Introduction
Arctigenin is a bioactive lignan and a member of the
Asteraceae family isolated from the seeds of Arctium lappa.
The roots of Arctium lappa, commonly known as greater
burdock, are consumed as a vegetable worldwide. Dried burdock roots
have been used in folk medicine as a diuretic, diaphoretic and
blood purifying agent (1).
Moreover, arctigenin has also been shown to have anti-viral
(2–4), anti-inflammatory (5) and immunomodulatory activities
(6). It is one of the major
ingredients in Essiac tea, which is used as an alternative
treatment for certain types of cancer (7). The anticancer effects of arctigenin
have also been reported in various human cancer cell lines
(8–16), in which arctigenin induced
apoptosis mainly via the mitochondrial pathway (8,10,12,15,16)
and cell cycle arrest. Reports of G0/G1 arrest in lung (13,14)
and bladder (16) cancer cells,
but G2/M arrest in colon cancer cells (17), suggest that arctigenin exerts
differential effects at the molecular level in different cell
types.
However, studies on the effects of arctigenin in
breast cancer cells are limited. Hsieh et al demonstrated
that arctigenin markedly inhibited the growth of estrogen receptor
(ER)α-negative MDA-MB-231 cells by triggering the mitochondrial
apoptotic pathway (8). Another
recent study demonstrated that signal transducer and activator of
transcription 3 (STAT3) was inhibited by arctigenin, leading to
apoptosis and tumor suppression of the same breast cancer cell line
(18). We have also previously
reported that arctigenin exerts anti-metastatic effects in breast
cancer cells, regardless of ERα expression (19). The effects of arctigenin on ERβ in
Th17 cells as an agonist have also been reported (20). However, to the best of our
knowledge, its effects on hormone-responsive breast cancer cells
have not yet been fully elucidated. Although both ER receptors
display extensive similarities, in that they bind to estradiol and
related compounds and initiate the transcription of DNA sequences
referred to as estrogen response elements (ERE), they are encoded
by distinct genes located on different chromosomes and have
significantly different primary sequences in their ligand binding
domains (21). Thus, ER subtypes
can bind certain ligands with different affinities, and these
ligands may also have different agonist or antagonist characters
downstream. Due to the diversity of estrogen target tissues, and
given that the levels and proportion of ERα and ERβ differ in
different target cells, there is some overlap between ERα and ERβ,
but also some differences with respect to ligand interaction or
activity that may be important in the biological actions at the
tissue level. In breast cancer, ERβ is considered to play a
protective role. ERβ is lost in the majority of breast tumors
(22) as a result of ERβ promoter
methylation and is a possible tumor suppressor gene. On the other
hand, ERα expression is measured in order to come to a clinical
decision on the treatment of course for patients with breast
cancer. Since it has been reported that arctigenin is a
phytoestrogen (23), there may be
a possibility that it could bind to ERα and exert an inhibitory or
proliferative effect in ERα-positive breast cancer cells.
Therefore, it is important to understand the effects of arctigenin
on ERα-positive cells.
Thus, in this study, to determine whether arctigenin
is safe for consumption by patients with ERα-positive breast
cancer, we investigated the effects of arctigenin on ERα-positive
MCF-7 human breast cancer cells. We examined its effects on cell
viability and explored the underlying mechanisms. We also examined
the effects of arctigenin on ERα expression in order to verify
whether it affects the cytotoxicity of tamoxifen.
Materials and methods
Cell culture
MCF-7 and MDA-MB-231 human breast cancer cells were
purchased from the Korean Cell Line Bank (Seoul, Korea). MCF-7
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
Welgene Daegu, Korea) supplemented with 10% fetal bovine serum-FBS
(ATCC, Rockville, MD, USA), 10 µg/ml insulin (Welgene) and
1% antibiotic-antimycotic solution (Welgene). Phenol red-free DMEM
(Welgene) containing 10% dextran-coated charcoal-stripped FBS and
10 nM estradiol (Sigma, St. Louis, MO, USA) was used only for the
experiments with tamoxifen (AG Scientific Inc., San Diego, CA,
USA). For use as positive controls of RIPK3, MDA-MB-231 cells were
cultured in DMEM supplemented with 10% FBS and
antibiotic-antimycotic solution.
SRB cell cytotoxicity assay
A total of 5,000 cells/well seeded in 96-well plates
were treated with conditioned medium containing 0, 1, 2.5, 5, 10,
25, 50, 100 and 200 µM arctigenin (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and 2% FBS for 24, 48 or 72 h. The
medium was removed, and the cells were fixed with 100 µl
ice-cold 20% trichloroacetic acid (Samchun Pure Chemical,
Pyongtaek, Korea) for 1 h at 4°C. The plates were then washed 5
times under slow-running tap water and allowed to air-dry
overnight. A solution of 50 µl 0.4% sulforhodamine B (SRB;
Sigma) was added for staining at room temperature for 30 min.
Unbound SRB was washed off with 1% acetic acid, and the plate was
air-dried. Bound SRB was solubilized by the addition of 100
µl of 10 mM Tris pH 10.5 to each well and shaken for 5 min
before reading the absorbance at 510 nm using a Multi-Detection
Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
Colony formation assay
A total of 700 cells/well seeded in 6-well plates
were treated with conditioned medium containing 0, 2.5, 50 and 200
µM arctigenin in 2% FBS, and were allowed to grow for a week
until the colonies of appropriate size were formed. The medium was
removed, and the cells were fixed with 1 ml 10% formaldehyde for 20
min at room temperature and washed with PBS. The colonies were
stained with 1 ml 0.01% crystal violet (Sigma) for 40 min at room
temperature and washed with PBS and air-dried. Light microscopic
images were captured and colonies were counted using ImageJ
software (National Institute of Mental Health, Bethesda, MD, USA),
as per the manufacturer's instructions.
Cell cycle analysis by flow
cytometry
For cell cycle analysis, the cells grown in medium
containing 0, 2.5, 50 and 200 µM arctigenin and 2% FBS for
72 h were trypsinized after washing with PBS, centrifuged at 100 ×
g for 3 min, and fixed in cold 70% ethanol. Following
centrifugation, the cells were washed with PBS containing 2% FBS
and stained in the dark with 20 µg/ml propidium iodide
(Sigma) and 40 µg/ml RNase A (Sigma) for 30 min at room
temperature. The cells were then analyzed using a FACS Calibur II
flow cytometer (BD Biosciences, San Jose, CA, USA).
Protein extraction and western blot
analysis
The cells were treated with conditioned medium
containing 0, 1, 2.5, 5, 10, 25, 50, 100 and 200 µM
arctigenin for 72 h. For treatment with inhibitors, the cells were
treated with 500 nM MG132 (Cayman Chemical, Ann Arbor, MI, USA), 10
µM chloroquine (Sigma), 20 mM NH4Cl (Stemcell
Technologies, Vancouver, Canada), 2.5 µM decitabine (AZA; LC
Laboratories, Woburn, MA, USA), or 5 nM trichostatin A (TSA;
ApexBio, Houston, TX, USA) 30 min prior to arctigenin treatment.
The cells were lysed with RIPA buffer (50 mM Tris-HCl pH 7.5 with
150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium
dodecyl sulphate-SDS and 2 mM EDTA) containing phosphatase and
protease inhibitor cocktail (GenDEPOT, Barker, TX, USA). In the
lysates, the debris was cleared off by centrifugation at 17,000 × g
for 20 min at 4°C and protein concentrations were determined using
bicinchoninic acid reagent (Sigma). The proteins were separated by
SDS-PAGE, and transferred onto polyvinylidene fluoride (PVDF)
membranes at 100 V for 40 min. The membranes were blocked in 5%
skim milk in Tris-buffered saline-Tween-20 buffer (10 mM Tris-HCl,
150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature. The
following primary monoclonal antibodies from Cell Signaling
Technology (Beverly, MA, USA): B-cell lymphoma-extra-large (Bcl-xL,
#2764), apoptosis-inducing factor (AIF, #5318), poly(ADP-ribose)
polymerase (PARP, #9542), caspase-7 (#9492), microtubule-associated
protein 1A/1B-light chain 3 (LC3A/B, #4108), estrogen receptor α
(ERα, #8644) phosphorylated forms of histone 2A.X (p-H2A.X, #9718),
ataxia telangiectasia mutated-Rad3-related (p-ATR, #2853),
phosphorylated and total forms of mechanistic target of rapamycin
(p-mTOR, #2971; and mTOR, #2972), ribosomal protein S6 kinase
beta-1 (p-S6K1, #9205; and S6K, #9202), ribosomal protein S6 (p-S6,
#4858; and S6, #2217), adenosine monophosphate-activated protein
kinase (p-AMPK, #2535; and AMPK, #2532), or from Santa Cruz
Biotechnology: RIPK3 (sc-374639), RAD51 (sc-8349), β-actin
(sc-69879) were diluted 3,000-fold and incubated with the blots
overnight at 4°C. Corresponding HRP-conjugated secondary antibody
from Santa Cruz Biotechnology goat anti-rabbit (sc-2004) or goat
anti-mouse (sc-2005) was diluted 5,000-fold, and incubated with the
blots for 2 h at room temperature. The blots were developed and
imaged using Luminescent Image Analyzer LAS-4000 (Fujifilm, Tokyo,
Japan). Densitometric analysis was performed using Scion Image
(Scion Corporation, Frederick, MD, USA) with data from at least 3
independent experiments.
RNA extraction and cDNA synthesis
RNA was isolated from the cells treated with
conditioned medium containing arctigenin or rapamycin (LC
Laboratories) using the easy-BLUE™ Total RNA Extraction kit (iNtRON
Biotechnology, Inc., Sungnam, Korea), which was performed in
accordance with the manufacturer's instructions. The RNA
concentrations were determined using a NanoDrop spectrophotometer
(Schimadzu Scientific Instruments, Columbia, MD, USA). A total of 1
µg of total RNA was reverse transcribed using a Primescript
Reverse Transcriptase (Takara, Shiga, Japan).
Real-time PCR
Real-time PCR reactions were performed in triplicate
in 10 µl total volumes with 1 µl of each primer (0.5
µM final concentrations) and 5 µl of SYBR-Green qPCR
2X master mix (Cell Safe, Suwon, Korea) in the Eco™ Real-Time PCR
system (Illumina Inc., San Diego, CA, USA). The PCR products were
verified by a melt curve analysis and relative intensity compared
with the controls. The primer sequences and annealing temperatures
for the target genes were as follows: progesterone receptor (PR)
forward, 5′-AAC TTG CAT GAT CTT GTC AAA CA-3′ and reverse, 5′-CAC
CAT CCC TGC CAA TAT CT-3′, 57°C; ERα forward, 5′-CAC ATG AGT AAC
AAA GGC ATG G-3′ and reverse, 5′-ATG AAG TAG AGC CCG CAG TG-3, 58°C
and GAPDH (amplified as an internal control) forward, 5′-ATC CCA
TCA CCA TCT TCC AG-3′ and reverse, 5′-TTC TAG ACG GCA GGT CAG
GT-3′.
Determination of combined drug
interactions
To determine the synergistic, additive, or
antagonistic effects of arctigenin in combination with tamoxifen,
various concentrations of tamoxifen (0, 5 and 10 µM) were
combined with various concentrations of arctigenin for the
determination of their effects on MCF-7 cell proliferation. The
association between the dose and effect for single agents and their
combinations were analyzed, as previously described by Chou and
Talalay (24) using the Compusyn
Version 1.0 free software package (Compusyn Inc., USA). The
combination index (CI) values were calculated for each dose and the
corresponding effect level, presented as the fraction affected
(Fa). The Fa-CI graph was plotted for a graphical representation of
drug interactions. The CI values provide a quantitative definition
for the additive effect (CI=1), the synergistic effect (CI<1)
and the antagonistic effect (CI>1) of drug combinations.
Statistical analysis
Statistical significance was determined using
one-way ANOVA and Tukey's post hoc test with SPSS V20.0 software
(SPSS, Inc., Chicago, IL, USA). The results are presented as the
means ± SD. P-values <0.05 were considered to indicate
statistically significant differences.
Results
Arctigenin is cytotoxic, not cytostatic,
to ERα-positive MCF-7 human breast cancer cells
First, we assessed the effects of arctigenin on the
viability of ERα-positive human breast cancer cells. The MCF-7
cells were grown in medium containing various concentrations of
arctigenin for 24, 48 or 72 h. SRB cell cytotoxicity assay revealed
that the effects of arctigenin were concentration- and
time-dependent. Treatment with arctigenin at a concentration as low
as 1 µM for 24 h significantly decreased the viability of
the MCF-7 cells, and treatment with arctigenin at 200 µM for
72 h inhibited cell viability by as much as 50% (Fig. 1A).
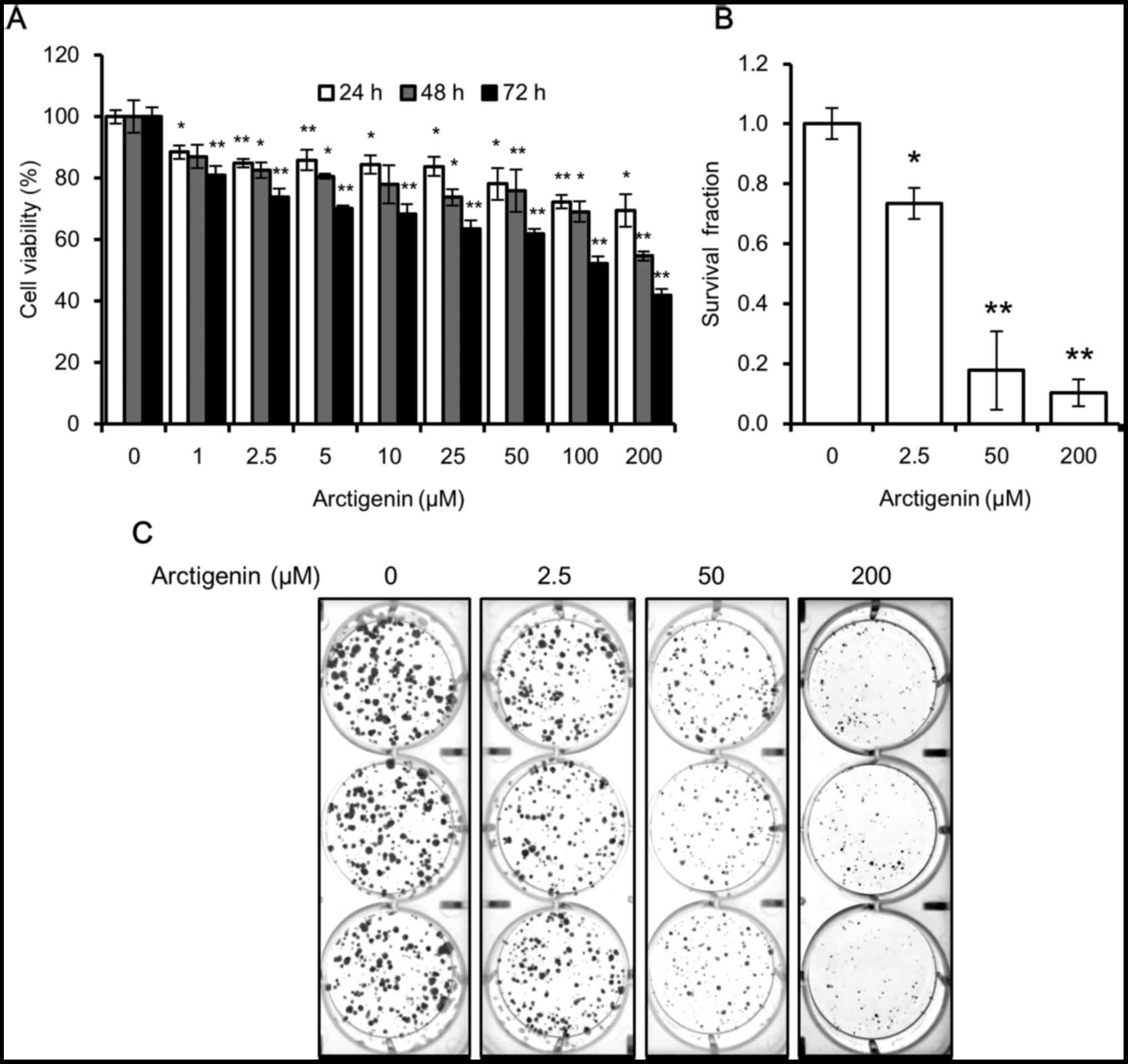 | Figure 1Arctigenin is cytotoxic to MCF-7
human breast cancer cells. (A) The viability of the MCF-7 cells
treated with 0, 1, 2.5, 5, 10, 25, 50, 100 and 200 µM
arctigenin for 24, 48, or 72 h measured by SRB assay was decreased
in a concentration- and time-dependent manner. (B) Quantification
of colony formation assay, using ImageJ software, of cells treated
with 0, 2.5, 50 and 200 µM arctigenin for 72 h and then
allowed to grow in arctigenin-free medium for 2 weeks confirmed
that arctigenin was not cytostatic but cytotoxic. (C)
Representative images of colony formation assay. Graphs represent
the means ± SD of at least 3 independent experiments. P-values were
compared to the controls treated with 0 µM arctigenin;
*P<0.05 and **P<0.01. |
However, from the SRB assay, we were unable to
determine whether arctigenin suppressed cell proliferation
(cytostatic effect) or induced cell death (cytotoxic effect).
Hence, we conducted a colony formation assay to confirm this. When
the cells were treated with arctigenin for 72 h and then allowed to
grow in arctigenin-free medium, the survival fraction of the cells
treated with 50 µM arctigenin was 20% that of control, and
this was even lower in the cells treated with 200 µM
arcti-genin (Fig. 1B and C).
Arctigenin suppressed the ability of the cells to form colonies and
confirmed the cytotoxic effects of arctigenin.
Arctigenin treatment does not induce the
apoptosis or necrop-tosis of MCF-7 human breast cancer cells
Since arctigenin was found to be cytotoxic, we
wished to determine whether the cytotoxic effects of arctigenin are
mediated through the induction of cell cycle arrest, as arctigenin
has previously been reported to affect the cell cycle (10,14,16).
Surprisingly, in contrast to the findings of other studies, the
results of cell cycle analysis revealed that treatment with
arctigenin for up to 72 h had no effect on the MCF-7 cells
(Fig. 2A).
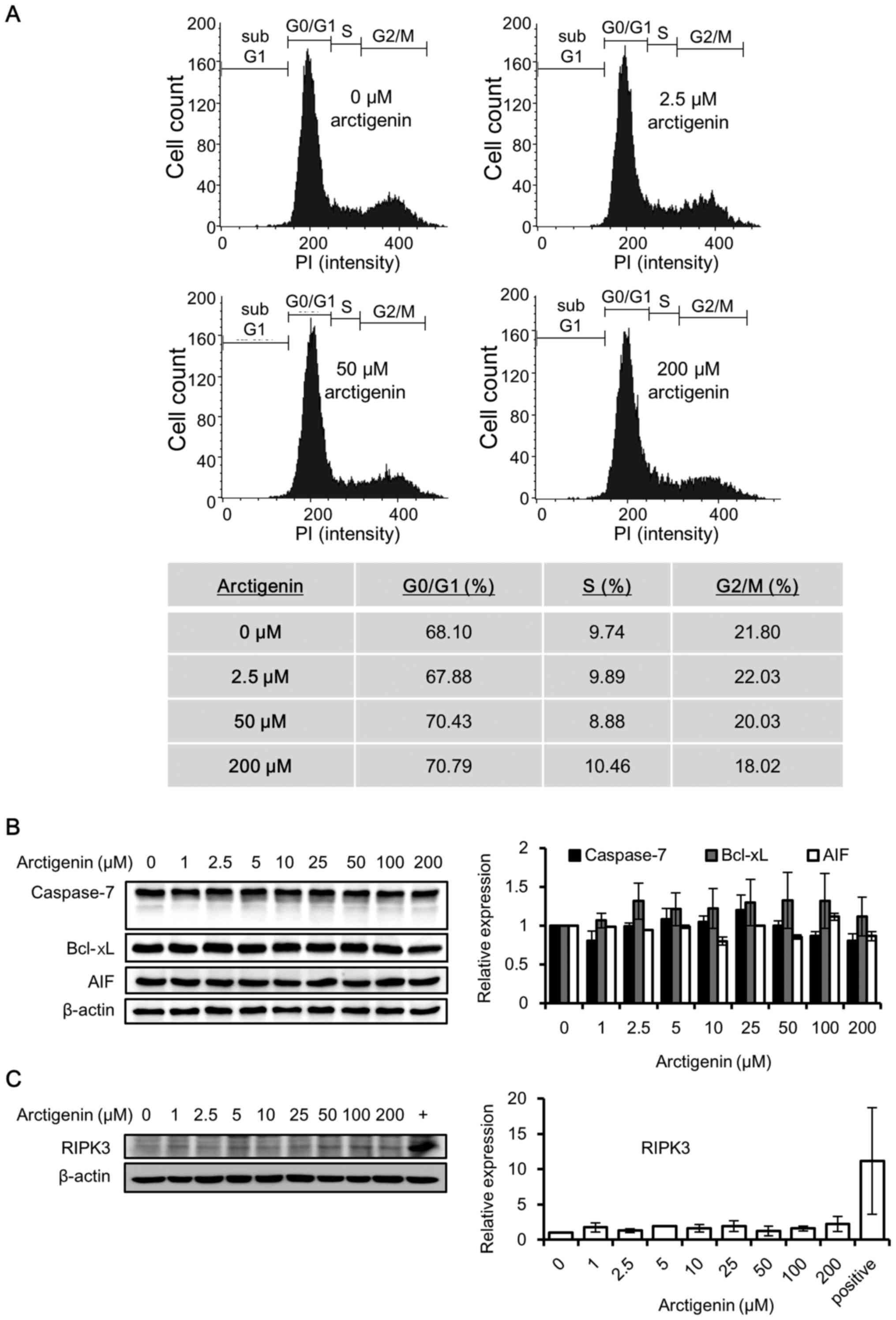 | Figure 2Arctigenin does not cause cell cycle
arrest, apoptosis, or the necroptosis of MCF-7 human breast cancer
cells. (A) Flow cytometric analysis and population distribution of
MCF-7 cells treated with 0, 2.5, 50 and 200 µM arctigenin
for 72 h. The x-axis represents the fluorescence intensity of
propidium iodide and the y-axis represents the number of
cells/channel. (B) Western blot analysis revealed that treatment of
the MCF-7 cells with arctigenin for 72 h did not alter the levels
of the apoptotic markers: The anti-apoptotic protein Bcl-xL,
apoptosis- inducing factor (AIF) and the effector caspase,
caspase-7. (C) Treatment with arctigenin for 72 h failed to induce
the expression of the necroptosis marker, RIPK3, confirming that
arctigenin does not cause the necroptosis of MCF-7 cells. (+)
MDA-MB-231 human breast cancer cells, previously reported (27) to express detectable levels of
RIPK3, were used as the positive control. Graphs represent the
means ± SD of at least 3 independent experiments. P-values were
compared to the controls treated with 0 µM arctigenin, and
the results revealed no statistical significance. |
Arctigenin has previously been reported to induce
the apoptosis of several cancer cell lines (8–12,14–16).
Since the MCF-7 cells do not express caspase-3 (25), we examined the effector caspase,
caspase-7, which is known to induce the apoptosis of MCF-7 cells.
The caspase-7 levels remained unaltered with arctigenin treatment,
and its activation in the form of cleaved caspase was not detected
(Fig. 2B). The expression levels
of Bcl-xL, the anti-apoptotic protein involved in preventing
apoptosis via the mitochondrial pathway, and AIF were also not
altered by treatment with arctigenin (Fig. 2B). Hence, our data indicated that
arctigenin did not induce the apoptosis of MCF-7 cells.
Necroptosis is a non-apoptotic form of cell death
that resembles necrosis morphologically; nonetheless, it can be
induced by the activation of death receptors. Several stimuli are
known to induce necroptosis via protein kinase receptor-interacting
serine/threonine-protein kinase 3 (RIPK3), which is now an accepted
marker for necroptosis (26).
Hence, in this study, we analyzed whether arctigenin induced the
necroptosis of the MCF-7 cells. Untreated MDA-MB-231 human breast
cancer cells were used as a positive control, since they have
previously been shown to express RIPK3 (27). Our results from western blot
analysis (Fig. 2C) revealed that
treatment with arctigenin at a concentration as high as 200
µM for up to 72 h did not induce RIPK3 expression.
Therefore, it was concluded that the cytotoxic effects of
arctigenin on the MCF-7 cells did not involve apoptosis or
necroptosis.
Cytotoxic effects of arctigenin on MCF-7
human breast cancer cells are caused by mTOR inhibition, leading to
autophagic cell death and DNA damage
Finally, we examined whether the arctigenin-induced
cell cytotoxicity was due to autophagic cell death. Autophagy is a
bulk recycling stress response in cells. It predominantly activates
the catabolic pathways that help cells survive under conditions in
which the nutrient or energy supply is low, or in which there are
other forms of stress. In the case of cancer cells, it can aid in
cell survival, as well as in the development of drug resistance
(28). However, it has also been
reported that cells undergo autophagy-induced cell death, if the
stress is beyond their comprehension. Previous studies have
indicated that arctigenin induces autophagy, which can lead to the
death of several cell lines (11,20,29–31).
Therefore, in this study, to examine the autophagic activity of
MCF-7 cells in response to arctigenin treatment, we analyzed the
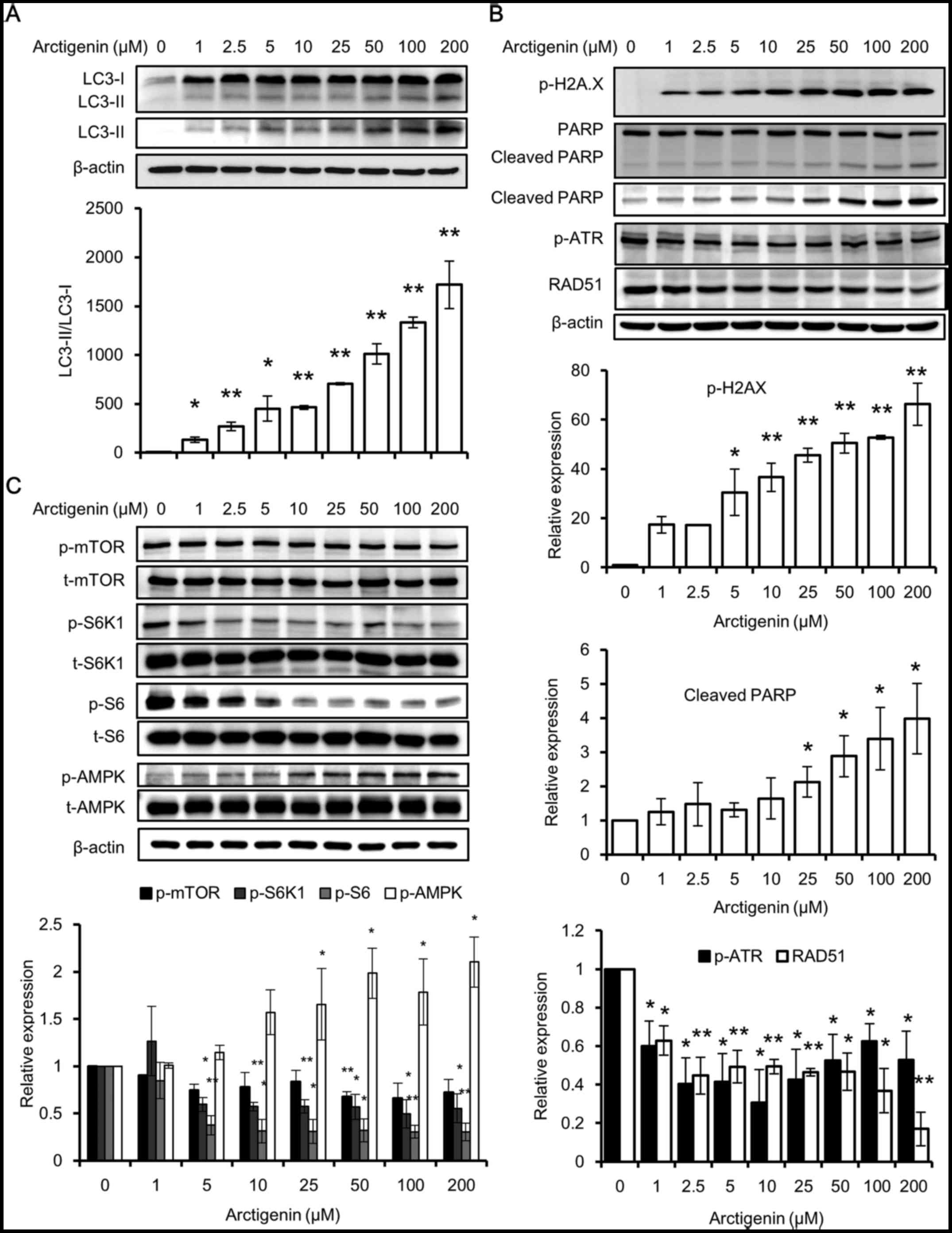
conversion of LC3 (LC3-I) into LC3-II. LC3 is a specific marker of
autophagolysosome formation, which occurs throughout the process of
autophagy (32), and a higher
ratio of LC3-II/LC3-I means a higher autophagic activity. Using
western blot analysis, we found that the untreated cells had no
detectable levels of LC3-II and very low levels of LC3-I. The
expression of both LC3 types along with the ratio of LC3-II/LC3-I
increased, in a concentration-dependent manner with arctigenin
treatment (Fig. 3A). This
indicated that treatment with arctigenin at a concentration as low
as 1 µM induced autophagy in MCF-7 cells.
The results of western blot analyses also revealed
that the phosphorylation of H2A.X and PARP cleavage was induced in
a concentration-dependent manner with arctigenin treatment
(Fig. 3B). H2A.X is a histone H2A
family protein, which is phosphorylated and recruited to sites of
double-strand DNA breaks and hence is considered a marker for DNA
damage (33). PARP is a DNA repair
enzyme cleaved by activated caspases. Its fragmentation is a marker
of apoptosis. The levels of the DNA repair response proteins, RAD51
(34) and phosphorylated ATR
(35), which are increased in
cases of DNA damage, were found to be decreased. This suggested
that the DNA repair response was inadequate (Fig. 3B). Although phosphorylated H2A.X
and cleaved PARP are both considered to be markers of apoptosis,
specifically they are markers of DNA damage and cell death
(33). In this study, apoptosis
was ruled out as the levels of apoptosis signaling proteins were
not affected, and therefore, we hypothesized that DNA damage may
have occurred due to autophagic cell death.
Autophagy is a highly conserved process that is
tightly regulated by several pathways. One of the well-studied
regulators of autophagy is the mTOR pathway, which directly
inhibits autophagy (28). The
induction of autophagy by arctigenin via the inhibition of the mTOR
pathway has previously been reported in other cell lines (11,20,29–31).
In line with the findings of these previous studies, we found that
arctigenin treatment inhibited downstream effector molecules of
mTOR by suppressing the activation of ribosomal protein S6 and its
kinase (Fig. 3C). A crosstalk
between mTOR and AMPK pathways has been well defined (28). The phosphorylation of AMPK is a
marker of low energy in cells and has also been found to be
activated prior to autophagy (36). Predictably, even in our study, we
detected the increased phosphorylation of AMPK with arctigenin
treatment (Fig. 3C). Taken
together, our data suggested that arctigenin inhibited downstream
effector molecules of the mTOR signaling pathway in the MCF-7
cells, resulting in autophagic cell death and DNA damage.
Inhibition of mTOR pathway by arctigenin
results in a decreased expression of ERα and its downstream
signaling
Although we found that arctigenin exerted cytotoxic
effects on ERα-positive MCF-7 cells, as it is a phytoestrogen, we
wished to determine whether it interferes with the cytotoxic
effects of tamoxifen. Tamoxifen therapy is most widely used in the
treatment of hormone-responsive cancers. It acts by binding to ERα
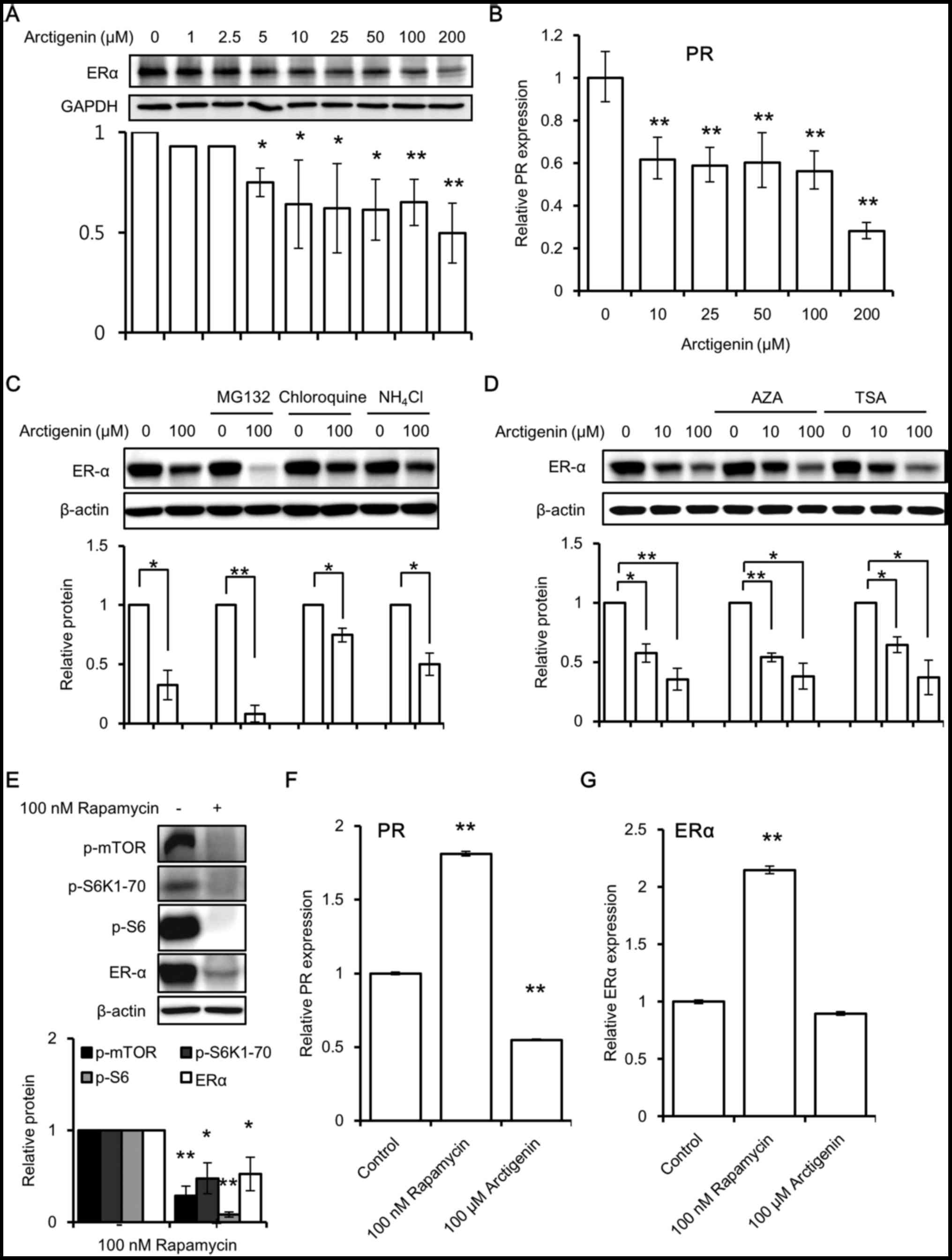
and blocking its downstream signaling (37). Upon arctigenin treatment, we
observed a dose-dependent decrease in ERα protein expression
(Fig. 4A) in the MCF-7 cells.
Predictably, a corresponding decrease in the downstream
transcription of the ERE responsive gene, PR (Fig. 4B) was also observed. Subsequently,
we wished to elucidate the mechanisms behind this effect.
First, we examined whether the arctigenin-induced
decrease in ERα expression was due to an increase in protein
degradation. In the absence of a substrate, the half-life of ERα is
up to 5 days. However, binding to estradiol accelerates the ERα
turnover and reduces its half-life to approximately 3 h (38). Although the exact mechanisms are
unclear to date, it is known that substrate-induced proteolysis,
via the ubiquitin proteasome pathway, is required for the
activation of ERα receptors (38).
In this study, the cells were treated with inhibitors of
proteasomal degradation (38)
(MG132) and lysosomal degradation (chloroquine and
NH4Cl) prior to arctigenin treatment. This did not
hinder the decrease in ERα expression induced by arctigenin
(Fig. 4C). This suggested that
arctigenin treatment did not influence protein degradation in the
MCF-7 cells.
Second, we investigated whether epigenetic
modifications play a role in the arctigenin-induced decrease in ERα
expression (39). It has been
reported that arctigenin induces histone modification in
ER-negative breast cancer cells (8). Therefore, we wished to determine
whether arctigenin decreased ERα expression by altering the enzymes
involved in DNA methylation or histone modifications. Pre-treatment
with inhibitors of DNA methylation (AZA) or histone deacetylation
(TSA) failed to have any effect on the arctigenin-induced
inhibition of ERα protein expression (Fig. 4D).
Finally, we examined whether mTOR inhibition would
result in a decreased ERα expression. Similar to treatment with
arctigenin, treatment with rapamycin, which is a specific mTOR
inhibitor, also inhibited ERα protein expression (Fig. 4E), suggesting that mTOR inhibition
could lead to a decreased ERα protein expression, probably due to
protein synthesis inhibition. However, in contrast to the effects
of arctigenin, the results of real-time PCR revealed that the mRNA
levels of the downstream ERE responsive gene, PR (Fig. 4F), and those of ERα (Fig. 4G) were found to be increased with
rapamycin treatment. It has been previously reported that growth
factors may inhibit PR expression via a non-genomic
PI3K/Akt/mTOR-dependent pathway that is independent of ER (40); the exact mechanisms responsible for
this remain to be elucidated. We hypothesized that removing this
inhibition with an mTOR inhibitor could lead to an increased
transcription of PR. However, we did not further pursue the
mechanisms of action of rapamycin, since it was beyond the scope of
this study. Arctigenin decreased the mRNA levels of PR, but had no
effect on those of ERα compared to the control. This suggests that
the mTOR and ER pathways are tightly regulated at multiple levels.
On the whole, our data indicated that arctigenin inhibited mTOR
downstream effector molecules, including protein synthesis, and
hence the synthesis of ERα.
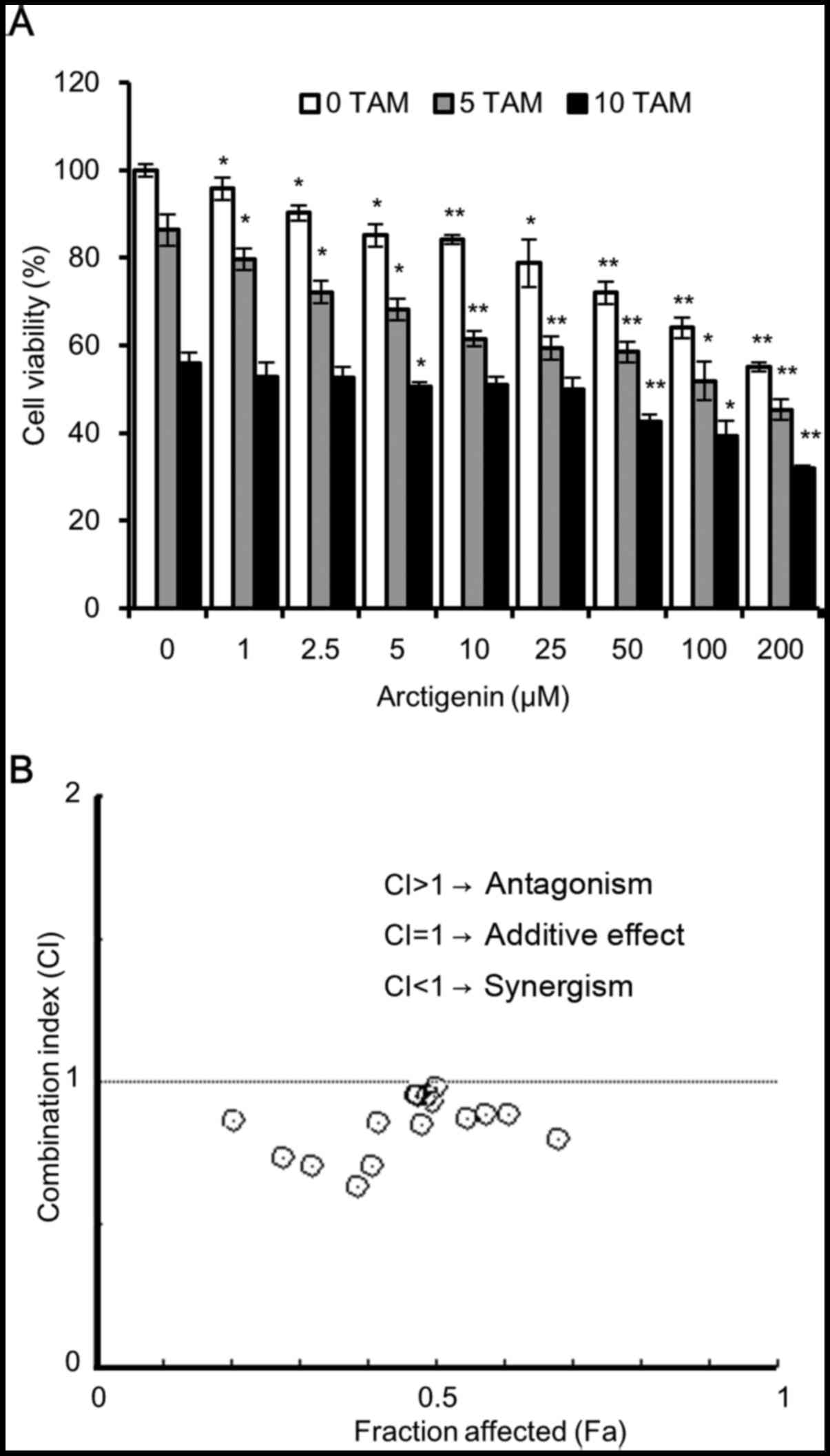
Co-treatment of the MCF-7 cells with
arctigenin and tamoxifen exerts a synergistic effect
Since we observed that arctigenin treatment led to a
decrease in ERα expression, it is possible that it could affect the
sensitivity of MCF-7 cells to tamoxifen. To assess cell
cytotoxicity, we treated the MCF-7 cells with various
concentrations of arctigenin in the presence of 5 or 10 µM
tamoxifen. The results of SRB assay revealed that both arctigenin
and tamoxifen inhibited MCF-7 cell viability, in a
concentration-dependent manner, with an enhanced effect from
combined treatment (Fig. 5A). To
quantitatively validate our observations, we used the method
described in the study by Chou and Talalay (24) and calculated the dose-effect
associations for each drug and their combinations using Compusyn
software. The combination index (CI) and the corresponding effect
level, designated as the fraction affected (Fa), were calculated
for each concentration, and the Fa-CI graph was plotted. The CI
values provide a quantitative definition for the additive effect
(CI=1), synergistic effect (CI<1), and antagonistic effect
(CI>1) in drug combinations. As shown in Fig. 5B, for most combinations of
tamoxifen and arctigenin, the CI was <1, indicating that
arctigenin treatment did not interfere with the sensitivity of the
cells to tamoxifen, but indeed had a synergistic effect. Hence, we
also propose that arctigenin may safely be used as an effective
supplement, even for patients undergoing endocrine therapy.
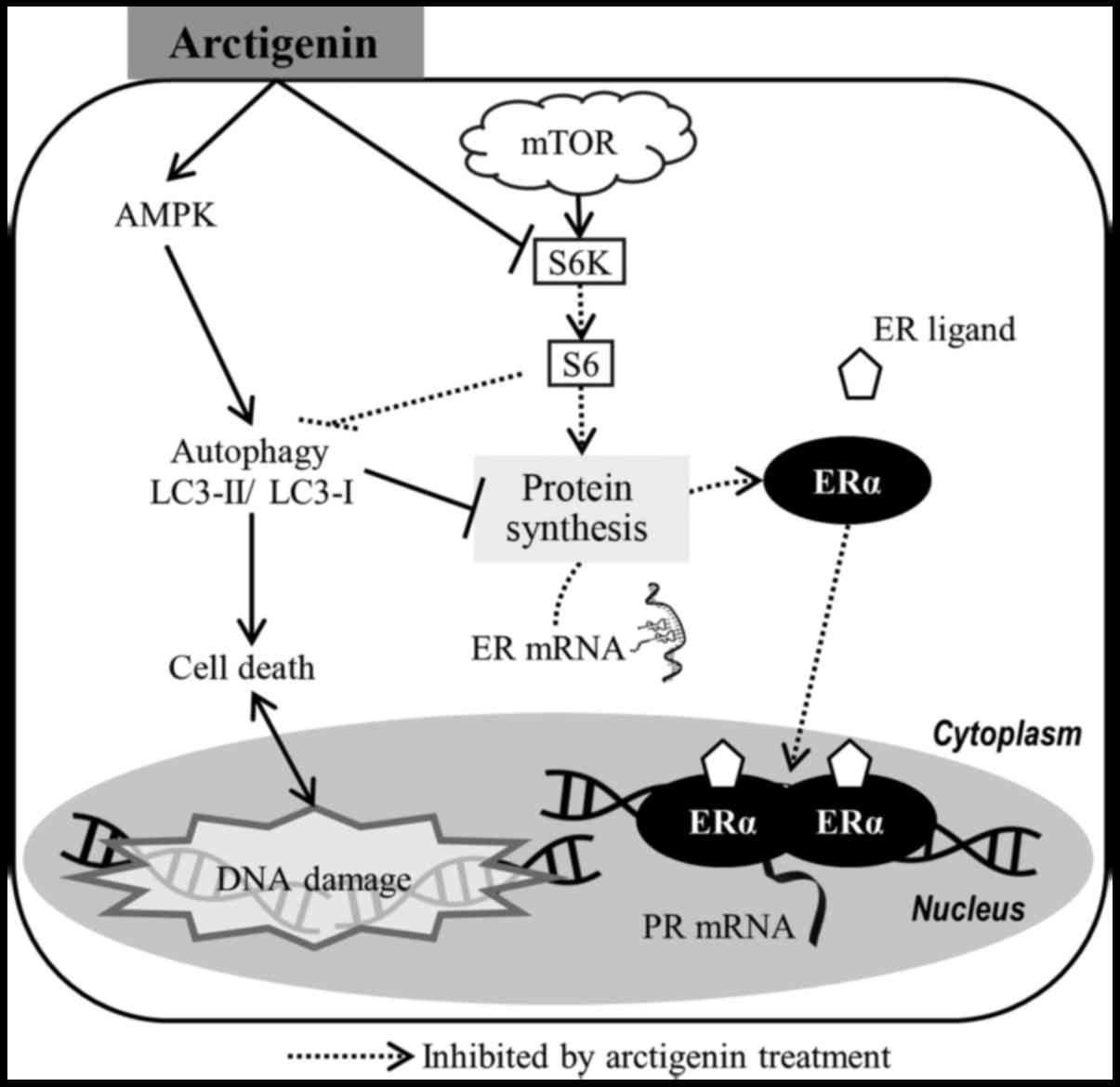
Taken together, our data suggest that the
phytoestrogen, arctigenin, inhibited downstream effector molecules
of the mTOR pathway in ER-positive MCF-7 human breast cancer cells,
leading to autophagy-induced cell death and the downregulation of
ERα (Fig. 6). This, however, did
not affect the sensitivity of the cells to tamoxifen.
Discussion
Phytoestrogens are plant-derived chemical compounds
with estrogenic activities (41).
Their affinities for estrogen receptors are at least
1,000-10,000-fold lower than those of estradiol; nonetheless, they
are capable of binding to estrogen receptors and initiating or
disrupting estrogen-dependent transcriptions, which could then lead
to cell proliferation or growth inhibition. The role of
phytoestrogens on breast cancer cells is a highly debated topic.
Although phytoestrogens have been found to have an agonistic affect
that can lead to cell proliferation in vitro (42), it has been found that their
consumption has the opposite effect, presenting with benefits of
cancer prevention (41). These
anomalies remain unresolved; however, the benefits of
phytoestrogens on breast cancer cells are likely mediated through
other pathways, and not through estrogen signaling. Since
arctigenin and its metabolites have been reported to have
estrogenic properties (23), an
investigation of the effects of arctigenin on ERα-positive breast
cancer cells is warranted.
In this study, we demonstrated that arctigenin was
cytotoxic to MCF-7 human breast cancer cells. Previously, several
studies have reported the antitumor effects of arctigenin by the
induction of apoptosis (8–12,14–16,18).
However, in this study, arctigenin did not trigger any apoptotic
signals. Phosphorylated H2A.X and cleaved PARP were found to be
induced by arctigenin. PARP cleavage in the arctigenin-treated
MCF-7 cells has been recently reported (43). However, since the levels of the
effector caspase, caspase-7 and upstream signals were not altered,
it was deduced that cell death may have involved non-apoptotic
pathways, and p-H2A.X and PARP cleavage were due to extensive DNA
damage. The possibility of arctigenin causing necroptosis was also
ruled out due to the absence of the necroptosis marker, RIPK3.
In this study, arctigenin was found to inhibit mTOR
downstream effector molecules. The inhibition of the mTOR pathway
is known to directly induce autophagy as mTOR inhibits autophagy
via Ulk1 (28), and suppresses
protein translation, cell growth and proliferation, all of which
influence tumorigenesis. It has previously been reported that
arctigenin inhibits the mTOR pathway, which leads to endoplasmic
reticulum stress (29) and
apoptosis (11) in liver cancer
cells, to an anti-colitis effect in immune cells (30), and to β-amyloid clearance in an
Alzheimer's disease model (31).
It has also been reported that arctigenin inhibits mTORC1
activation by binding to ERβ in Th17 cells (20). Hence, our data also suggest that
the mTOR pathway may be an important target site for the effects of
arctigenin.
Moreover, arctigenin was also found to significantly
downregulate ERα protein expression and downstream transcription of
PR, which is an ERE responsive gene. We ruled out the role of
protein degradation and epigenetic modifications in the
arctigenin-induced decrease in ERα protein expression using
specific inhibitors. The downregulation of ERα protein expression
and its transcriptional activity by the mTOR inhibitor, everolimus,
has previously been reported (44). We also compared the effects of
arctigenin on MCF-7 cells to another well-known mTOR inhibitor,
rapamycin, and found a decreased ERα protein expression. Hence, we
inferred that the decrease in ERα expression with arctigenin
treatment may have been a consequence of the inhibition of the mTOR
pathway molecules, particularly due to the decreased activation of
ribosomal protein S6, which is required for protein synthesis.
However, in contrast to arctigenin, rapamycin increased the mRNA
levels of ERα and those of the downstream transcription of ERE
gene. Although the exact mechanisms involved are unclear,
PI3K/Akt/mTOR has been reported to inhibit ERE transcription. There
have been reports suggesting that during IGF-I signaling, the
removal of this inhibition with an mTOR inhibitor could lead to
increased transcription of PR (40). This indicates that the ER and mTOR
pathways are inter-related and are regulated at several levels, and
the effects of each drug need to be evaluated individually to
better ascertain their pharmacological effects. Our data suggest
that for patients with breast cancer, arctigenin may be a better
inhibitor of mTOR compared with rapamycin, as it does not increase
ERE transcription, which could lead to cell proliferation.
The decrease in ERα protein expression compelled us
to determine whether arctigenin interferes with the effect of
tamoxifen, and found that arctigenin in combination with tamoxifen
treatment resulted in a synergistic effect. Hence, according to our
data, the effects of arctigenin on breast cancer are likely a
double-edged sword. Arctigenin inhibited both ER and mTOR
downstream effector molecules. The benefits of inhibiting ER
signaling in cancer therapy have been shown by the use of
tamoxifen. More recently, fulvestrant, a 'real' anti-estrogen, is
gaining popularity as a means to treat breast cancer therapy for
its effectiveness in binding, blocking, and degrading the ER
(45). We demonstrated that
arctigenin also decreased ER protein expression and blocked
down-stream signaling. Moreover, we showed that arctigenin
inhibited mTOR downstream effector molecules. Preclinical studies
have shown that ER-positive breast cancer cells require
hyperactivation of the PI3K/Akt/mTOR pathway for an adaptation to
hormone-independent growth after long-term estrogen deprivation.
There is evidence to indicate that the combined inhibition of ER
and the mTOR pathway may be beneficial in the early lines of
treatment (46). Drug combination
studies have shown that mTOR inhibitors exert synergistic effects
with anti-estrogens, including tamoxifen (47,48),
and blocking both pathways not only enhances the antitumor
activity, but also reverses the resistance to endocrine therapy.
Finally, since arctigenin is a plant extract, it may be safe for
human consumption, as it has no notable side-effects. Burdock root,
which is rich in arctigenin, has been consumed as a vegetable and
in the form of tea, with no reported side-effects. This may explain
the success of Essiac tea, which is an herbal remedy for breast
cancer; its main ingredient is Burdock root (7). The antitumor effects of arctigenin on
mice with breast cancer tumor xenografts have also been reported
(18). Hence, it is worth noting
that arctigenin may have potential benefits, including its efficacy
as a natural alternative to conventional hormone-responsive
therapy.
In conclusion, the phytoestrogen, arctigenin,
inhibited mTOR downstream effector molecules in ER-positive MCF-7
human breast cancer cells, leading to autophagy-induced cell death
and down regulation of ERα. Therefore, we suggest that arctigenin
may not only be safe for consumption by patients with
hormone-sensitive cancers, but its synergistic effects with
tamoxifen may also make this a viable and effective
co-treatment.
Glossary
Abbreviations
Abbreviations:
|
ER
|
estrogen receptor
|
|
PR
|
progesterone receptor
|
|
Bcl-xL
|
B-cell lymphoma-extra large
|
|
AIF
|
apoptosis-inducing factor
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
ADP
|
adenosine diphosphate
|
|
ATR
|
ATM-Rad3-related
|
|
ATM
|
ataxia telangiectasia mutated
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
AMPK
|
adenosine monophosphate-activated
protein kinase
|
|
mTOR
|
mechanistic target of rapamycin
|
|
S6K1
|
ribosomal protein S6 kinase beta-1
|
|
S6
|
ribosomal protein S6
|
|
4EBP1
|
eukaryotic translation initiation
factor 4E-binding protein 1
|
|
LC3
|
microtubule-associated protein
1A/1B-light chain 3
|
|
PCR
|
polymerase chain reaction
|
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education
(2015R1D1A1A01058841).
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
TM, KSL, SK and KSN designed the experiments; TM
performed the experiments; TM, KSL, SK and KSN analyzed and
discussed the data; TM wrote the article. All authors have reviewed
and approved the manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Chan YS, Cheng LN, Wu JH, Chan E, Kwan YW,
Lee SM, Leung GP, Yu PH and Chan SW: A review of the
pharmacological effects of Arctium lappa (burdock).
Inflammopharmacology. 19:245–254. 2011. View Article : Google Scholar
|
|
2
|
Hayashi K, Narutaki K, Nagaoka Y, Hayashi
T and Uesato S: Therapeutic effect of arctiin and arctigenin in
immunocompetent and immunocompromised mice infected with influenza
A virus. Biol Pharm Bull. 33:1199–1205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swarup V, Ghosh J, Mishra MK and Basu A:
Novel strategy for treatment of Japanese encephalitis using
arctigenin, a plant lignan. J Antimicrob Chemother. 61:679–688.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim Y, Hollenbaugh JA, Kim DH and Kim B:
Novel PI3K/Akt inhibitors screened by the cytoprotective function
of human immunodeficiency virus type 1 Tat. PLoS One. 6:e217812011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kang HS, Lee JY and Kim CJ:
Anti-inflammatory activity of arctigenin from Forsythiae Fructus. J
Ethnopharmacol. 116:305–312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JY and Kim CJ: Arctigenin, a
phenylpropanoid dibenzylbutyrolactone lignan, inhibits type I-IV
allergic inflammation and pro-inflammatory enzymes. Arch Pharm Res.
33:947–957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zick SM, Sen A, Feng Y, Green J, Olatunde
S and Boon H: Trial of Essiac to ascertain its effect in women with
breast cancer (TEA-BC). J Altern Complement Med. 12:971–980. 2006.
View Article : Google Scholar
|
|
8
|
Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai
EM and Hsu YL: Arctigenin, a dietary phytoestrogen, induces
apoptosis of estrogen receptor-negative breast cancer cells through
the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic Biol
Med. 67:159–170. 2014. View Article : Google Scholar
|
|
9
|
Huang K, Li LA, Meng YG, You YQ, Fu XY and
Song L: Arctigenin promotes apoptosis in ovarian cancer cells via
the iNOS/NO/STAT3/survivin signalling. Basic Clin Pharmacol
Toxicol. 115:507–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jeong JB, Hong SC, Jeong HJ and Koo JS:
Arctigenin induces cell cycle arrest by blocking the
phosphorylation of Rb via the modulation of cell cycle regulatory
proteins in human gastric cancer cells. Int Immunopharmacol.
11:1573–1577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang X, Zeng L, Huang J, Zhou H and Liu
Y: Arctigenin, a natural lignan compound, induces apoptotic death
of hepatocellular carcinoma cells via suppression of PI3-K/Akt
signaling. J Biochem Mol Toxicol. 29:458–464. 2015. View Article : Google Scholar
|
|
12
|
Kim JY, Hwang JH, Cha MR, Yoon MY, Son ES,
Tomida A, Ko B, Song SW, Shin-ya K, Hwang YI, et al: Arctigenin
blocks the unfolded protein response and shows therapeutic
antitumor activity. J Cell Physiol. 224:33–40. 2010.PubMed/NCBI
|
|
13
|
Susanti S, Iwasaki H, Inafuku M, Taira N
and Oku H: Mechanism of arctigenin-mediated specific cytotoxicity
against human lung adenocarcinoma cell lines. Phytomedicine.
21:39–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang HQ, Jin JJ and Wang J: Arctigenin
enhances chemosensitivity to cisplatin in human nonsmall lung
cancer H460 cells through downregulation of survivin expression. J
Biochem Mol Toxicol. 28:39–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Zhao F and Liu K: Induction of
apoptosis of the human leukemia cells by arctigenin and its
mechanism of action. Yao Xue Xue Bao. 43:542–547. 2008.In Chinese.
PubMed/NCBI
|
|
16
|
Yang S, Ma J, Xiao J, Lv X, Li X, Yang H,
Liu Y, Feng S and Zhang Y: Arctigenin anti-tumor activity in
bladder cancer T24 cell line through induction of cell-cycle arrest
and apoptosis. Anat Rec (Hoboken). 295:1260–1266. 2012. View Article : Google Scholar
|
|
17
|
Kang K, Lee HJ, Yoo JH, Jho EH, Kim CY,
Kim M and Nho CW: Cell and nuclear enlargement of SW480 cells
induced by a plant lignan, arctigenin: Evaluation of cellular DNA
content using fluorescence microscopy and flow cytometry. DNA Cell
Biol. 30:623–629. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng T, Cao W, Shen W, Zhang L, Gu X, Guo
Y, Tsai HI, Liu X, Li J, Zhang J, et al: Arctigenin inhibits STAT3
and exhibits anticancer potential in human triple-negative breast
cancer therapy. Oncotarget. 8:329–344. 2017.
|
|
19
|
Maxwell T, Chun SY, Lee KS, Kim S and Nam
KS: The anti-metastatic effects of the phytoestrogen arctigenin on
human breast cancer cell lines regardless of the status of ER
expression. Int J Oncol. 50:727–735. 2017. View Article : Google Scholar
|
|
20
|
Wu X, Tong B, Yang Y, Luo J, Yuan X, Wei
Z, Yue M, Xia Y and Dai Y: Arctigenin functions as a selective
agonist of estrogen receptor β to restrict mTORC1 activation and
consequent Th17 differentiation. Oncotarget. 7:83893–83906. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bardin A, Boulle N, Lazennec G, Vignon F
and Pujol P: Loss of ERbeta expression as a common step in
estrogen-dependent tumor progression. Endocr Relat Cancer.
11:537–551. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Skliris GP, Munot K, Bell SM, Carder PJ,
Lane S, Horgan K, Lansdown MR, Parkes AT, Hanby AM, Markham AF, et
al: Reduced expression of oestrogen receptor beta in invasive
breast cancer and its re-expression using DNA methyl transferase
inhibitors in a cell line model. J Pathol. 201:213–220. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie LH, Ahn EM, Akao T, Abdel-Hafez AA,
Nakamura N and Hattori M: Transformation of arctiin to estrogenic
and antiestrogenic substances by human intestinal bacteria. Chem
Pharm Bull (Tokyo). 51:378–384. 2003. View Article : Google Scholar
|
|
24
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang Y, Yan C and Schor NF: Apoptosis in
the absence of caspase 3. Oncogene. 20:6570–6578. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vandenabeele P, Galluzzi L, Vanden Berghe
T and Kroemer G: Molecular mechanisms of necroptosis: An ordered
cellular explosion. Nat Rev Mol Cell Biol. 11:700–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu X, Wu MY, Jiang M, Zhi Q, Bian X, Xu
MD, Gong FR, Hou J, Tao M, Shou LM, et al: TNF-α sensitizes
chemotherapy and radiotherapy against breast cancer cells. Cancer
Cell Int. 17:132017. View Article : Google Scholar
|
|
28
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu Y, Sun XX, Ye JM, He L, Yan SS, Zhang
HH, Hu LH, Yuan JY and Yu Q: Arctigenin alleviates ER stress via
activating AMPK. Acta Pharmacol Sin. 33:941–952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu X, Dou Y, Yang Y, Bian D, Luo J, Tong
B, Xia Y and Dai Y: Arctigenin exerts anti-colitis efficacy through
inhibiting the differentiation of Th1 and Th17 cells via an
mTORC1-dependent pathway. Biochem Pharmacol. 96:323–336. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu Z, Yan J, Jiang W, Yao XG, Chen J,
Chen L, Li C, Hu L, Jiang H and Shen X: Arctigenin effectively
ameliorates memory impairment in Alzheimer's disease model mice
targeting both β-amyloid production and clearance. J Neurosci.
33:13138–13149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoshioka A, Miyata H, Doki Y, Yamasaki M,
Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y and Monden M:
LC3, an autophagosome marker, is highly expressed in
gastrointestinal cancers. Int J Oncol. 33:461–468. 2008.PubMed/NCBI
|
|
33
|
Chanoux RA, Yin B, Urtishak KA, Asare A,
Bassing CH and Brown EJ: ATR and H2AX cooperate in maintaining
genome stability under replication stress. J Biol Chem.
284:5994–6003. 2009. View Article : Google Scholar :
|
|
34
|
Koehn H, Magan N, Isaacs RJ and Stowell
KM: Differential regulation of DNA repair protein Rad51 in human
tumour cell lines exposed to doxorubicin. Anticancer Drugs.
18:419–425. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maier P, Hartmann L, Wenz F and Herskind
C: Cellular pathways in response to ionizing radiation and their
targetability for tumor radiosensitization. Int J Mol Sci. 17:1–32.
2016. View Article : Google Scholar
|
|
36
|
Meijer AJ and Codogno P: Autophagy:
Regulation by energy sensing. Curr Biol. 21:R227–R229. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shagufta and Ahmad I: Tamoxifen a
pioneering drug: An update on the therapeutic potential of
tamoxifen derivatives. Eur J Med Chem. 143:515–531. 2018.
View Article : Google Scholar
|
|
38
|
Lonard DM, Nawaz Z, Smith CL and O'Malley
BW: The 26S proteasome is required for estrogen receptor-alpha and
coactivator turnover and for efficient estrogen receptor-alpha
transactivation. Mol Cell. 5:939–948. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pinzone JJ, Stevenson H, Strobl JS and
Berg PE: Molecular and cellular determinants of estrogen receptor
alpha expression. Mol Cell Biol. 24:4605–4612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui X, Zhang P, Deng W, Oesterreich S, Lu
Y, Mills GB and Lee AV: Insulin-like growth factor-I inhibits
progesterone receptor expression in breast cancer cells via the
phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin
pathway: Progesterone receptor as a potential indicator of growth
factor activity in breast cancer. Mol Endocrinol. 17:575–588. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rice S and Whitehead SA: Phytoestrogens
and breast cancer–promoters or protectors? Endocr Relat Cancer.
13:995–1015. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Matsumura A, Ghosh A, Pope GS and Darbre
PD: Comparative study of oestrogenic properties of eight
phytoestrogens in MCF7 human breast cancer cells. J Steroid Biochem
Mol Biol. 94:431–443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee J, Imm JY and Lee SH: β-catenin
mediates anti-adipogenic and anticancer effects of arctigenin in
preadipocytes and breast cancer cells. J Agric Food Chem.
65:2513–2520. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lui A, New J, Ogony J, Thomas S and
Lewis-Wambi J: Everolimus downregulates estrogen receptor and
induces autophagy in aromatase inhibitor-resistant breast cancer
cells. BMC Cancer. 16:4872016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nathan MR and Schmid P: A review of
fulvestrant in breast cancer. Oncol Ther. 5:17–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miller TW, Hennessy BT, González-Angulo
AM, Fox EM, Mills GB, Chen H, Higham C, García-Echeverría C, Shyr Y
and Arteaga CL: Hyperactivation of phosphatidylinositol-3 kinase
promotes escape from hormone dependence in estrogen
receptor-positive human breast cancer. J Clin Invest.
120:2406–2413. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chumsri S, Sabnis G, Tkaczuk K and Brodie
A: mTOR inhibitors: Changing landscape of endocrine-resistant
breast cancer. Future Oncol. 10:443–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ghayad SE, Bieche I, Vendrell JA, Keime C,
Lidereau R, Dumontet C and Cohen PA: mTOR inhibition reverses
acquired endocrine therapy resistance of breast cancer cells at the
cell proliferation and gene-expression levels. Cancer Sci.
99:1992–2003. 2008.PubMed/NCBI
|