B and C). Annotations of those proxy variants also suggested
either synonymous mutations or the lack of clear biological
function. As not all variants in Table
I were on the same chromosome, and most linked proxy pairs were
outside the immediate proximity (only 12/170 were within 1,000 bp;
data available upon request), potential associations on allele
frequencies of SNPs in Table I by
those proxy variants most likely had no effect on disease
outcomes.
Discussion
Roughly 10% of the SNPs genotyped in this study
displayed critical roles in liver functions and various cancer
phenotypes. For instance, AMACR is an ubiquitously expressed
enzyme responsible for bile branched-chain fatty acid metabolism
(27) that undergoes increases in
mRNA and protein expressions in colorectal and prostate cancers as
well as pre-cancerous hyperplastic polyps in the large intestine
(28,29). Co-increases of COX-2 expression and
AMACR could also potentially lead to immune suppression,
neoplastic changes and tumor invasion (30). As polymorphisms in AMACR
have been implicated in prostate and colorectal tumorigenesis
(28), its possible contribution
in HCV infection-induced disease phenotypes shall not be
overlooked. Similarly, despite the lack of prior evidence
suggesting direct association of ARHGAP8 to HCV-induced HCC,
the loss of Rho-suppressing ARHGAP7 has been implicated in
multiple cancers including HCC (31). Regulatory features of RhoGAP via
Rho has been said to attribute to reductions in hepatic fibrosis,
suggesting that a loss of function variant may induce liver
cirrhosis in a manner independent from HCV infection. Adding to the
ability for statins to attenuate liver fibrosis in chronic HCV
infections (32), inter-regulatory
feedback of Rho acylation by members of the ARHGAP family, for
instance ARHGAP8, may well attribute to the aberrant
dysregulation, via HCV infections, during HCC development (33).
While mutations in DNA mismatch or strand-break
repairs have always been intimately associated with tumor
development, the presence of risk alleles in XPC, a gene
critical in nucleotide excision repair (34), further highlights the importance of
exogenous genomic editing by HCV in HCC. XPC has been said
to elevate the rise of skin cancer 1,000-fold and escalating organ
neoplasms as much as 10-fold (35)
through its role in xeroderma pigmentosum, a pre-cancerous
condition. From this, it is likely that defects in XPC may
also increase one's susceptibility to HCC. Additionally,
aberrations in FANCA, a known tumor suppressor (36) as well as BRCA2, a DNA repair
gene, can have serious repercussions leading to oncogenesis, most
notably breast cancer, both in Japanese and other ethnic cohorts
(37–40). Among these reports, candidate
8 also belongs to the group of 25 breast cancer potential
risk alleles, sufficiently justifying the need for further
investigation into its mechanistic role in HCV-induced HCC.
Likewise, mutations in PMS2 may be similarly implicated in
response to HCV virulence as the gene was most remarkably known for
its role in DNA mismatch repair and links to microsatellite
instability as in hereditary nonpolyposis colorectal cancer
(41–43).
Our genotyping results also revealed some
curiosities on the role of functional surrogates in HCV-induced
oncogenesis, particularly through potential risk alleles in
ADAMTS16. ADAMTS16 belongs to a family of secreted
metalloproteases that tends to be fairly tissue limited, and the
discovery of its association to HCV was unexpected as
ADAMTS16 was primarily expressed only in the lung and brain;
with that said, recent literature did suggest the possibility of
high-quantity shuttling of ADAMTS13 and ADAMTS19 to
unexpected destinations, such as tumors and nearby tissues in cases
of osteosarcoma, melanoma and colon cancer (44). Surfacing of ADAMTS16 in HCC
samples could also infer the occurrence of a similar phenomenon by
mutations or other mechanisms. Interestingly, while some in the
ADAMTS family were implicated in diseases such as angio-inhibition
(45) and Ehlers-Danlos syndrome
(46), most remain relatively
unknown and inconclusive (44,47,48).
As such, risk alleles in ADAMTS16 as well as the observation
that these metalloproteases were typically downregulated in cancer
as a consequence of promoter hypermethylation (49), would suggest that polymorphisms in
ADAMTS16 may potentially push the gene to displace and
redistribute itself and other members in the family, possibly via
extracellular matrix remodeling (50), and become oncogenic. Additionally,
not all conditions were genetically predisposed to the same
polymorphisms (Table III) or
reacted identically to the virulence of HCV. For example, a
mutation to 3 would more likely drive onset in HCC over LC or CH,
while subjects with a mutation to 6 would perhaps present HCC
phenotypes differently. Odds ratios (OR >1, P<0.05) also
inferred differential susceptibility for subjects with mutations in
XPC and BRCA2, as XPC showed elevated odds for
CH and BRCA for all subgroups. Just as the BRCA genes
helped physicians to assess breast cancer susceptibility, the
combination of BRCA and perhaps a combination of several
aforementioned SNPs could also help elucidate risks to HCC by the
process of elimination.
| Table IIIStratified genotypic comparisons of
significant SNPs among disease phenotypes. |
Table III
Stratified genotypic comparisons of
significant SNPs among disease phenotypes.
| ID Symbol dbSNP
rsID | Alleles | Grouping (or
JSNP/NCBI set ID) | Genotype
|
|---|
| All P-value | HCC P-value | LC P-value | CH P-value |
|---|
| 1 | T/G | HCC | – | – | 0.511 | 0.259 |
| AMACR | | LC | – | – | – | 0.092 |
| rs34677 | | CH | – | – | – | – |
| | NCBI 12675253 | 0.032 | 0.106 | 0.147 | 0.002 |
| | NCBI 68931918 | ** | ** | ** | ** |
| | JSNP
IMS-JST038996 | ** | 0.017 | 0.002 | 0.003 |
| 2 | T/C | HCC | – | – | 0.712 | 0.017 |
| ARHGAP8 | | LC | – | – | – | 0.159 |
| rs2071762 | | CH | – | – | – | – |
| | NCBI 12527661 | 0.111 | 0.988 | 0.818 | 0.053 |
| | NCBI 69271108 | 0.065 | 0.862 | 0.659 | 0.014 |
| | NCBI 71647951 | 0.111 | 0.988 | 0.818 | 0.053 |
| | JSNP
IMS-JST007439 | 0.012 | 0.056 | 0.102 | 0.029 |
| 3 | T/C | HCC | – | – | 0.59 | 0.316 |
| KDR | | LC | – | – | – | 0.646 |
| rs2305948 | | CH | – | – | – | – |
| | NCBI 48430022 | 0.647 | 0.374 | 0.438 | 0.603 |
| | NCBI 68899172 | 0.651 | 0.401 | 0.461 | 0.599 |
| | JSNP
IMS-JST063410 | 0.158 | 0.044 | 0.302 | 0.389 |
| 4 | A/C | HCC | – | – | 0.338 | 0.689 |
| SELE | | LC | – | – | – | 0.338 |
| rs5361 | | CH | – | – | – | – |
| | NCBI 48428825 | 0.188 | 0.091 | 0.413 | 0.091 |
| | NCBI 68784305 | 0.201 | 0.095 | 0.424 | 0.095 |
| | JSNP | 0.001 | 0.001 | 0.145 | 0.001 |
| 5 | A/C | HCC | – | – | 0.001 | 0.054 |
| XPC | | LC | – | – | – | 0.086 |
| rs2228001 | | CH | – | – | – | – |
| | NCBI 44411054 | 0.011 | 0.523 | 0.092 | 0.656 |
| | NCBI 66856826 | 0.01 | 0.365 | 0.108 | 0.847 |
| | NCBI 68853585 | 0.012 | 0.326 | 0.185 | 0.555 |
| | JSNP
IMS-JST086794 | 0.003 | 0.001 | 0.371 | 0.165 |
| 6 | T/C | HCC | – | – | 0.117 | 0.061 |
| FANCA | | LC | – | – | – | 0.386 |
| rs2239359 | | CH | – | – | – | – |
| | NCBI 16702260 | 0.169 | 0.366 | 0.191 | 0.581 |
| | NCBI 43872290 | – | – | – | – |
| | NCBI 66860952 | 0.169 | 0.366 | 0.191 | 0.581 |
| | NCBI 69355410 | 0.169 | 0.366 | 0.191 | 0.581 |
| | JSNP
IMS-JST010482 | 0.087 | 0.029 | 0.504 | 0.488 |
| 7 | T/C | HCC | – | – | ** | 0.5 |
| LRRN3 | | LC | – | – | – | ** |
| rs17439799 | | CH | – | – | – | – |
| | NCBI 24455640 | ** | 0.845 | ** | 0.926 |
| | JSNP | ** | 0.035 | ** | 0.001 |
| 8 | T/G | HCC | – | – | 0.313 | 0.993 |
| BRCA2 | | LC | – | – | – | 0.274 |
| rs766173 | | CH | – | – | – | – |
| | NCBI 5586314 | 0.019 | 0.008 | ** | 0.01 |
| | NCBI 66862596 | 0.013 | 0.005 | ** | 0.006 |
| | NCBI 69130493 | 0.013 | 0.005 |
<0.001 | 0.003 |
| 9 | G/C | HCC | – | – | 0.518 | 0.147 |
| PCDHGB7 | | LC | – | – | – | 0.351 |
| rs17208397 | | CH | – | – | – | – |
| | NCBI 24621861 | 0.004 | 0.026 | 0.04 | 0.002 |
| | NCBI 68951534 | 0.004 | 0.026 | 0.04 | 0.002 |
| 10 | T/C | HCC | – | – | 0.354 | 0.573 |
| ERCC4 | | LC | – | – | – | 0.815 |
| rs1799801 | | CH | – | – | – | – |
| | NCBI 52072800 | 0.156 | 0.027 | 0.237 | 0.07 |
| | NCBI 66861998 | 0.156 | 0.027 | 0.237 | 0.07 |
| 11 | T/C | HCC | – | – | 0.011 | 0.061 |
| PMS2 | | LC | – | – | – | 0.388 |
| rs1805321 | | CH | – | – | – | – |
| | NCBI 5259684 | ** | ** | ** | ** |
| 12 | A/G | HCC | – | – | 0.258 | 0.018 |
|
ADAMTS16 | | LC | – | – | – | 0.682 |
| rs16875054 | | CH | – | – | – | – |
| | NCBI 48414945 | ** | ** | 0.074 | 0.007 |
| | NCBI 66315014 | ** | ** | 0.074 | 0.007 |
| | NCBI 68925406 | ** | ** | 0.074 | 0.007 |
In some situations nearby variants may act in
concert and contribute jointly to particular phenotypes; to examine
the possibility of these nonrandom associations, we next explored
possible interactions and the presence of common functionally
active proxy variants associated with polymorphisms identified in
this study. While the presence of a small interaction revealed the
presence of common interacting partners with the 10 candidate
genes, for instance SMARCA4, PPP2CA and XPO1
(Fig. 1A), linkage disequilibrium
analysis painted a slightly different picture, alternatively
suggesting that these candidates were genotypically pairwise
independent, and contributed cumulatively to the outcome of each
disease. Additionally, multivariate analysis across genders and
ages revealed no strong covariances among the candidates (data not
shown). Interestingly, enrichment analysis highlighted strong
connections to DNA repair and post-translational modifications, two
biologically orthogonal functions, for the candidate genes and
their interacting partners (table available upon request).
Enrichment in the two nonparallel pathways would thus posit an
overall additive effect that reflected the candidates' genotypic
independence. While defects in DNA repair were commonplace in
cancer as a consequence of unregulated division, no mechanistically
direct explanation could answer the functional enrichment in
protein modification as a consequence; as such, the hypothesis that
both pathways, via functional alteration by the candidates
genotyped, acted in concert to achieve this additive effect
reflected in the association of HCV to HCC, appeared to be a
plausible one and hopefully could be further pondered.
We also analyzed changes in protein local secondary
and quaternary structures in order to attribute the impact of
non-synonymous mutations to the connection between HCV and HCC to
ascertain the underlying routes to disease progression. Although
DNA repair mechanisms may be biochemically similar across different
types of cancer, focused studies on BRCA2 and FANCA
still remain relatively unexplored in HCV-induced HCC. Especially
since hepatic tissues are constantly under oxidative stress, genome
repair during acute and chronic HCV infection and the oncogenesis
of HCC are critical in understanding disease progression. We
observed that ADAMTS16, SELE and KDR all
carried polymorphisms with potentially damaging mutations; as a
matter of fact, both ADAMTS16 and SELE were
associated with HCC and CH, suggesting a possible path of disease
progression upon HCV infection from CH to HCC via these mutations
(Table I). Nonetheless, the fact
that KDR also manifested a damaging mutation and was
associated with HCC would also suggest a possible role for the
gene, as KDR-mediated mechanisms could support HCV infection
(51), subsequently directing
cells to the carcinogenesis of HCC.
A difficulty in establishing genetic connections
between diseases and HCV infections is that the virus can remain
dormant in the liver, leaving the patient with normal liver
functions and no symptoms. As such, disease progression in early
stages becomes retarded. These associated SNPs can, in theory,
display no phenotypical effect during early onset, which can delay
diagnosis and treatment altogether. As both acute and chronic HCV
infections could transform healthy livers, the full and exact
extent of the infection would be difficult to gauge. HCV carriers
may present normal ALT levels from normal liver panel screenings
and the amount of fat deposits in the tissue may also mask the
presence of HCVs, liver biopsies are likely to be inconclusive in
these cases for determining the stage of infection. As such,
extended genotyping studies beyond the one described herein,
alongside functional characterizations of BRCA2 and
FANCA, may be a practical option clinically for
diagnosis.
Among the different genotyping technologies,
MassARRAY proved to be a capable choice, demonstrating high
sensitivities in various applications such as human papillomavirus
genotyping for cervical cancer (52) or classification of ectopic crypts
in colorectal cancers (53) via
KRAS and BRAF mutations. Although we were unable to
call every SNP on the initial test, we nonetheless obtained call
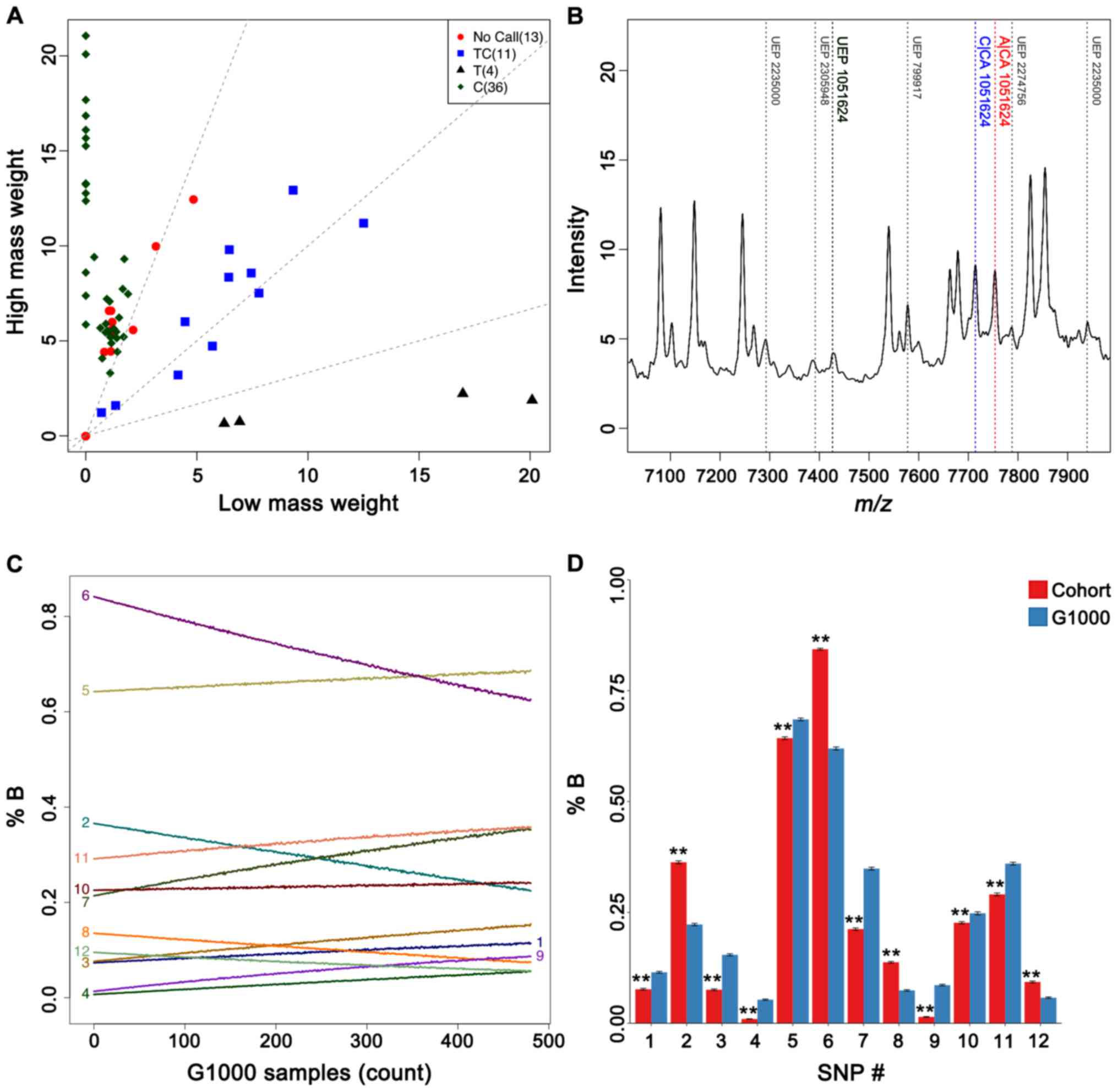
rates surpassing 90% in duplicate typing of most SNPs, suggesting
reasonably minimal genotyping errors (Fig. 2A and B). Mass spectrometry-based
genotyping offers excellent sensitivity in analyte detection and is
thus an ideal approach for characterizing specimens from tissue
depositories, where amounts of DNA may be very limited. Extension
(1-bp) coupled with mass spectrometric-based characterization by
MassARRAY was a suitable choice in assessing candidate SNPs for
studies of similar scale. We do, however, recognize that such a
method is limited to the number of SNP candidates determined a
priori, so the discovery of novel SNPs can be more challenging.
Nonetheless, for cancers and precancerous conditions dependent on a
small of oncogenic drivers, this method is a sensible and
attractive option at the clinical setting.
The MassARRAY method allowed us to maintain a
reasonable subject size of 480, although the relatively lower
number of LC cases might have marginally reduced the statistical
power in this study. While increasing the number of test subjects
could certainly be beneficial in improving the statistical power,
such an approach was not always possible in practice. In
consideration for the potential loss in statistical power, we
performed two validation simulations to see whether these
differences in allele frequencies were still preserved if subsets
of our cohort were 'diluted' with a different population sample. By
taking genotypes from randomly selected G1000 individuals and
mixing them with (also randomly selected) subjects in the cohort,
we assembled subsets of 480 individuals and checked the frequencies
of the candidate risk allele. Plotting such changes as a function
of different numbers of G1000 individuals (Fig. 2C) revealed moderate frequency
shifts for all of the candidate polymorphisms. Frequencies of these
risk alleles thus appeared to be condition-specific, as they were
certainly susceptible to dilution with subjects from a different
and considerably more heterogeneous population of various
ethnicities and health states. Additionally, bootstrapping our
cohorts to smaller subsets of 100 individuals for direct comparison
to sets of equal sizes in the G1000 population also confirmed
differences in allele frequencies (Fig. 2D). While such tests could not truly
offset benefits of increasing cohort sizes, results nonetheless
demonstrated a reasonable level of statistical robustness
considering the size of our cohort.
Genotyping more candidates, e.g. Tolloid-like 1
protein (54), from overlooked
biochemical pathways (for instance, stress responses, inflammation
and cell cycles) could allow us to further understand the manner in
which HCV infections evolve phenotypically. Characterizing changes
in known and nearby polymorphisms in genes implicated in insulin
biosynthesis, inflammation and adipose tissue remodeling, etc.,
would be highly useful in deciphering risk factors of obesity to
HCV-induced HCC; in a similar vein, alcohol metabolism and various
other pathways related to opioid metabolism would also be highly
useful in risk assessment of substance abuse to HCC. Additionally,
this type of candidate expansion would be particularly more
beneficial in analyzing ethnically more heterogeneous populations.
As most candidate SNPs, with the exception of AMACR
(rs34677), exhibited pairwise independence across non-ethnically
Japanese datasets, our results hinted that HCV-induced liver
disease progressed differently from other infection-driven
oncogenesis such as the case with Helicobacter pylori and
stomach adenocarcinoma. Based on the differences across different
datasets, we would recommend increasing the number of candidates
for genotyping to validate this hypothesis. Nonetheless, our
approach here could still serve as a useful tool for early
diagnosis based on liver disease examination.
An ongoing discussion in liver cancer is the
possibility of oncogenic addiction, in which cancer progression is
often controlled by a handful of driver genes subsequently leading
to uncontrollable growth and eventual transformation to tumor.
Horizontally integrating SNP-based oncogenesis information from one
cancer type to another can be a useful approach in deciphering the
puzzle. For liver diseases that may ultimately lead to cancer,
identifying function-altering SNPs is a useful tool for
facilitating earlier diagnosis and expediting treatment.
Furthermore, for cancers which the theory of oncogenic addiction
well applies, the ability to identify driver mutations quickly also
has significant therapeutic implications. Based on our previous
knowledge of SNPs in liver diseases and oncogenes, we utilized a
targeted SNP-screening approach using a MassARRAY-based protocol
from a set of 131 SNPs, and were able to identify 12 positions with
implications ranging from disease onset, survival rates and in
other metrics. Additionally, we were also able to highlight the
significance of BRCA2 and FANCA germ-line mutations
and associate them to liver cancers. Most of these HCV-dependent
mutation candidates were non-synonymous, and their association with
other risk factors further suggested their potential roles as
biomarkers for the liver conditions described in this study.
It is however important to note that several factors
such as age have not been well explored in this study, as
illuminated by the relatively smaller sample sizes of subjects
under 50 years old in all three disease groups; additionally, at 80
samples, the statistical power for SNPs associated to cirrhosis may
also be lower than the other two groups. Other factors such as
hepatitis B infection, substance use or obesity could also
influence disease progression. It is now common knowledge that
sharing syringe needles is a common way that HCV is transmitted
between persons, and that alcohol abuse has been said to worsen
chronic HCV progression (55).
Obesity, mostly through nonalcoholic fatty liver disease and type 2
diabetes, can also have an effect on the outcome of liver cancer
(56). In the Japanese population,
hepatitis B infection attributed to ~16% of HCC cases (2), a fraction ~1/5 of HCV infections;
while relatively minor compared to HCV, hepatitis B infections
still present a great public health concern and future genotyping
studies should also consider the inclusion of genes potentially
implicated in these infections. Along this note, it is also
important to note that the status of HCV infections such as virus
titers, genotypes and history of prior treatments may also affect
the progression of liver cancer and thus critical to be taken into
consideration in the cohort selection. Nonetheless, the
incorporation of in silico screening and whole-exome
sequencing data to identify and refine the list of SNP candidates
for future MassARRAY studies should provide definitive improvements
for more reliable characterization of disease associations and
medium-size clinical cohort studies.
Acknowledgments
This study was supported by the Academic Frontier
Project for Private Universities of 2006, Grant-in-Aid for
Scientific Research (B) JP26290060 and Research on Innovative Areas
(JP26112517, JP25134718 and JP16H01579) whilst being conducted as
part of the BioBank Japan Project sponsored by the Ministry of
Education, Culture, Sports, Science and Technology as well as the
Japan Agency for Medical Research and Development.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
SNP
|
single nucleotide polymorphisms
|
|
HCV
|
hepatitis C virus
|
|
CH
|
chronic hepatitis
|
|
LC
|
liver cirrhosis
|
|
PCR
|
polymerase chain reaction
|
|
DNA
|
deoxyribose nucleic acid
|
|
JSNP
|
Japanese Single Nucleotide
Polymorphisms databank
|
|
TMM
|
Tohoku Medical Megabank
|
|
AMACR
|
α-methylacyl-CoA racemase
|
|
ARHGAP
|
Rho GTPase-activating proteins
|
|
ADAMTS
|
a disintegrin and metalloproteinase
with thrombospondin motifs
|
|
G1000
|
the 1000 Genomes Project
|
References
|
1
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen SL and Morgan TR: The natural history
of hepatitis C virus (HCV) infection. Int J Med Sci. 3:47–52. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yoshizawa H: Hepatocellular carcinoma
associated with hepatitis C virus infection in Japan: Projection to
other countries in the foreseeable future. Oncology. 62(Suppl 1):
8–17. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colombo M and Iavarone M: Role of
antiviral treatment for HCC prevention. Best Pract Res Clin
Gastroenterol. 28:771–781. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alavian SM and Haghbin H: Relative
importance of hepatitis B and C viruses in hepatocellular carcinoma
in EMRO countries and the Middle East: A systematic review. Hepat
Mon. 16:e351062016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang L, Marcell L and Kottilil S: Systemic
manifestations of hepatitis C infection. Infect Agent Cancer.
11:292016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kato N, Ji G, Wang Y, Baba M, Hoshida Y,
Otsuka M, Taniguchi H, Moriyama M, Dharel N, Goto T, et al:
Large-scale search of single nucleotide polymorphisms for
hepatocellular carcinoma susceptibility genes in patients with
hepatitis C. Hepatology. 42:846–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gu X, Qi P, Zhou F, Ji Q, Wang H, Dou T,
Zhao Y and Gao C: An intronic polymorphism in the
corticotropin-releasing hormone receptor 2 gene increases
susceptibility to HBV-related hepatocellular carcinoma in Chinese
population. Hum Genet. 127:75–81. 2010. View Article : Google Scholar
|
|
9
|
Segat L, Milanese M, Pirulli D, Trevisiol
C, Lupo F, Salizzoni M, Amoroso A and Crovella S: Secreted protein
acidic and rich in cysteine (SPARC) gene polymorphism association
with hepatocellular carcinoma in Italian patients. J Gastroenterol
Hepatol. 24:1840–1846. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang KS, Lee CM, Teng HC, Huang MJ and
Huang CS: UDP-glucuronosyltransferase 1A7 polymorphisms are
associated with liver cirrhosis. Biochem Biophys Res Commun.
366:643–648. 2008. View Article : Google Scholar
|
|
11
|
Kim YS, Cheong JY, Cho SW, Lee KM, Hwang
JC, Oh B, Kim K, Lee JA, Park BL, Cheong HS, et al: A functional
SNP of the Interleukin-18 gene is associated with the presence of
hepatocellular carcinoma in hepatitis B virus-infected patients.
Dig Dis Sci. 54:2722–2728. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanaka Y, Nishida N, Sugiyama M, Kurosaki
M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S,
et al: Genome-wide association of IL28B with response to pegylated
interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat
Genet. 41:1105–1109. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nalpas B, Lavialle-Meziani R, Plancoulaine
S, Jouanguy E, Nalpas A, Munteanu M, Charlotte F, Ranque B, Patin
E, Heath S, et al: Interferon gamma receptor 2 gene variants are
associated with liver fibrosis in patients with chronic hepatitis C
infection. Gut. 59:1120–1126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tillmann HL, Thompson AJ, Patel K, Wiese
M, Tenckhoff H, Nischalke HD, Lokhnygina Y, Kullig U, Göbel U,
Capka E, et al: German Anti-D Study Group: A polymorphism near
IL28B is associated with spontaneous clearance of acute hepatitis C
virus and jaundice. Gastroenterology. 139:1586–1592. 1592.e12010.
View Article : Google Scholar
|
|
15
|
GBD 2015 Disease and Injury Incidence and
Prevalence Collaborators: Global, regional, and national incidence,
prevalence and years lived with disability for 310 disease and
injuries, 1990–2015: a systematic analysis for the Global Burden of
Disease Study 2015. Lancet. 388:1545–1602. 2016. View Article : Google Scholar
|
|
16
|
Clendenen TV, Rendleman J, Ge W, Koenig
KL, Wirgin I, Currie D, Shore RE, Kirchhoff T and
Zeleniuch-Jacquotte A: Genotyping of single nucleotide
polymorphisms in DNA isolated from serum using sequenom MassARRAY
technology. PLoS One. 10:e01359432015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirakawa M, Tanaka T, Hashimoto Y, Kuroda
M, Takagi T and Nakamura Y: JSNP: A database of common gene
variations in the Japanese population. Nucleic Acids Res.
30:158–162. 2002. View Article : Google Scholar :
|
|
18
|
Haga H, Yamada R, Ohnishi Y, Nakamura Y
and Tanaka T: Gene-based SNP discovery as part of the Japanese
Millennium Genome Project: Identification of 190,562 genetic
variations in the human genome Single-nucleotide polymorphism. J
Hum Genet. 47:605–610. 2002. View Article : Google Scholar
|
|
19
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar :
|
|
20
|
Kuriyama S, Yaegashi N, Nagami F, Arai T,
Kawaguchi Y, Osumi N, Sakaida M, Suzuki Y, Nakayama K, Hashizume H,
et al: The Tohoku Medical Megabank Project: Design and Mission. J
Epidemiol. 26:493–511. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yue P, Melamud E and Moult J: SNPs3D:
Candidate gene and SNP selection for association studies. BMC
Bioinformatics. 7:1662006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura Y: The BioBank Japan Project.
Clin Adv Hematol Oncol. 5:696–697. 2007.PubMed/NCBI
|
|
23
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grantham R: Amino acid difference formula
to help explain protein evolution. Science. 185:862–864. 1974.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Machiela MJ and Chanock SJ: LDlink: A
web-based application for exploring population-specific haplotype
structure and linking correlated alleles of possible functional
variants. Bioinformatics. 31:3555–3557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Auton A, Brooks LD, Durbin RM, Garrison
EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and
Abecasis GR; 1000 Genomes Project Consortium: A global reference
for human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levin AM, Zuhlke KA, Ray AM, Cooney KA and
Douglas JA: Sequence variation in alpha-methylacyl-CoA racemase and
risk of early-onset and familial prostate cancer. Prostate.
67:1507–1513. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daugherty SE, Platz EA, Shugart YY, Fallin
MD, Isaacs WB, Chatterjee N, Welch R, Huang WY and Hayes RB:
Variants in the alpha-Methylacyl-CoA racemase gene and the
association with advanced distal colorectal adenoma. Cancer
Epidemiol Biomarkers Prev. 16:1536–1542. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
FitzGerald LM, Thomson R, Polanowski A,
Patterson B, McKay JD, Stankovich J and Dickinson JL: Sequence
variants of alpha-methylacyl-CoA racemase are associated with
prostate cancer risk: A replication study in an ethnically
homogeneous population. Prostate. 68:1373–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pruthi RS, Derksen E and Gaston K:
Cyclooxygenase-2 as a potential target in the prevention and
treatment of genitourinary tumors: A review. J Urol. 169:2352–2359.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ng IO, Liang ZD, Cao L and Lee TK: DLC-1
is deleted in primary hepatocellular carcinoma and exerts
inhibitory effects on the proliferation of hepatoma cell lines with
deleted DLC-1. Cancer Res. 60:6581–6584. 2000.PubMed/NCBI
|
|
32
|
Trebicka J and Schierwagen R: Statins, Rho
GTPases and KLF2: New mechanistic insight into liver fibrosis and
portal hypertension. Gut. 64:1349–1350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Khan FS, Ali I, Afridi UK, Ishtiaq M and
Mehmood R: Epigenetic mechanisms regulating the development of
hepatocellular carcinoma and their promise for therapeutics.
Hepatol Int. 11:45–53. 2017. View Article : Google Scholar
|
|
34
|
Haiman CA, Hsu C, de Bakker PI, Frasco M,
Sheng X, Van Den Berg D, Casagrande JT, Kolonel LN, Le Marchand L,
Hankinson SE, et al: Comprehensive association testing of common
genetic variation in DNA repair pathway genes in relationship with
breast cancer risk in multiple populations. Hum Mol Genet.
17:825–834. 2008. View Article : Google Scholar
|
|
35
|
de Boer J and Hoeijmakers JH: Nucleotide
excision repair and human syndromes. Carcinogenesis. 21:453–460.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Krupa R, Sliwinski T, Morawiec Z,
Pawlowska E, Zadrozny M and Blasiak J: Association between
polymorphisms of the BRCA2 gene and clinical parameters in breast
cancer. Exp Oncol. 31:250–251. 2009.PubMed/NCBI
|
|
37
|
Carreira A, Hilario J, Amitani I, Baskin
RJ, Shivji MK, Venkitaraman AR and Kowalczykowski SC: The BRC
repeats of BRCA2 modulate the DNA-binding selectivity of RAD51.
Cell. 136:1032–1043. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johnson N, Fletcher O, Palles C, Rudd M,
Webb E, Sellick G, dos Santos Silva I, McCormack V, Gibson L,
Fraser A, et al: Counting potentially functional variants in BRCA1,
BRCA2 and ATM predicts breast cancer susceptibility. Hum Mol Genet.
16:1051–1057. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ishitobi M, Miyoshi Y, Ando A, Hasegawa S,
Egawa C, Tamaki Y, Monden M and Noguchi S: Association of BRCA2
polymorphism at codon 784 (Met/Val) with breast cancer risk and
prognosis. Clin Cancer Res. 9:1376–1380. 2003.PubMed/NCBI
|
|
40
|
Sliwinski T, Krupa R, Majsterek I, Rykala
J, Kolacinska A, Morawiec Z, Drzewoski J, Zadrozny M and Blasiak J:
Polymorphisms of the BRCA2 and RAD51 genes in breast cancer. Breast
Cancer Res Treat. 94:105–109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Peltomäki P: Deficient DNA mismatch
repair: A common etiologic factor for colon cancer. Hum Mol Genet.
10:735–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yuan ZQ, Gottlieb B, Beitel LK, Wong N,
Gordon PH, Wang Q, Puisieux A, Foulkes WD and Trifiro M:
Polymorphisms and HNPCC: PMS2-MLH1 protein interactions diminished
by single nucleotide polymorphisms. Hum Mutat. 19:108–113. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Umar A, Boland CR, Terdiman JP, Syngal S,
de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ,
Hamelin R, et al: Revised Bethesda Guidelines for hereditary
nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite
instability. J Natl Cancer Inst. 96:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cal S, Obaya AJ, Llamazares M, Garabaya C,
Quesada V and López-Otín C: Cloning, expression analysis, and
structural characterization of seven novel human ADAMTSs, a family
of metalloproteinases with disintegrin and thrombospondin-1
domains. Gene. 283:49–62. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vázquez F, Hastings G, Ortega MA, Lane TF,
Oikemus S, Lombardo M and Iruela-Arispe ML: METH-1, a human
ortholog of ADAMTS-1, and METH-2 are members of a new family of
proteins with angio-inhibitory activity. J Biol Chem.
274:23349–23357. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Colige A, Sieron AL, Li SW, Schwarze U,
Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, et
al: Human Ehlers-Danlos syndrome type VII C and bovine
dermatosparaxis are caused by mutations in the procollagen I
N-proteinase gene. Am J Hum Genet. 65:308–317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rodriguez-Lopez J, Pombo-Suarez M,
Loughlin J, Tsezou A, Blanco FJ, Meulenbelt I, Slagboom PE, Valdes
AM, Spector TD, Gomez-Reino JJ, et al: Association of an sSNP in
ADAMTS14 to some osteoarthritis phenotypes. Osteoarthritis
Cartilage. 17:321–327. 2009. View Article : Google Scholar
|
|
48
|
Hu X, Chen H, Jin M, Wang X, Lee J, Xu W,
Zhang R, Li S and Niu J: Molecular cytogenetic characterization of
undifferentiated embryonal sarcoma of the liver: A case report and
literature review. Mol Cytogenet. 5:262012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kumar S, Rao N and Ge R: Emerging Roles of
ADAMTSs in Angiogenesis and Cancer. Cancers (Basel). 4:1252–1299.
2012. View Article : Google Scholar
|
|
50
|
Bonnans C, Chou J and Werb Z: Remodelling
the extracellular matrix in development and disease. Nat Rev Mol
Cell Biol. 15:786–801. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Goldman O, Han S, Sourisseau M, Dziedzic
N, Hamou W, Corneo B, D'Souza S, Sato T, Kotton DN, Bissig KD, et
al: KDR identifies a conserved human and murine hepatic progenitor
and instructs early liver development. Cell Stem Cell. 12:748–760.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Basu P, Chandna P, Bamezai RNK, Siddiqi M,
Saranath D, Lear A and Ratnam S: MassARRAY spectrometry is more
sensitive than PreTect HPV-Proofer and consensus PCR for
type-specific detection of high-risk oncogenic human papillomavirus
genotypes in cervical cancer. J Clin Microbiol. 49:3537–3544. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim MJ, Lee EJ, Chun SM, Jang SJ, Kim DS,
Lee DH and Youk EG: The significance of ectopic crypt formation in
the differential diagnosis of colorectal polyps. Diagn Pathol.
9:2122014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Matsuura K, Sawai H, Ikeo K, Ogawa S, Iio
E, Isogawa M, Shimada N, Komori A, Toyoda H, Kumada T, et al
Japanese Genome-Wide Association Study Group for Viral Hepatitis:
Genome-wide association study identifies TLL1 variant associated
with development of hepatocellular carcinoma after eradication of
hepatitis C virus infection. Gastroenterology. 152:1383–1394. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Peters MG and Terrault NA: Alcohol use and
hepatitis C. Hepatology. 36(Suppl 1): S220–S225. 2002.PubMed/NCBI
|
|
56
|
Aleksandrova K, Stelmach-Mardas M and
Schlesinger S: Obesity and liver cancer. Recent Results Cancer Res.
208:177–198. 2016. View Article : Google Scholar : PubMed/NCBI
|