Introduction
Cancer is a relevant global public health problem,
considered as the most important barrier to increasing global life
expectancy in the 21st century (1). Each year, ~88% of people diagnosed
with lung cancer have a death outcome, accounting for 2,093,876 new
cases and 1,761,007 deaths worldwide in 2018 (1). Despite the development of early
diagnosis methods and new treatment modalities, the 5-year survival
rate of patients with lung cancer remains poor, increasing only by
3.7% between 1985 and 2004 (2).
In advanced stages, when surgical resection is not possible,
chemotherapy using platinum drugs, including cisplatin, is the
current standard treatment for non-small cell lung cancer (NSCLC)
(3,4), although it is strongly associated
with intrinsic and acquired resistance (5).
The common strategy used to improve the sensitivity
to platinum compounds and overcome their resistance is the
combination with radiotherapy, antibodies, selective inhibitors or
already prescribed drugs (3).
Studies exploring the molecular profile characterization of lung
cancer have allowed the development of targeted therapies, such as
monoclonal antibodies against vascular endothelial growth factor,
epidermal growth factor receptor (6), programmed cell death 1 ligand 1
(7), anaplastic lymphoma kinase
(8), proto-oncogene
tyrosine-protein kinase ROS1 (9)
and serine/threonine-protein kinase B-raf (10) inhibitors, replacing or enhancing
basic cytotoxic therapies (11).
Repurposing drugs already approved by the Food and
Drug Administration can speed up therapeutic management due to
overcoming the steps of new drug development. Metformin, a
well-known oral antidiabetic drug, is widely associated with a
decreased cancer risk (12) and
increases the chemotherapeutic effects for different types of
cancer, including endometrial cancer (13,14), osteosarcoma (15), hepatocarcinoma (16), non-small cell lung cancer
(17) and gastric cancer
(18). Metformin decreases the
proliferation of lung cancer cells treated with cisplatin compared
with cisplatin treatment alone (19) and sensitizes cells to tyrosine
kinase inhibitors (20) and
crizotinib treatment through the insulin growth factor 1 (IGF1)
signaling pathway (21). The
mechanisms by which metformin exerts antineoplastic effects remain
unclear, but the AMPK-driven inhibition of mTOR seems to be
required for its antimitotic activity (22).
The serine/threonine kinase mTOR is a widely
evolutionarily conserved protein essential for cellular metabolism,
acting as a sensor for the availability of nutrients and growth
factors (23). Overactivation of
the mTOR signaling pathway contributes to several disorders
(24) and is associated with a
poor cancer prognosis (25,26). Most cases of lung cancer have a
mutation in liver kinase B1 (LKB1), such as A549 cells (27), which leads to the partial
impairment of AMPK and overactivation of mTOR signaling (28). Studies have revealed that
cisplatin sensitivity is associated with mTOR inhibition,
especially in LKB1- and KRAS-mutant cancer (29-31). Despite the overactivation of mTOR
signaling, the lack of LKB1 can also sensitize to metformin due to
the inability to restore energy homeostasis (32,33). This makes mTOR signaling an
important regulatory mechanism in lung cancer progression and
metformin treatment.
The present study aimed to investigate the effects
of metformin in cisplatin resistance and regulation of metabolic
cancer pathways, including mTOR signaling, in lung cancer. The
proteomes upon metformin treatment in the context of resistance and
sensitivity to cisplatin in A549 cells were compared, revealing new
possible molecular strategies and targets for NSCLC treatment to
overcome cisplatin resistance.
Materials and methods
Cell culture
A549 cells were maintained in HAM-F12 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.), penicillin (100 U/ml) and
streptomycin (100 µg/ml). Cells were maintained at 37°C in a
humidified atmosphere containing 5% CO2. Cells were
treated at 37°C with 10 µM cisplatin (Sigma-Aldrich; Merck
KGaA) for 72 h to generate the resistant population [CIS vs.
control (C)] and were exposed to 10 mM metformin (Sigma-Aldrich;
Merck KGaA) (MET or CISMET) or 100 nM rapamycin at 37°C
(Sigma-Aldrich; Merck KGaA) (RAPA or CISRAPA) for 24, 48 or 72 h
according to different assays.
MTT viability assay
A549 cells were seeded at a density of
3×104 cells/well for the C group (sensitive cells) and
9×104 cells/well for the CIS population (resistant
cells) in 96-well plates and treated with cisplatin for 72 h and
metformin or rapamycin for another 72 h. After incubation, 12 mM
MTT solution was added to each well for 2 h at 37°C. The culture
medium was aspirated and the formazan crystals were solubilized
with a solution of 1M HCl:isopropanol (1:25) for 15 min at 37°C,
and the absorbance of each sample was measured at 570 nm.
Clonogenic assay and microscopy
A549 cells were seeded at low density in 6-well
plates (3×102 cells/well), treated with 10 mM metformin
or 100 nM rapamycin for 72 h and incubated for 7 days at 37°C.
After incubation, cells were washed with PBS and stained with 3 ml
methylene blue dye (0.3% in 50% ethanol) for 30 min at room
temperature. The plates were washed with deionized water and
staining was measured at an absorbance of 590 nm by eluting with
10% acetic acid. The size of the colonies was calculated using
ImageJ software v1.53 (National Institutes of Health). Colonies
>0.01 pixel2 were considered as hits by the ImageJ
software and were then manually divided into size ranges using
Excel (Microsoft Corporation). The size ranges were hits <1
pixel2, between 1 and 25 pixel2 and >25
pixel2. Images of the cells for morphology analysis were
captured using a light microscope coupled to a camera
(magnification, ×40; Leica Microsystems, Inc.), using scale bars of
100 µm.
Cell cycle and size analysis
A549 cells were seeded in 6-well plates at a density
of 3×105 cells/well and treated with 10 µM
cisplatin for 72 h and 10 mM metformin or 100 nM rapamycin for 24 h
for cell size analysis or 72 h for cell cycle analysis. After
treatment, cells were washed with 1X PBS (0.137 M NaCl and 0.05 M
NaH2PO4, pH 7.4) and resuspended in 500
µl HAM-F12 medium. Cells were centrifuged at 300 × g for 5
min at room temperature and resuspended in 100 µl propidium
iodide (PI) solution (0.1% Triton X-100, 20 µg/ml PI and 10
µg/ml RNAse in PBS) for cell cycle analysis or 100 µl
PBS for cell size analysis. The PI fluorescence and cell profile
were determined using a BD Accuri™ C6 flow cytometer and analyzed
using the BD Accuri™ software v1.0.264.21 (BD Biosciences).
Western blotting
Protein extracts obtained by lysing cells (50 mM
Tris-Cl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease
inhibitor cocktail and phosphatase inhibitor cocktail) were
quantified using a bicinchoninic acid assay and samples containing
20-40 µg of protein were separated by SDS-PAGE (8 and 10%
gels). The gels were electrotransferred to 0.45-µm
nitrocellulose membranes (Bio-Rad Laboratories, Inc.) and the
membranes were incubated for 2 h at room temperature with 5%
non-fat powdered milk dissolved in TBS-Tween (TBST; 50 mM Tris-Cl,
pH 7.5; 150 mM NaCl; 0.1% Tween-20) to saturate unspecific binding
sites, followed by an overnight incubation at 4°C with primary
antibodies. Membranes were washed 3 times with TBST and then
incubated with HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich;
Merck KGaA; cat. no. AP308P; 1:2,000) and goat anti-rabbit IgG
(Sigma-Aldrich; Merck KGaA; cat. no. AP307P; 1:5,000) secondary
antibodies for 1 h at room temperature. Protein bands were
visualized using Pierce™ ECL Western Blotting Substrate (Thermo
Fisher Scientific, Inc.) and densitometry was performed using
ImageJ software v1.53. Primary antibodies (all 1:2,000) against
mTOR (cat. no. 2972), phospho-mTOR (Ser2448; cat. no. 2971), AMPKα
(cat. no. 5831), phospho-AMPKα (Thr172; cat. no. 50081), p70-S6K1
(cat. no. 2708), phospho-p70-S6K1 (Thr389; cat. no. 9234), S6 (cat.
no. 2317), phospho-S6 (Ser240/244; cat. no. 2215) and GAPDH (cat.
no. 2118) were purchased from Cell Signaling Technology, Inc.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from A549 cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
The cDNA was synthesized using the High Capacity cDNA Reverse
Transcription kit with 2,000 ng of total RNA according to the
manufacturer's protocol (Thermo Fisher Scientific, Inc.). The qPCR
reaction was performed with SYBR Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). qPCR was performed
with an initial denaturation step at 95°C for 10 min and then 40
cycles of 95°C for 15 sec and 60°C for 1 min. A melting curve was
performed after the PCR from 60°C to 95°C. The 2−ΔΔCq
method was used for quantification (34). β-actin was used as the normalizing
gene. Samples for this reaction were added in triplicates in a
96-well plate (MicroAmp; Applied Biosystems; Thermo Fisher
Scientific, Inc.) for amplification and reading in the Step One
Plus Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The primers used were designed using
Primer-BLAST (35) (National
Institutes of Health): Superoxide dismutase 2 (SOD2) forward,
5′-AAG GAA CGG GGA CAC TTA CAA A-3′ and reverse, 5′-AGC AGT GGA ATA
AGG CCT GTT G-3′; Annexin 4 (ANXA4) forward, 5′-CAG AGG AAC AAC CAG
GAA CTT G-3′ and reverse, 5′-CAA GCA GAA GTT CTT CGA GGC-3′; and
β-actin forward, 5′-GCC GCC AGC TCA CCA T-3′ and reverse, 5′-CCA
CGA TGG AGG GGA AGA C-3′.
Stable isotope labeling by amino acids in
cell culture (SILAC)
For heavy (H)- or light (L)-lysine labeling
experiments, A549 cells were maintained in a T25 flask with SILAC™
HAMF12 medium (Thermo Fisher Scientific, Inc.) supplemented with
10% dialyzed FBS without lysine and arginine. To obtain H and L
conditions, SILAC™ HAMF12 medium was supplemented with
13C6 L-lysine-2HCl (H) or 12C6 L-lysine-2HCl
(L) and 12C6 L-arginine-2HCl (H and L). The final
concentration of amino acids were 0.46 mM (lysine) and 0.47 mM
(arginine). After 5 passages, C and CIS cells were treated in
triplicate with 10 mM metformin for 72 h (CxMET representing the
Ctrl population; CISxCISMET representing the CIS population),
washed two times with PBS in a 6-well plate and collected using 100
µl lysis buffer as aforementioned.
The mixed H-/L-labeled protein sample (27.5
µg of total protein per group) was separated by
electrophoresis in reducing 10% SDS-PAGE, heated for 10 sec in a
microwave in Coomassie Brilliant Blue stain (Sigma-Aldrich; Merck
KGaA; 2% Coomassie in 50% ethanol and 2.5% acetic acid) and
incubated for 1 h at room temperature. After overnight incubation
of the gel for background de-staining, each gel lane was sliced
into 10 pieces. The excised sections were unstained twice with a
de-staining solution (50% ethanol and 2.5% acetic acid in purified
water) in 1.5-ml microtubes to start the gel bands digestion
protocol using trypsin. The percentage of isotopic label
incorporation was tested according to the following equation:
(Ratio H/L x 100)/(Ratio H/L + 1) (36).
Trypsin digestion, mass spectrometry (MS)
and data analysis
The protein bands were reduced, alkylated and
digested with trypsin overnight at 37°C. An aliquot of the peptide
mixture was separated using a 2-40% acetonitrile gradient in 0.1%
formic acid using an analytical PicoFrit Column (20 cm x ID75
µm; 5-µm particle size; New Objective, Inc.), at a
flow rate of 300 nl/min over 35 min. Peptides were analysed using
the EASY-nLC II (Proxeon Biosystems) coupled to LTQ Orbitrap Velos
(Thermo Fisher Scientific, Inc.), with electrospray ionization in
positive mode and set up in the data-dependent acquisition mode.
Full scan MS spectra (m/z 300-1,600) were acquired in the Orbitrap
analyzer after accumulation to a target value of 1×106.
Resolution in the Orbitrap was set to r=60,000, and the 20 most
intense peptide ions with charge states ≥2 were sequentially
isolated to a target value of 5,000 and fragmented in the linear
ion trap by low-energy collision-induced dissociation (normalized
collision energy of 35%). Three independent experiments were
performed.
Bioinformatics analysis
For SILAC data analysis, the raw files were
processed using MaxQuant 2012 version 1.3.0.5 (https://www.maxquant.org) and the MS/MS spectra were
searched using the Andromeda search engine against the Uniprot
Human Protein Database (37)
(release 17 February 2016; 91,974 sequences; 36,693,332 amino acid
residues). The initial maximal allowed mass tolerance was set to 20
ppm for precursor, 6 ppm in the main search afterward and then to
0.5 Da for fragment ions. Enzyme specificity was set to trypsin
with a maximum of two missed cleavages. Carbamidomethylation of
cysteine (57.021464 Da) was set as a fixed modification, and
oxidation of methionine (15.994915 Da) and protein N-terminal
acetylation (42.010565 Da) were selected as variable modifications.
The minimum peptide length was set to 6 amino acids, including
heavy label Lys6. For protein quantification, a minimum of two
ratio counts was set and the 'requantify' and 'match between runs'
functions were enabled. The false discovery rates (FDRs) of peptide
and protein were both set to 0.01. Data processing was performed
using Perseus v.1.2.7.4 available in the MaxQuant database
(https://www.maxquant.org/). First,
reverse and contaminant entries were excluded from further
analysis. A protein ratio intensity between H and L was used to
compare differential protein expression in the total extract from C
and MET cells, and CIS and CISMET treated cells. The protein ratios
were calculated from the median of all normalized peptide ratios
using unique peptides or peptides assigned to the protein group
with the highest number of peptides (razor peptides).
For statistical analysis of differentially expressed
proteins, the ratios were converted into log2 and Student's
unpaired t-test was applied on the C and CMET treated groups and
CIS and CISMET treated groups. All MS raw files and search
parameter settings associated with the present study are available
for download via the PRIDE data repository at https://www.ebi.ac.uk/pride/archive/
(accession no. PXD017645). After the t-test, volcano plots were
constructed to each replicate separately using the Volcano Plot
Plugin of the OriginLab 2019b software (v9.65; OriginLab)
considering P<0.05[Log10(0.05)=1.3010] and log2 ratio
>±1.0 (fold change of CxMET and CISxCISMET ratio H/L normalized)
as exclusion parameters. Heatmaps were generated on Morpheus online
(https://software.broadinstitute.org/morpheus)
considering common proteins in both groups with P<0.05. The
heatmap represents the standardized values to robust z-score
clustered by Euclidian distance using the average linkage method.
Proteome networks were generated using the Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING) v11 (https://string-db.org/), using the significant
proteins list after t-test analysis and setting a High Confidence
value of 0.700 as a parameter. FDRs of Gene Ontology (GO)
classification were calculated using STRING v11 for biological
processes and performed as previously described by Szklarczyk et
al (38). Biological
processes were arbitrarily chosen so as not to overlap redundant
classifications and obtain the largest number of classified
targets. The FDR of GO and the P-value of the protein-protein
interaction (PPI) enrichment were automatically calculated by
STRING and the detailed description of the enrichment algorithm has
been previously described (39).
Survival rates of patients with lung adenocarcinoma
and lung squamous cell carcinoma were assessed using The Cancer
Genome Atlas Pan-Lung Cancer database (40) (dbGaP Study Accession:
phs000488.v1.p1) for ANXA4 and SOD2 in the cBioPortal software v3
5.0 (https://www.cbioportal.org/) (41). Each sample was defined as altered
or unaltered for each gene based on the Onco Query Language, which
considers non-synonymous mutations, fusions, amplifications and
deep deletions (https://www.cbioportal.org/oql).
Statistical analysis
Statistical analyses were assessed using GraphPad
Prism 8.01 software (GraphPad Software, Inc.) applying Student's
unpaired t-test or one-way ANOVA followed by Tukey's post-hoc test
when n sample was homogeneous between groups and Bonferroni's test
when not meeting that condition. P<0.05 was considered to
indicate a statistically significant difference. For proteomics
analysis, Student's t-tests were performed using Perseus.
Results
Metformin (MET) decreases cell viability
and size without changes in the cell cycle
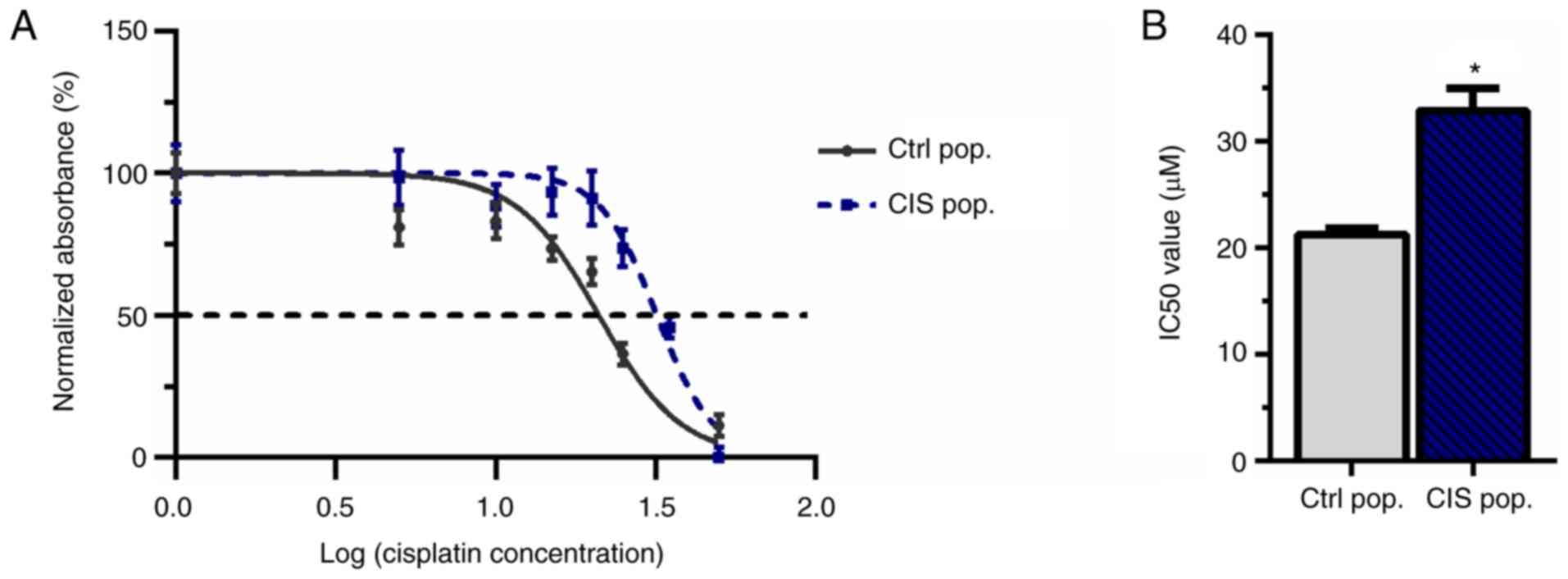
To demonstrate that 10 µM cisplatin was able
to create resistance in A549 cells, an MTT assay was performed to
calculate the IC50. This cisplatin concentration was
adopted since it had been previously reported to confer resistance
to new cisplatin exposure in melanoma (42). A549 cells were subjected to 10
µM cisplatin treatment for 72 h (called CIS population or
CIS pop) and subsequent doses of cisplatin (5, 10, 15, 20, 25, 35
and 50 µM) for another 72 h. The CIS pop presented a
significant increase in the IC50 compared with the
untreated control population (Ctrl pop), suggesting the acquisition
of resistance (21.24 µM for Ctrl pop vs. 32.86 µM for
CIS pop; Fig. 1A and B).
To investigate the effects of MET in lung cancer
cells, MTT assay was performed (Fig.
2B) in the CIS pop compared with the Ctrl pop. MET decreased
cell viability more extensively in the Ctrl pop (71.2% for 24 h and
64.2% for 72 h exposure) compared with in the CIS pop (84.5% for 24
h and 83.5% for 72 h) (Fig. 2B).
Additionally, the viability of cells treated with MET was
significantly decreased compared with that of cells treated with
rapamycin (RAPA) in the Ctrl pop after 72 h (64.2 vs. 95.5%;
Fig. 2B).
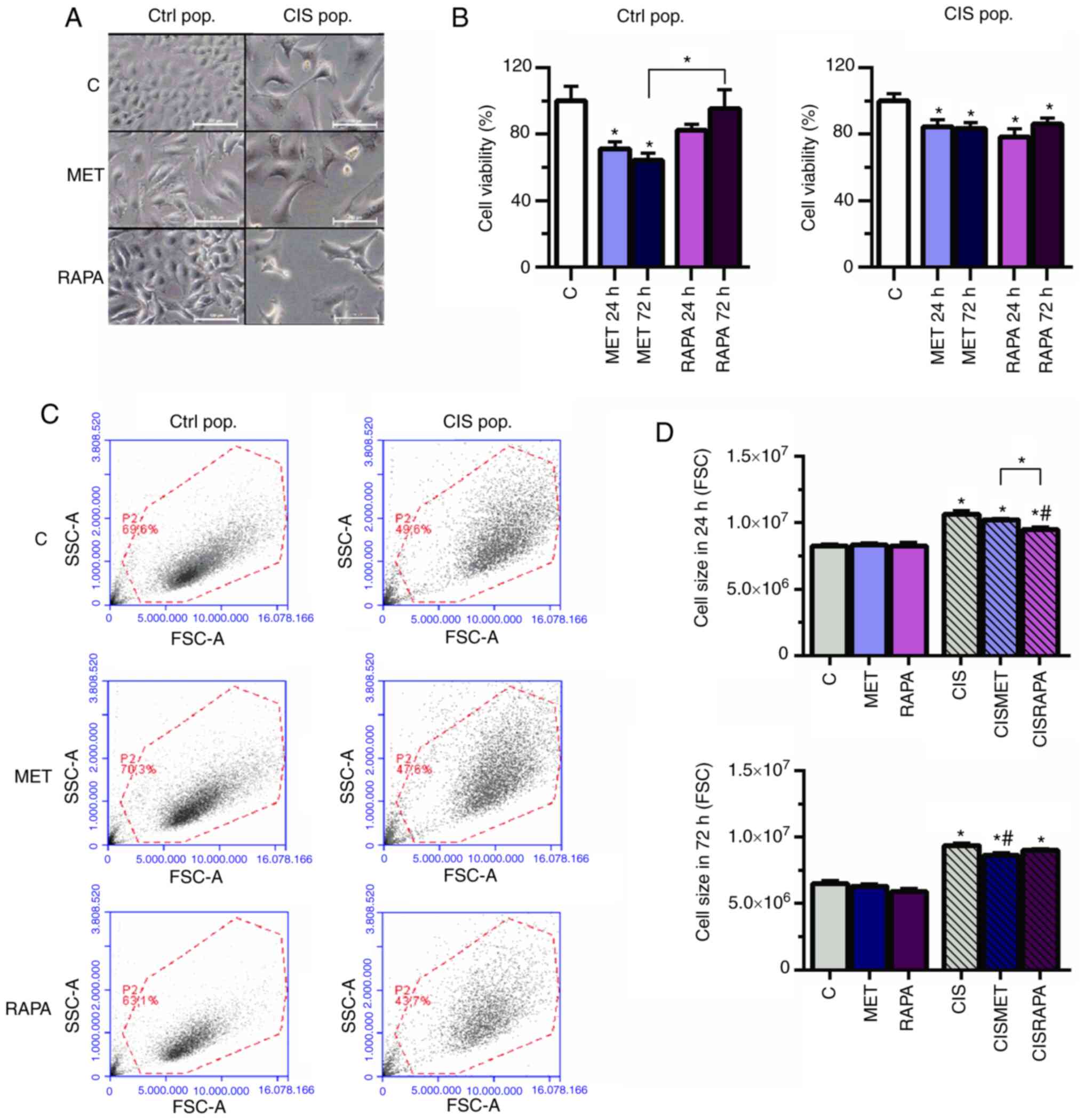 | Figure 2MET decreases cell viability and size
in Ctrl and CIS-treated A549 cells. A549 cells exposed to 10
µM CIS (CIS pop) or not (Ctrl pop) for 72 h were
subsequently exposed to 10 mM MET or 100 nM RAPA for 24 and 72 h.
(A) Cell size increased in CIS-treated A549 cells and decreased in
cells treated with 10 mM MET or 100 nM RAPA after 72 h, as seen by
microscopy. Scale bar, 100 µm. (B) MET and RAPA
significantly decreased cell viability in the Ctrl and CIS pop
after 72 h. (C and D) CIS altered cell size after 24 and 72 h, as
analyzed by flow cytometry, and was then restored by MET after 72 h
and by RAPA after 24 h. *P<0.05 vs. C;
#P<0.05 vs. CIS. CIS, cisplatin; Ctrl/C, control;
pop, population; MET, metformin; RAPA, rapamycin; FSC, forward
scatter; SSC, side scatter. |
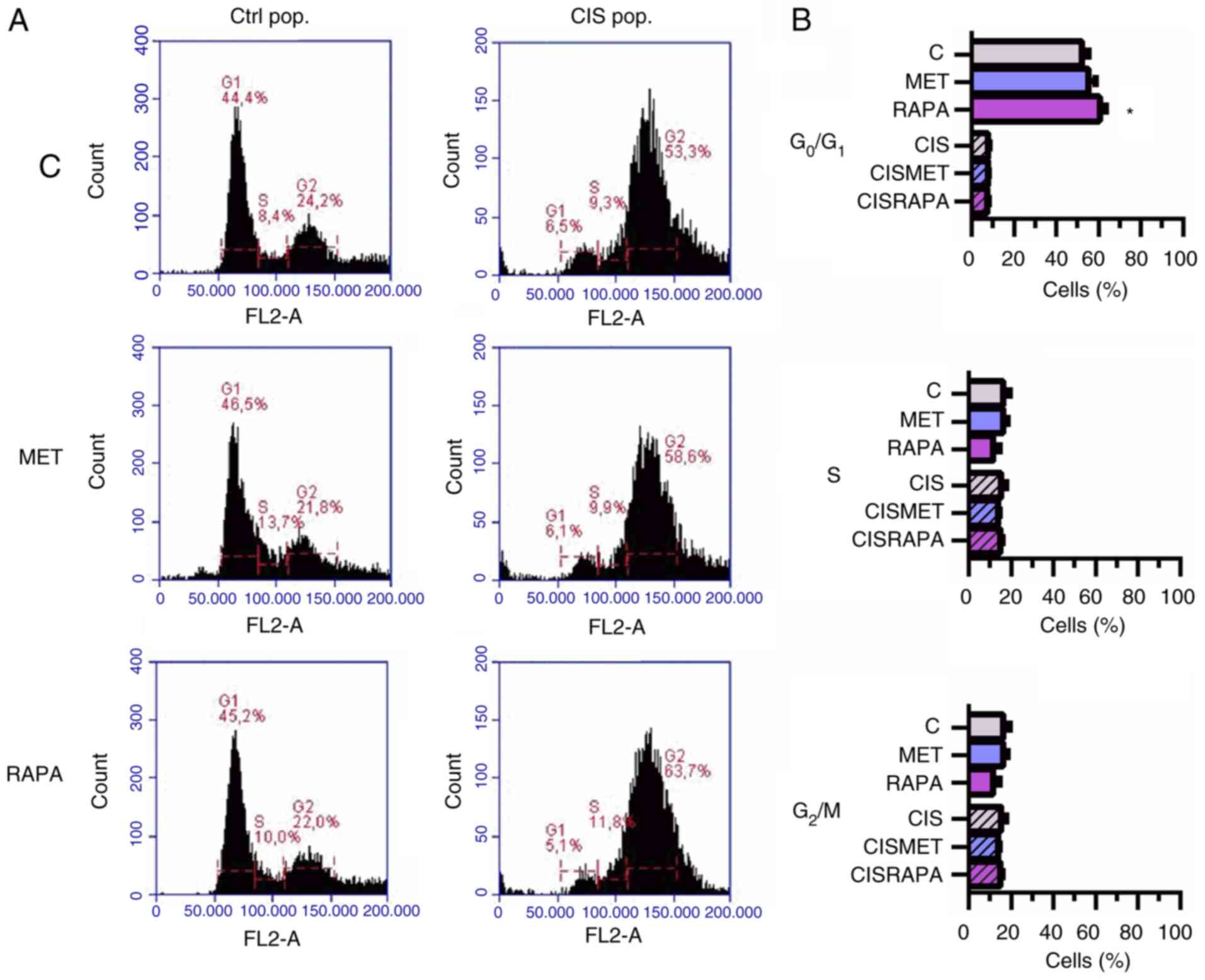
Cisplatin led to increased cell size and
granularity, as seen by light microscopy and forward scatter and
side scatter flow cytometry parameters (Fig. 2A and C, respectively), conferring
more heterogeneity to the CIS pop compared with the Ctrl pop. In
the CIS pop, flow cytometry data revealed that RAPA significantly
decreased cell size after 24 h, while MET significantly decreased
cell size after 72 h treatment, both compared with cisplatin alone
(Fig. 2D). However, only RAPA led
to G0/G1 cell cycle arrest after 72 h
treatment (Fig. 3A and B). The
present data suggested that 72 h of MET treatment decreased cell
viability and size in cisplatin-sensitive and -resistant cells
without cell cycle impairment. In summary, MET decreased lung
cancer cell viability and size without significantly changing the
cell cycle.
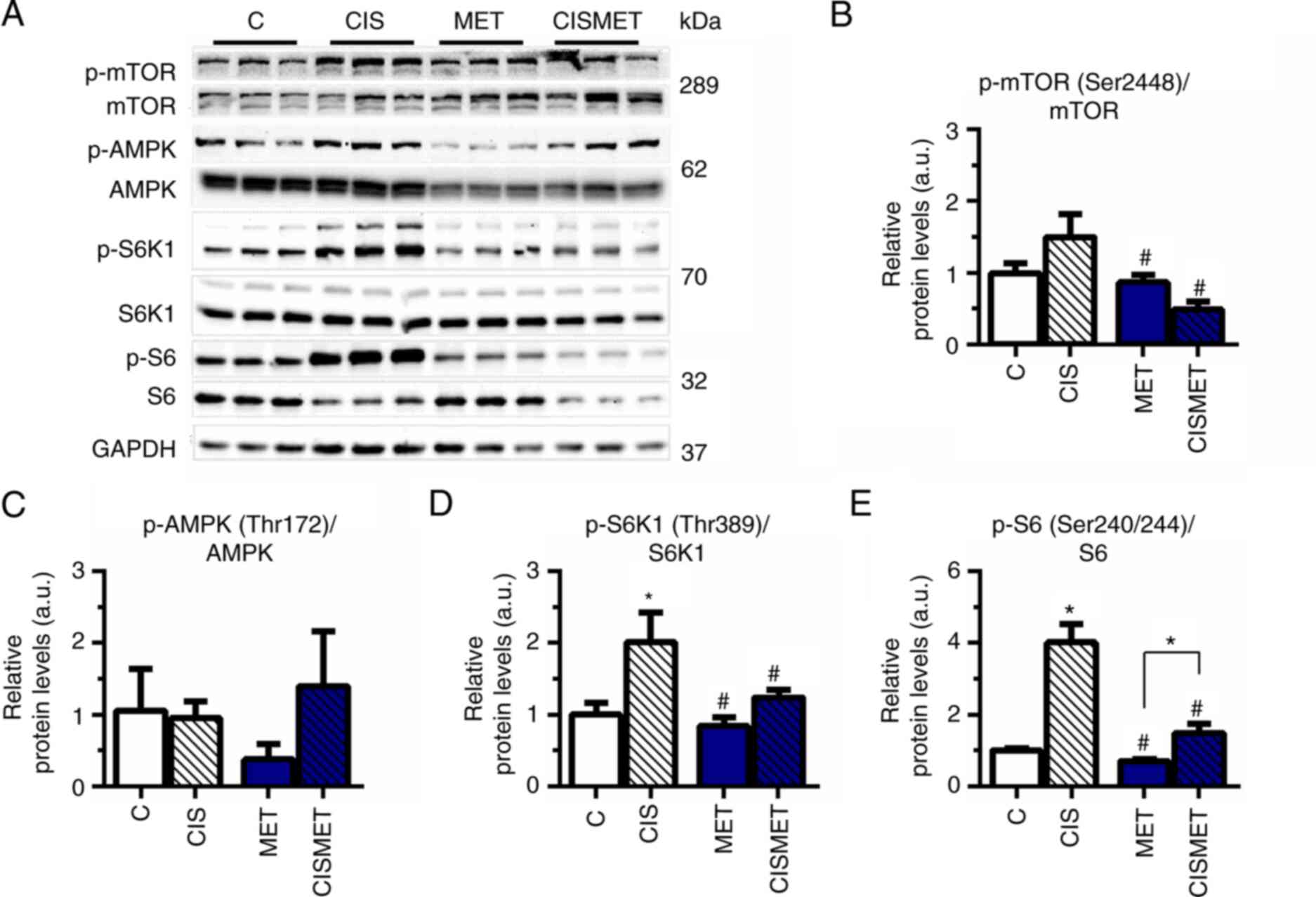
MET reverts mTOR activation induced by
cisplatin in A549 cells
The mTOR signaling pathway status was evaluated by
western blotting after MET treatment in Ctrl and CIS pops. The
analysis revealed that the mTOR signaling pathway was overactivated
after cisplatin treatment, with a significant increase in S6K1 and
S6 phosphorylation (Fig. 4A, D and
E) compared with the control group. Subsequent MET treatment
significantly decreased mTOR, S6K1 and S6 activation compared with
CIS treatment, corroborating the aforementioned decreases in cell
viability and size. A non-significant difference was observed in
AMPK phosphorylation after MET treatment in A549 cells (Fig. 4C). The present results indicated
robust activation of the mTOR signaling pathway after cisplatin
treatment in A549 cells.
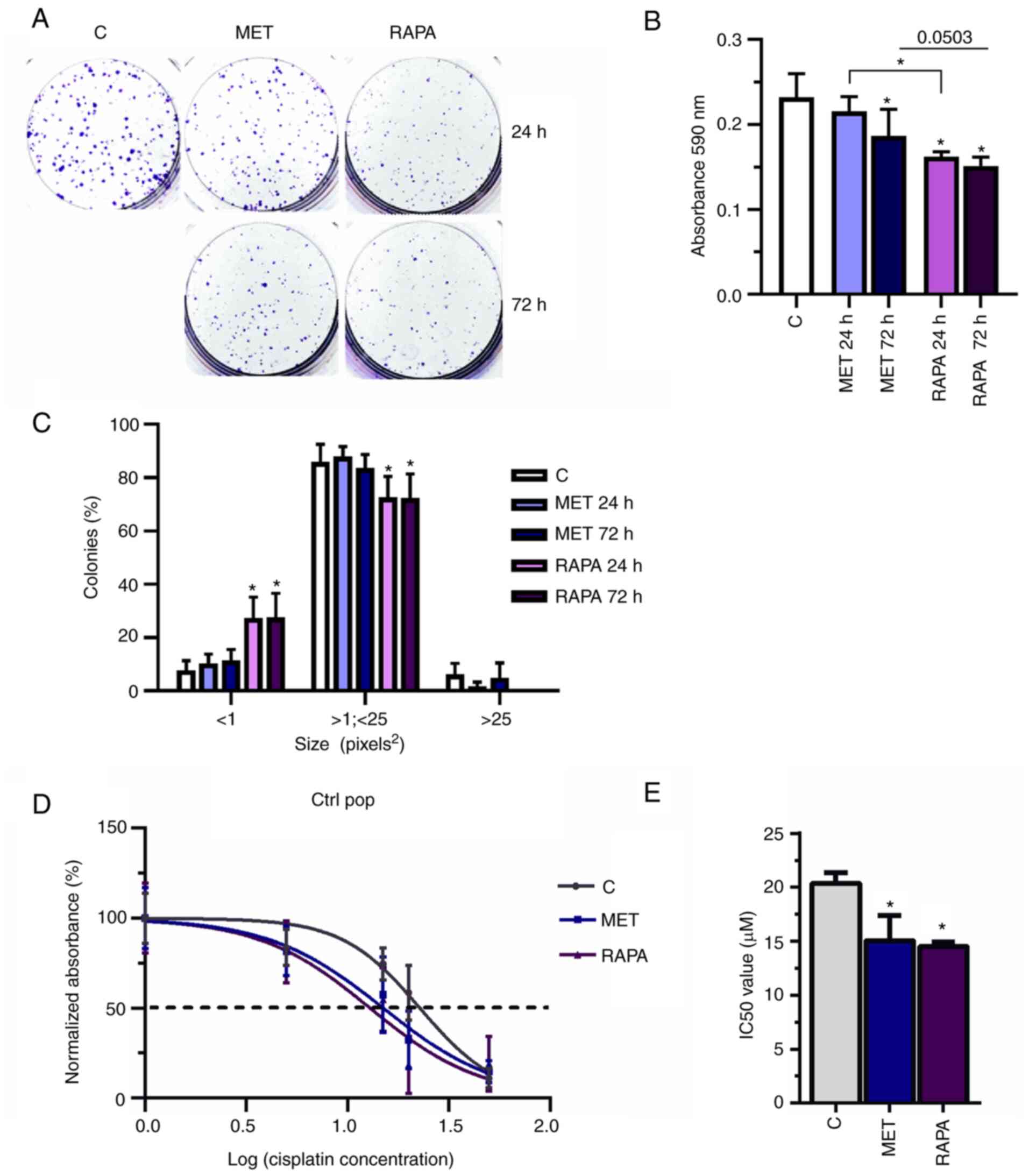
MET and RAPA decrease colony formation
and sensitize A549 cells to cisplatin
To determine the colony formation potential of MET-
and RAPA-treated cells, A549 cells exposed to 10 mM MET or 100 nM
RAPA for 24 or 72 h were evaluated in a clonogenic cell assay
(Fig. 5A). Compared with the
control group, MET and RAPA treatments significantly decreased the
absorbance of colonies formed (MET at 72 h and RAPA at 24 and 72 h)
compared with the control group (Fig.
5B), but only RAPA decreased the size of colonies compared with
the control group (Fig. 5C). Both
MET and RAPA significantly decreased the IC50 in Ctrl
pop cells (IC50=15.4 and 14.5 µM, respectively,
vs. 20.4 µM for Ctrl; Fig. 5D
and E). Thus, the decrease of IC50 indicated that
previous treatment with MET or RAPA may potentiate and sensitize
cells to cisplatin treatment.
Proteomics analysis reveals MET treatment
profile in Ctrl and CIS populations
In addition to the mTOR signaling pathway, MET
exerts important changes in crucial pathways involved in cancer
(22). To investigate other
possible molecular mechanisms involved in cisplatin sensitivity
induced by MET, a proteomics analysis of A549 cells after MET
treatment was performed, and the proteomes in control and
cisplatin-resistant cells were compared using the SILAC approach
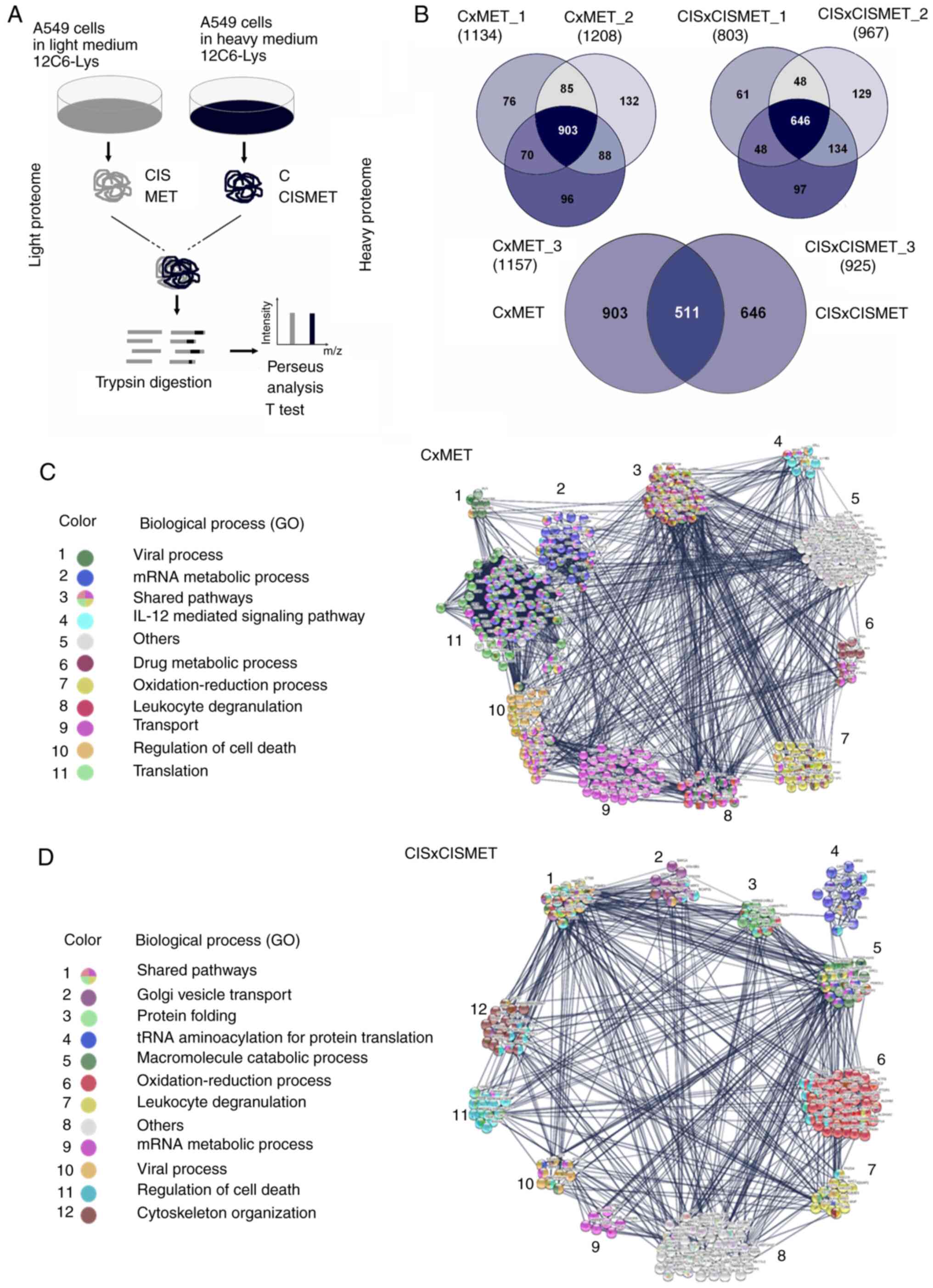
(CxMET and CISxCISMET; Fig. 6A).
A total of 903 proteins were quantified in CxMET and 646 in
CISxCISMET, with 511 common proteins (Fig. 6B). The Student's t-test analysis
indicated 361 differentially expressed and statistically
significant proteins (P<0.05) for CxMET ratios and 254 for
CISxCISMET ratios (Tables SI and
SII). These proteins were classified and grouped based on their
involvement in biological processes (Fig. 6C and D). GO terms and FDRs
generated by STRING analysis are presented in Table I. PPI enrichment P-values were
<1.00−16 for CxMET and CISxCISMET networks (data not
shown).
 | Table IBiological processes, GO numbers and
FDRs associated with each proteomic network. |
Table I
Biological processes, GO numbers and
FDRs associated with each proteomic network.
| Biological
processes | GO no. | CxMET | CISxCISMET | Upregulated | Downregulated |
|---|
| Viral process | 0016032 |
8.62×1011 |
6.97×1007 | - | - |
| Regulation of mRNA
metabolic process | 1903311 | - | - | - | 0.0283 |
| mRNA metabolic
process | 0016071 |
3.57×1035 | 0.0076 | - | - |
| IL-12 mediated
signaling pathway | 0035722 |
1.94×1007 | - | - | 0.0019 |
| Translation | 0006412 |
2.30×1042 |
2.83×1007 |
6.65×1005 | - |
| Regulation of cell
death | 0010941 |
3.78×1007 |
3.02×1009 | - | - |
| Transport | 0006810 |
8.36×1027 | - | - | 0.0290 |
| Leukocyte
degranulation | 0043299 |
4.18×1006 |
5.24×1010 | - | - |
| Oxidation-reduction
process | 0055114 |
4.55×1015 |
1.76×1020 |
9.19×1006 | 0.0021 |
| Drug metabolic
process | 0017144 |
3.11×1013 | - | - | - |
| Golgi vesicle
transport | 0048193 | - |
6.64×1005 | - | - |
| Protein
folding | 0006457 | - |
3.41×1013 | 0.00014 | - |
| Macromolecule
catabolic process | 0009057 | - | 0.00032 | - | - |
| Cytoskeleton
organization | 0007010 | - | 0.00055 | - | - |
| Negative regulation
of apoptotic process | 0043066 | - | - | - | 0.0019 |
| Regulated
exocitosis | 0045055 | - | - |
4.45×1006 | - |
| Regulation of
apoptotic process/apoptosis | 0042981 | - | - | 0.0048 | - |
In the CxMET group, MET regulated 'viral process',
'mRNA metabolic process', 'IL-12 mediated signaling pathway', 'drug
metabolic process', 'oxidation-reduction process', 'leukocyte
degranulation', 'transport', 'regulation of cell death' and
'translation' (Fig. 6C). In the
CISxCISMET group, MET regulated 'Golgi vesicle transport', 'protein
folding', 'tRNA aminoacylation for protein translation',
'macromolecule catabolic process', 'oxidation-reduction process',
'leucocyte degranulation', 'mRNA metabolic process', 'viral
process', 'regulation of cell death' and 'cytoskeleton
organization' (Fig. 6D). The data
revealed five shared pathways between groups ('mRNA metabolic
process', 'oxidation-reduction process', 'leukocyte degranulation',
'viral process' and 'regulation of cell death'). The raw list of
proteins identified by Perseus in both groups is presented in
Tables SI and SII. Therefore,
these molecular pathways may be potentially important signatures of
the mechanisms of action of MET in lung cancer.
MET alters translation, oxidative stress,
apoptosis and metabolic pathways in control and cisplatin-resistant
A549 cells
Volcano plots displayed the 1,448 proteins in CxMET
and 1,158 in CISxCISMET (Fig.
7A), both separated by magnitude of evidence (P-value) and
change (fold change of log2 ratio values) [cut-off values,
log10(0.05)=1.3010; log2 ratio=1.00]. A total of 186 and 184
proteins were significantly downregulated and upregulated in MET
compared with C, respectively, and 102 and 167 proteins were
significantly downregulated and upregulated in CISMET compared with
CIS, respectively. Applying the cut-off P-value, in CxMET the
volcano plot expressed 3 significantly downregulated proteins,
including ANXA4, and 12 significantly upregulated proteins,
including SOD2. In CISxCISMET, the volcano plot expressed 7
significantly down-regulated proteins and 12 significantly
upregulated proteins. Subsequently, 99 common significant proteins
between CxMET and CISxCISMET analysis (ACADVL, ACTC1, ACTN1, ACTN4,
AIFM1, AK3, AKR1C1, ALDH1A1, ALDH6A1, ANXA1, ANXA2, ANXA4, ARCN1,
ARF3/ARF1, ARF4, C1orf57/NTPCR, CALD1, CALM2/CALM1, CANX, CCT8,
CFL1, CPLX2, CRYAB, CS, CSRP1, CTSB, CTSD, DDX46, DYNC1H1,
DYNC1LI2, ECH1, EIF4A1, EPRS, ETFA, ETFB, FARSB, FH, FKBP3, FLNA,
G6PD, GAA, GAPDH, GCN1L1, GRPEL1, GSTP1, HADHA, HMGA1, HNRNPA2B1,
HNRNPL, HSP90AA1, HSPA8, HSPD1, IARS, IGF2BP1, ILF2, ILF3, ITGB1,
LARS, LDHA, LDHB, LRRFIP1, MARCKS, MDH2, MSN, MYH9, NCL, NPC2,
NPM1, PDHB, PGD, PKM2, PLOD2, PPIF, PTBP1, PTGR1, PYCR1,
RAB2A/RAB2B, RPL19, RPL3, RPLP0, RPS17, SERPINE1, SF3A1, SLC39A7,
SOD2, SRI, STIP1, SUB1, SUCLG2, TCP1, TGM2, TKT, TPM4, TRAP1, TUFM,
UBC, UGDH, VCL and ZYX), altered after MET treatment and expressed
by standardized values, were grouped in clusters by Euclidian
distance using the average linkage in a heatmap (Fig. 7B). These proteins were then
classified into two different networks, according to their
upregulation or downregulation in resistant cells compared with
control cells (Fig. 7C and
Table I). Cisplatin resistance
decreased proteins associated with 'transport' and 'mRNA metabolic
process', while it upregulated proteins involved in 'translation'
(Fig. 7C), corroborating the
aforementioned activation of the mTOR signaling pathway. Regulation
of 'oxidation-reduction process' and apoptotic processes were
common biological processes found to be upregulated or
downregulated by cisplatin treatment (Fig. 7C).
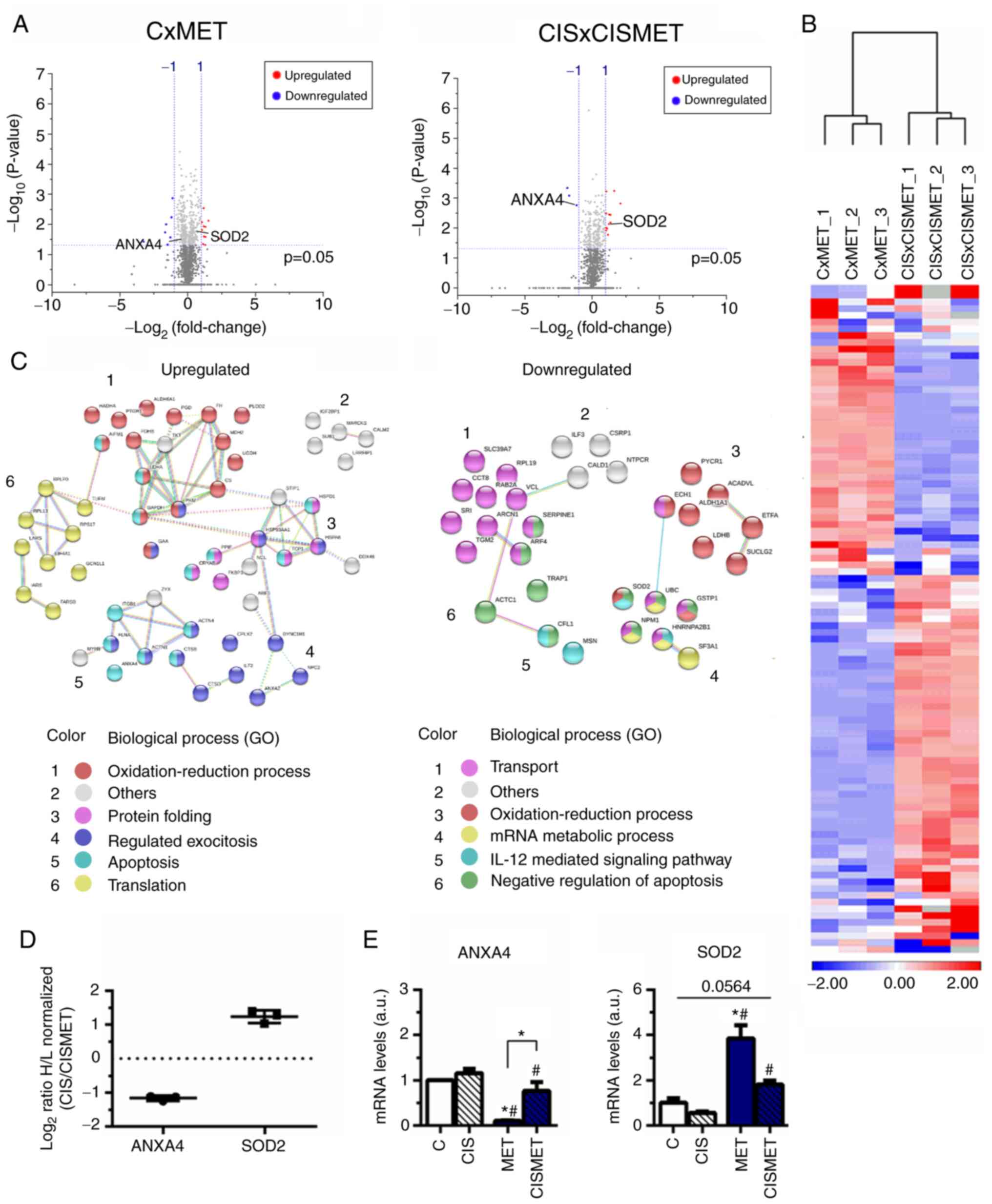 | Figure 7Proteomics analysis revealing
upregulated and downregulated targets after MET treatment in
control and CIS-resistant A549 cells. (A) Volcano plots of the
proteins significantly downregulated and upregulated with a cut-off
of P<0.05, represented by Log10 (0.05) value of
threshold (1.301), and normalized H/L ratio >±1 for both groups.
(B) Heatmap of the common significant proteins with P<0.05. (C)
Upregulated and downregulated proteins in CISxCISMET compared with
CxMET were classified using the Search Tool for the Retrieval of
Interacting Genes/Proteins according to GO biological processes.
(D) Normalized H/L ratios of two selected proteins from the
proteomics analysis, SOD2 and ANXA4. (E) Reverse
transcription-quantitative PCR reported altered expression levels
of ANXA4 and SOD2 after MET treatment. *P<0.05 vs. C;
#P<0.05 vs. CIS. CIS, cisplatin; C, control; MET,
metformin; GO, Gene Ontology; ANXA4, annexin 4; SOD2, superoxide
dismutase 2; H/L, heavy to light ratio; a.u., arbitrary units. |
Two proteins reported by the proteomics analysis
were chosen for further validation: ANXA4, involved in apoptosis
(43), and SOD2, involved in
oxidative stress pathways (44),
which presented decreased and increased expression levels in the
CIS versus CISMET analysis, respectively (Fig. 7D). The mRNA expression levels of
these targets were further evaluated by RT-qPCR, revealing that MET
significantly decreased ANXA4 expression compared with control, CIS
and CISMET groups (Fig. 7E).
Additionally, MET significantly increased SOD2 expression compared
with control and CIS groups (Fig.
7E). Furthermore, treatment with MET in the Cis pop (CISMET
group) significantly decreased ANXA4 expression and increased SOD2
expression compared with the CIS group (Fig. 7E). RT-qPCR data corroborated the
proteomics analysis presented in the heatmap. A complementary
analysis of the survival rates of patients with lung cancer using
the cBioPortal revealed that alterations in the ANXA4 gene
decreased the median survival time after initial treatment from
43.9 months to 19.5 months (Fig.
S1). The altered group for SOD2 is represented by 10 patients
(plus 2 deceased), with 5 patients with deep deletions of the gene
(homodeleted) and 5 patients with missense mutations (G141C, L176R,
R123H, G126A and S127F) (data not shown). The altered group for
ANXA4 is represented by 12 patients (plus 5 deceased), with 8
patients with gene amplification and 4 patients with missense
mutations (W188L, G33C, L316V and Q52E) (data not shown). Overall,
MET decreased ANXA4 expression and increased SOD2 expression in
A549 cells, validating the presented proteomics data.
Discussion
Over the last years, MET has been widely used as an
antidiabetic agent and has been characterized to present antitumor
properties and several advantages in cancer therapy (45). The mechanisms by which MET
decreases tumor progression and presents chemosensitivity abilities
are not fully described, but it seems that AMPK-driven inhibition
of mTOR is an important regulating axis in the tumorigenic process
(22). The mTOR signaling pathway
is also described as a pathway with implications in tumor
development and progression, metastasis and chemoresistance
(46). The present study
indicated that MET may potentially affect lung cancer progression
and cisplatin chemosensitivity, and may be associated with mTOR
signaling and other pathways, such as translation, oxidative stress
and apoptosis.
In the present study, MET decreased the viability in
Ctrl and cisplatin-treated cells, as described in breast (47,48), ovarian (49) and lung cancer cell lines (50). Stronger effects of MET on
viability may be possible due to its extensive and embracing
mechanisms of action compared with the point target effects of
RAPA, as previously reported in pancreatic cancer cells (51). Although other studies have
reported cell cycle arrest after MET treatment in different cell
lines (52-54), no changes were observed in the
present study in the A549 cell cycle. However, it was confirmed
that cisplatin led to cell cycle arrest in the G2/M
phase, which is a characteristic of platinum drugs (55), and that RAPA led to
G0/G1 arrest, which has been also previously
reported (56-58). Additionally, the current study
indicated that MET reverted the increase in cell size induced by
cisplatin after 72 h, which was consistent with decreased mTOR
signaling. Wang et al (59) reported that cell cycle arrest
induced by MET in myeloma is dependent on the mTOR signaling
pathway, possibly via intact LKB1-AMPK axis also observed in A549
cells (60). mTOR inhibition
seems to be an important strategy to improve cisplatin sensitivity,
mediating the chemotherapy resistance in KRAS-mutant lung cancer
(31). Several studies have
demonstrated that cisplatin resistance induces activation of the
mTOR/Akt signaling pathway and decreases apoptosis, whereas
inhibitors of the mTOR/Akt signaling pathway sensitize and enhance
the effects of cisplatin in different cancer cell lines, including
lung cancer (61) and
hepatocarcinoma cells (62). In
esophageal squamous cell carcinoma (ESCC) xenografts, a small
interfering RNA against mTOR significantly increased apoptosis when
combined with cisplatin (63).
Moreover, patients with endometrial cancer treated with MET
presented decreased levels of plasma IGF-1 and PI3K, phospho-Akt,
phospho-S6K1 and phospho-4EBP1 in biopsy specimens, reinforcing its
antiproliferative potential (64). In accordance with the
aforementioned studies, the present study confirmed in vitro
that MET decreases cell viability and clonogenic potential,
sensitizes cells to cisplatin by decreasing the IC50 and
is associated with decreased mTOR signaling, indicating that MET
may be a potential coadjuvant in NSCLC therapy.
Galluzzi et al (65,66) defined mechanisms of cisplatin
resistance and its associated targets in different signaling
pathways. Cisplatin resistance is generally multifactorial,
characterized by successive molecular alterations, including the
binding of cisplatin to its targets, increased repair mechanisms,
decreased apoptosis and stimulation of pro-survival mechanisms
(67,68). Considering cisplatin resistance
and tumor progression, it is important to target more than one
molecular mechanism to efficiently circumvent cisplatin resistance
(65,66).
Understanding the MET-induced proteomic changes in
sensitive and resistant contexts provides important information
about specific modifications acquired by cells (67). Regarding the multifactorial
resistance profile, the present study aimed to investigate other
potential signaling pathways involved in cisplatin sensitivity
induced by MET in addition to mTOR signaling. In both sensitive and
resistant cells, MET altered transcriptional processes, regulated
apoptosis, oxidation-reduction processes and proteins associated
with leukocyte degranulation, with 99 common significantly altered
targets, of which some have been previously described: AK3
(68), ALDH1A1 (69), ANXA2 and 4 (70,71), CFL1 (72), CRYAB (73), filamin A (FLNA) (74), GSTP1 (75), HMGA1 and G6PD (76), hnRNPA2B1 (77), HSP90 (78), IGF2BP1 (79), integrin b1 (ITGB1) (80), MYH9 (81), PKM2 (82-84), STIP1 (85), TGM2 (86), TKT (87) and TRAP1 (88).
In the present study, comparing sensitive and
resistant contexts, MET decreased several oncogenes, such as CD29,
FLNA, CTSD, MSN and ANXA4. Despite IL-12-mediated signaling having
been associated with antitumor effects (89), the pathway component MSN has been
largely associated with tumor progression (90-92). The present proteomic analysis
revealed that MET increased apoptosis in cisplatin-resistant cells,
as previously reported (33,53,93), by decreasing anti-apoptotic
proteins such as TRAP1, CFL1 and SOD2. On the other hand, potential
tumor progressors, such as CD29, cathepsin D (CTSD), FLNA and
ANXA4, were upregulated. CD29, also known as ITGB1, is a
transmembrane cell surface receptor that has been associated with
metastasis, tumor migration and drug resistance (94,95). ITGB1 is associated with resistance
to gefitinib in NSCLC (96) and
its knockdown overcomes erlotinib resistance in lung cancer cells
and decreases the activation of Akt after erlotinib treatment
(97). FLNA acts as a scaffold
for cancer-associated signaling pathways and is associated with the
aggressive pattern and poor survival outcomes in patients with
NSCLC treated with platinum-based drugs, such as cisplatin
(98). Furthermore, FLNA may
interact with other oncogenes, such as Akt, K-RAS, TRAF2, NIK and
14-3-3σ (99-102). CTSD is an intracellular aspartic
protease of the pepsin superfamily associated with inhibition of
SERPINE1 and is a tumor marker for invasion and metastasis
(103,104). Overexpression of CTSD promotes
breast cancer cell migration, invasion and metastasis through
intercellular cell adhesion molecule-1 both in vitro and
in vivo (105).
For the validation of the proteomics analysis, two
targets were chosen, SOD2 (upregulated) and ANXA4 (downregulated),
whose roles in cisplatin resistance are already known and
well-documented. SOD2, a superoxide scavenger, may be directly
involved in carcinogenesis by protecting cells against increased
levels of reactive oxygen species (ROS) (106). It has been previously described
that SOD2 can protect against DNA damage-inducing agents,
especially in radiation (107-109). ROS act as mediators of DNA
damage and SOD2, an endogenous antioxidant, protects the cells from
DNA damage by scavenging reactive molecules, such as superoxide
(110). One of the mechanisms
described for the action of cisplatin is the depletion of
antioxidant molecules to tilt the redox balance towards oxidative
stress, which facilitates DNA damage (65). Additionally, cisplatin treatment
increases ROS content in human lung cancer cells, including A549
cells (111). On the other hand,
the overexpression of SOD2 in mitochondria enhance the survival of
HeLa cells and contribute to cisplatin resistance in human ESCC and
oral squamous cell carcinoma cell lines (110,112).
ANXA4 is largely involved in the proliferation,
platinum resistance and migration in different types of cancer
cells, such as ovarian (43,113) and endometrial cancer cells
(114). ANXA4 overexpression is
associated with tumor cell invasion and poor prognosis in patients
with gallbladder cancer (115).
Furthermore, overexpression of ANXA4 confers carboplatin resistance
in ovarian carcinoma cells (114). ANXA4-knockdown increases
sensitivity to platinum-based drugs both in vitro and in
vivo (70,116) and the gain of its expression can
restore cisplatin resistance in mesothelioma cells (116), which reinforces the beneficial
effects of MET in decreasing ANXA4 expression. According to the
present survival analysis, alterations in ANXA4 may decrease
patient survival. Since treatment with MET significantly decreased
ANXA4 expression, ANXA4 may be explored as a potential marker for
survival and cisplatin responsiveness in lung cancer.
In conclusion, the present study demonstrated that
MET sensitized cells to cisplatin treatment and decreased
clonogenic survival and viability in A549 lung cancer cells. The
mTOR signaling pathway was overactivated after cisplatin treatment,
which was restored by MET regardless of the LKB1-AMPK axis.
Therefore, MET may be able to improve the chemotherapeutic effects
of cisplatin in A549 cells by decreasing mTOR signaling and
modulating apoptosis, translation-associated processes and
oxidative pathways, thus providing potential new therapeutic
targets to circumvent cisplatin resistance in NSCLC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request. Additionally, the proteomic datasets generated
and/or analyzed during the current study are available in the PRIDE
repository (https://www.ebi.ac.uk/pride/). The mass spectrometry
proteomics data have been deposited in the ProteomeXchange
Consortium (http://proteomecentral.proteomexchange.org) via the
PRIDE partner repository (117)
with the dataset identifier PXD017645 (https://www.ebi.ac.uk/pride/archive/projectPXD017645).
Authors' contributions
APM, ICBP and FRS performed the cell experiments
and revised the manuscript. APM, ICBP, AFPL, DCG, BAP and RRD
performed mass spectrometry experiments and proteomics data
analysis. GFP performed bioinformatics analysis. APM wrote the
manuscript and headed the execution of all experiments. APM, FMS,
RMNB, TCT, LPDM, DCG, AFPL and RC designed the experiments and
revised the manuscript. APM and FMS confirmed the authenticity of
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to acknowledge the Mass
Spectrometry Laboratory at Brazilian Biosciences National
Laboratory (Campinas, Brazil) for their support with mass
spectrometry analysis.
Funding
The present study was funded by the São Paulo Research
Foundation (FAPESP; grant nos. 2012/13558-7, 2018/14818-9,
2016/06457-0 and 2015/22814-5; fellowship nos. 2016/02483-7,
2017/04269-5, 2019/00607-9, 2015/003111 and 2015/16601-9) and by
the National Council for Scientific and Technological Development
(grant no. 447553/2014-3).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ridge CA, McErlean AM and Ginsberg MS:
Epidemiology of lung cancer. Semin Intervent Radiol. 30:93–98.
2013. View Article : Google Scholar :
|
|
3
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar
|
|
4
|
Talebian Yazdi M, Schinkelshoek MS, Loof
NM, Taube C, Hiemstra PS, Welters MJ and van der Burg SH: Standard
radiotherapy but not chemotherapy impairs systemic immunity in
non-small cell lung cancer. Oncoimmunology. 5:e12553932016.
View Article : Google Scholar
|
|
5
|
Sarin N, Engel F, Kalayda GV, Mannewitz M,
Cinatl J Jr, Rothweiler F, Michaelis M, Saafan H, Ritter CA, Jaehde
U and Frötschl R: Cisplatin resistance in non-small cell lung
cancer cells is associated with an abrogation of cisplatin-induced
G2/M cell cycle arrest. PLoS One. 12:e01810812017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang Z, Hackshaw A, Feng Q, Fu X, Zhang Y,
Mao C and Tang J: Comparison of gefitinib, erlotinib and afatinib
in non-small cell lung cancer: A meta-analysis. Int J Cancer.
140:2805–2819. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garon EB, Rizvi NA, Hui R, Leighl N,
Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L,
et al: Pembrolizumab for the treatment of non-small-cell lung
cancer. N Engl J Med. 372:2018–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Uchibori K, Inase N, Araki M, Kamada M,
Sato S, Okuno Y, Fujita N and Katayama R: Brigatinib combined with
anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated
non-small-cell lung cancer. Nat Commun. 8:147682017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Facchinetti F, Rossi G, Bria E, Soria JC,
Besse B, Minari R, Friboulet L and Tiseo M: Oncogene addiction in
non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat
Rev. 55:83–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Planchard D, Kim TM, Mazieres J, Quoix E,
Riely G, Barlesi F, Souquet PJ, Smit EF, Groen HJ, Kelly RJ, et al:
Dabrafenib in patients with BRAF(V600E)-positive advanced
non-small-cell lung cancer: A single-arm, multicentre, open-label,
phase 2 trial. Lancet Oncol. 17:642–650. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar
|
|
13
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation-implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar
|
|
14
|
Dong L, Zhou Q, Zhang Z, Zhu Y, Duan T and
Feng Y: Metformin sensitizes endometrial cancer cells to
chemotherapy by repressing glyoxalase I expression. J Obstet
Gynaecol Res. 38:1077–1085. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duo J, Ma Y, Wang G, Han X and Zhang C:
Metformin synergistically enhances antitumor activity of histone
deacetylase inhibitor trichostatin a against osteosarcoma cell
line. DNA Cell Biol. 32:156–164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fujita H, Hirose K, Sato M, Fujioka I,
Fujita T, Aoki M and Takai Y: Metformin attenuates hypoxia-induced
resistance to cisplatin in the HepG2 cell line. Oncol Lett.
17:2431–2440. 2018.
|
|
17
|
Lin CC, Yeh HH, Huang WL, Yan JJ, Lai WW,
Su WP, Chen HH and Su WC: Metformin enhances cisplatin cytotoxicity
by suppressing signal transducer and activator of transcription-3
activity independently of the liver kinase B1-AMP-activated protein
kinase pathway. Am J Respir Cell Mol Biol. 49:241–250. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu G, Fang W, Xia T, Chen Y, Gao Y, Jiao
X, Huang S, Wang J, Li Z and Xie K: Metformin potentiates rapamycin
and cisplatin in gastric cancer in mice. Oncotarget. 6:12748–1262.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Teixeira SF: Metformin synergistically
enhances antiproliferative effects of cisplatin and etoposide in
NCI-H460 human lung cancer cells. J Bras Pneumol. 39:644–649. 2013.
View Article : Google Scholar
|
|
20
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, Jiang J and He Y: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Wang Y, Peng T, Zhang K, Lin C, Han
R, Lu C and He Y: Metformin restores crizotinib sensitivity in
crizotinib-resistant human lung cancer cells through inhibition of
IGF1-R signaling pathway. Oncotarget. 7:34442–34452. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pernicova I and Korbonits M:
Metformin-mode of action and clinical implications for diabetes and
cancer. Nat Rev Endocrinol. 10:143–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu GY and Sabatini DM: mTOR at the nexus
of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol.
21:183–203. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tavares MR, Pavan IC, Amaral CL,
Meneguello L, Luchessi AD and Simabuco FM: The S6K protein family
in health and disease. Life Sci. 131:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012. View Article : Google Scholar
|
|
26
|
Amaral CL, Freitas LB, Tamura RE, Tavares
MR, Pavan IC, Bajgelman MC and Simabuco FM: S6Ks isoforms
contribute to viability, migration, docetaxel resistance and tumor
formation of prostate cancer cells. BMC Cancer. 16:6022016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhong D, Guo L, de Aguirre I, Liu X, Lamb
N, Sun SY, Gal AA, Vertino PM and Zhou W: LKB1 mutation in large
cell carcinoma of the lung. Lung Cancer. 53:285–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kullmann L and Krahn MP: Controlling the
master-upstream regulation of the tumor suppressor LKB1. Oncogene.
37:3045–3057. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang G and Liu CT: Knockdown of SALL4
overcomes cisplatin-resistance through AKT/mTOR signaling in lung
cancer cells. Int J Clin Exp Pathol. 11:634–641. 2018.PubMed/NCBI
|
|
30
|
Teng X, Fan XF, Li Q, Liu S, Wu DY, Wang
SY, Shi Y and Dong M: XPC inhibition rescues cisplatin resistance
via the Akt/mTOR signaling pathway in A549/DDP lung adenocarcinoma
cells. Oncol Rep. 41:1875–1882. 2019.PubMed/NCBI
|
|
31
|
Liang SQ, Bührer ED, Berezowska S, Marti
TM, Xu D, Froment L, Yang H, Hall SRR, Vassella E, Yang Z, et al:
mTOR mediates a mechanism of resistance to chemotherapy and defines
a rational combination strategy to treat KRAS-mutant lung cancer.
Oncogene. 38:622–636. 2019. View Article : Google Scholar
|
|
32
|
Algire C, Amrein L, Bazile M, David S,
Zakikhani M and Pollak M: Diet and tumor LKB1 expression interact
to determine sensitivity to anti-neoplastic effects of metformin in
vivo. Oncogene. 30:1174–1182. 2011. View Article : Google Scholar
|
|
33
|
Moro M, Caiola E, Ganzinelli M, Zulato E,
Rulli E, Marabese M, Centonze G, Busico A, Pastorino U, de Braud
FG, et al: Metformin enhances cisplatin-induced apoptosis and
prevents resistance to cisplatin in Co-mutated KRAS/LKB1 NSCLC. J
Thorac Oncol. 13:1692–1704. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Ye J, Coulouris G, Zaretskaya I,
Cutcutache I, Rozen S and Madden TL: Primer-BLAST: A tool to design
target-specific primers for polymerase chain reaction. BMC
Bioinformatics. 13:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bremang M, Cuomo A, Agresta AM, Stugiewicz
M, Spadotto V and Bonaldi T: Mass spectrometry-based identification
and characterisation of lysine and arginine methylation in the
human proteome. Mol Biosyst. 9:2231–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cox J, Neuhauser N, Michalski A, Scheltema
RA, Olsen JV and Mann M: Andromeda: A peptide search engine
integrated into the MaxQuant environment. J Proteome Res.
10:1794–1805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43(Database Issue): D447–D452. 2015. View Article : Google Scholar
|
|
39
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41(Database Issue): D808–D815. 2013. View Article : Google Scholar :
|
|
40
|
Campbell JD, Alexandrov A, Kim J, Wala J,
Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et
al: Distinct patterns of somatic genome alterations in lung
adenocarcinomas and squamous cell carcinomas. Nat Genet.
48:607–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roesch A, Vultur A, Bogeski I, Wang H,
Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA,
Philipp SE, et al: Overcoming intrinsic multidrug resistance in
melanoma by blocking the mitochondrial respiratory chain of
slow-cycling JARID1Bhigh cells. Cancer Cell. 23:811–825. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mogami T, Yokota N, Asai-Sato M, Yamada R,
Koizume S, Sakuma Y, Yoshihara M, Nakamura Y, Takano Y, Hirahara F,
et al: Annexin A4 is involved in proliferation, chemo-resistance
and migration and invasion in ovarian clear cell adenocarcinoma
cells. PLoS One. 8:e803592013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li N, Huang HQ and Zhang GS: Association
between SOD2 C47T polymorphism and lung cancer susceptibility: A
meta-analysis. Tumor Biol. 35:955–959. 2014. View Article : Google Scholar
|
|
45
|
Morales DR and Morris AD: Metformin in
cancer treatment and prevention. Annu Rev Med. 66:17–29. 2015.
View Article : Google Scholar
|
|
46
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sharma A, Bandyopadhayaya S, Chowdhury K,
Sharma T, Maheshwari R, Das A, Chakrabarti G, Kumar V and Mandal
CC: Metformin exhibited anticancer activity by lowering cellular
cholesterol content in breast cancer cells. PLoS One.
14:e02094352019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee JO, Kang MJ, Byun WS, Kim SA, Seo IH,
Han JA, Moon JW, Kim JH, Kim SJ, Lee EJ, et al: Metformin overcomes
resistance to cisplatin in triple-negative breast cancer (TNBC)
cells by targeting RAD51. Breast Cancer Res. 21:1152019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dang JH, Jin ZJ, Liu XJ, Hu D, Wang J, Luo
Y and Li LL: Metformin in combination with cisplatin inhibits cell
viability and induces apoptosis of human ovarian cancer cells by
inactivating ERK 1/2. Oncol Lett. 14:7557–7564. 2017.
|
|
50
|
Riaz MA, Sak A, Erol YB, Groneberg M,
Thomale J and Stuschke M: Metformin enhances the radiosensitizing
effect of cisplatin in non-small cell lung cancer cell lines with
different cisplatin sensitivities. Sci Rep. 9:12822019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang JW, Zhao F and Sun Q: Metformin
synergizes with rapamycin to inhibit the growth of pancreatic
cancer in vitro and in vitro. Oncol Lett. 15:1811–1816.
2018.PubMed/NCBI
|
|
52
|
Jin DH, Kim Y, Lee B, Han J, Kim HK, Shim
YM and Kim DH: Metformin induces cell cycle arrest at the G1 phase
through E2F8 suppression in lung cancer cells. Oncotarget.
8:101509–101519. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Queiroz EAIF, Puukila S, Eichler R,
Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB
and Khaper N: Metformin induces apoptosis and cell cycle arrest
mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast
cancer cells. PLoS One. 9. pp. e982072014, View Article : Google Scholar
|
|
54
|
Xie W, Wang L, Sheng H, Qiu J, Zhang D,
Zhang L, Yang F, Tang D and Zhang K: Metformin induces growth
inhibition and cell cycle arrest by upregulating MicroRNA34a in
renal cancer cells. Med Sci Monit. 23:29–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lundholm L, Hååg P, Zong D, Juntti T, Mörk
B, Lewensohn R and Viktorsson K: Resistance to DNA-damaging
treatment in non-small cell lung cancer tumor-initiating cells
involves reduced DNA-PK/ATM activation and diminished cell cycle
arrest. Cell Death Dis. 4:e4782013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chatterjee A, Mukhopadhyay S, Tung K,
Patel D and Foster DA: Rapamycin-induced G1 cell cycle arrest
employs both TGF-β and Rb pathways. Cancer Lett. 360:134–140. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lu Z, Peng K, Wang N, Liu HM and Hou G:
Downregulation of p70S6K enhances cell sensitivity to rapamycin in
esophageal squamous cell carcinoma. J Immunol Res.
2016:78289162016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Song J, Wang X, Zhu J and Liu J: Rapamycin
causes growth arrest and inhibition of invasion in human
chondrosarcoma cells. J BUON. 21:244–251. 2016.PubMed/NCBI
|
|
59
|
Wang Y, Xu W, Yan Z, Zhao W, Mi J, Li J
and Yan H: Metformin induces autophagy and G0/G1 phase cell cycle
arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways.
J Exp Clin Cancer Res. 37:632018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhong DS, Sun LL and Dong LX: Molecular
mechanisms of LKB1 induced cell cycle arrest. Thorac Cancer.
4:229–233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang Y, Bao C, Mu Q, Chen J, Wang J, Mi
Y, Sayari AJ, Chen Y and Guo M: Reversal of cisplatin resistance by
inhibiting PI3K/Akt signal pathway in human lung cancer cells.
Neoplasma. 63:362–370. 2016. View Article : Google Scholar
|
|
62
|
Sheng J, Shen L, Sun L, Zhang X, Cui R and
Wang L: Inhibition of PI3K/mTOR increased the sensitivity of
hepatocellular carcinoma cells to cisplatin via interference with
mitochondrial-lysosomal crosstalk. Cell Prolif. 52:e126092019.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hou G, Yang S, Zhou Y, Wang C, Zhao W and
Lu Z: Targeted inhibition of mTOR signaling improves sensitivity of
esophageal squamous cell carcinoma cells to cisplatin. J Immunol
Res. 2014:8457632014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhao Y, Sun H, Feng M, Zhao J, Zhao X, Wan
Q and Cai D: Metformin is associated with reduced cell
proliferation in human endometrial cancer by inbibiting
PI3K/AKT/mTOR signaling. Gynecol Endocrinol. 34:428–432. 2018.
View Article : Google Scholar
|
|
65
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar
|
|
66
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pandey A and Mann M: Proteomics to study
genes and genomes. Nature. 405:837–846. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chang X, Ravi R, Pham V, Bedi A,
Chatterjee A and Sidransky D: Adenylate kinase 3 sensitizes cells
to cigarette smoke condensate vapor induced cisplatin resistance.
PLoS One. 6:e208062011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wei Y, Wu S, Xu W, Liang Y, Li Y, Zhao W
and Wu J: Depleted aldehyde dehydrogenase 1A1 (ALDH1A1) reverses
cisplatin resistance of human lung adenocarcinoma cell A549/DDP.
Thorac Cancer. 8:26–32. 2017. View Article : Google Scholar :
|
|
70
|
Morimoto A, Serada S, Enomoto T, Kim A,
Matsuzaki S, Takahashi T, Ueda Y, Yoshino K, Fujita M, Fujimoto M,
et al: Annexin A4 induces platinum resistance in a chloride-and
calcium-dependent manner. Oncotarget. 5:7776–7787. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Feng X, Liu H, Zhang Z, Gu Y, Qiu H and He
Z: Annexin A2 contributes to cisplatin resistance by activation of
JNK-p53 pathway in non-small cell lung cancer cells. J Exp Clin
Cancer Res. 36:1232017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Becker M, De Bastiani MA, Müller CB,
Markoski MM, Castro MA and Klamt F: High cofilin-1 levels correlate
with cisplatin resistance in lung adenocarcinomas. Tumor Biology.
35:1233–1238. 2014. View Article : Google Scholar
|
|
73
|
Wittig R, Nessling M, Will RD, Mollenhauer
J, Salowsky R, Münstermann E, Schick M, Helmbach H, Gschwendt B,
Korn B, et al: Candidate genes for cross-resistance against
DNA-damaging drugs. Cancer Res. 62:6698–6705. 2002.PubMed/NCBI
|
|
74
|
Zeller C, Dai W, Steele NL, Siddiq A,
Walley AJ, Wilhelm-Benartzi CS, Rizzo S, van der Zee A, Plumb JA
and Brown R: Candidate DNA methylation drivers of acquired
cisplatin resistance in ovarian cancer identified by methylome and
expression profiling. Oncogene. 31:4567–4576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang M, Li Y, Shen X, Ruan Y, Lu Y, Jin X,
Song P, Guo Y, Zhang X, Qu H, et al: CLDN6 promotes chemoresistance
through GSTP1 in human breast cancer. J Exp Clin Cancer Res.
36:1572017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang R, Tao F, Ruan S, Hu M, Hu Y, Fang
Z, Mei L and Gong C: The TGFβ1-FOXM1-HMGA1-TGFβ1 positive feedback
loop increases the cisplatin resistance of non-small cell lung
cancer by inducing G6PD expression. Am J Transl Res. 11:6860–6876.
2019.
|
|
77
|
Wang JM, Liu BQ, Zhang Q, Hao L, Li C, Yan
J, Zhao FY, Qiao HY, Jiang JY and Wang HQ: ISG15 suppresses
translation of ABCC2 via ISGylation of hnRNPA2B1 and enhances drug
sensitivity in cisplatin resistant ovarian cancer cells. Biochim
Biophys Acta Mol Cell Res. 1867:1186472020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Di Martino S, Amoreo CA, Nuvoli B, Galati
R, Strano S, Facciolo F, Alessandrini G, Pass HI, Ciliberto G,
Blandino G, et al: HSP90 inhibition alters the chemotherapy-driven
rearrangement of the oncogenic secretome. Oncogene. 37:1369–1385.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Qin X, Sun L and Wang J: Restoration of
microRNA-708 sensitizes ovarian cancer cells to cisplatin via
IGF2BP1/Akt pathway. Cell Biol Int. 41:1110–1118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Xu Z, Zou L, Ma G, Wu X, Huang F, Feng T,
Li S, Lin Q, He X, Liu Z and Cao X: Integrin β1 is a critical
effector in promoting metastasis and chemo-resistance of esophageal
squamous cell carcinoma. Am J Cancer Res. 7:531–542. 2017.
|
|
81
|
Li Y, Liu X, Lin X, Zhao M, Xiao Y, Liu C,
Liang Z, Lin Z, Yi R, Tang Z, et al: Chemical compound
cinobufotalin potently induces FOXO1-stimulated cisplatin
sensitivity by antagonizing its binding partner MYH9. Signal
Transduct Target Ther. 4:482019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Depeng S, Wu J, Guo L, Xu Y, Liu L and Lu
J: Metformin increases sensitivity of osteosarcoma stem cells to
cisplatin by inhibiting expression of PKM2. Int J Oncol.
50:1848–1856. 2017. View Article : Google Scholar
|
|
83
|
Zhu H, Wu J, Zhang W, Luo H, Shen Z, Cheng
H and Zhu X: PKM2 enhances chemosensitivity to cisplatin through
interaction with the mTOR pathway in cervical cancer. Sci Rep.
6:307882016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C
and Chen X: PKM2 inhibitor shikonin overcomes the cisplatin
resistance in bladder cancer by inducing necroptosis. International
J Biol Sci. 14:1883–1891. 2018. View Article : Google Scholar
|
|
85
|
Krafft U, Tschirdewahn S, Hess J, Harke
NN, Hadaschik BA, Nyirády P, Szendröi A, Szücs M, Módos O, Olah C,
et al: STIP1 tissue expression is associated with survival in
chemotherapy-treated bladder cancer patients. Pathol Oncol Res.
26:1243–1249. 2020. View Article : Google Scholar
|
|
86
|
Li C, Cai J, Ge F and Wang G: TGM2
knockdown reverses cisplatin chemoresistance in osteosarcoma. Int J
Molr Med. 42:1799–1808. 2018.
|
|
87
|
Yang H, Wu XL, Wu KH, Zhang R, Ju LL, Ji
Y, Zhang YW, Xue SL, Zhang YX, Yang YF and Yu MM: MicroRNA-497
regulates cisplatin chemosensitivity of cervical cancer by
targeting transketolase. Am J Cancer Res. 6:2690–2699.
2016.PubMed/NCBI
|
|
88
|
Matassa DS, Amoroso MR, Lu H, Avolio R,
Arzeni D, Procaccini C, Faicchia D, Maddalena F, Simeon V,
Agliarulo I, et al: Oxidative metabolism drives
inflammation-induced platinum resistance in human ovarian cancer.
Cell Death Differ. 23:1542–1554. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yue T, Zheng X, Dou Y, Zheng X, Sun R,
Tian Z and Wei H: Interleukin 12 shows a better curative effect on
lung cancer than paclitaxel and cisplatin doublet chemotherapy. BMC
Cancer. 16:6652016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Barros FBA, Assao A, Garcia NG, Nonogaki
S, Carvalho AL, Soares FA, Kowalski LP and Oliveira DT: Moesin
expression by tumor cells is an unfavorable prognostic biomarker
for oral cancer. BMC Cancer. 18:532018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Alam F, Mezhal F, El Hasasna H, Nair VA,
Aravind SR, Saber Ayad M, El-Serafi A and Abdel-Rahman WM: The role
of p53-microRNA 200-Moesin axis in invasion and drug resistance of
breast cancer cells. Tumor Bio. 39:10104283177146342017.
|
|
92
|
Wang Q, Lu X, Zhao S, Pang M, Wu X, Wu H,
Hoffman RM, Yang Z and Zhang Y: Moesin expression is associated
with glioblastoma cell proliferation and invasion. Anticancer Res.
37:2211–2218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang J, Gao Q, Wang D, Wang Z and Hu C:
Metformin inhibits growth of lung adenocarcinoma cells by inducing
apoptosis via the mitochondria-mediated pathway. Oncol Lett.
10:1343–1349. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Seguin L, Desgrosellier JS, Weis SM and
Cheresh DA: Integrins and cancer: Regulators of cancer stemness,
metastasis, and drug resistance. Trends Cell Biol. 25:234–240.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Blandin AF, Renner G, Lehmann M,
Lelong-Rebel I, Martin S and Dontenwill M: β1 integrins as
therapeutic targets to disrupt hallmarks of cancer. Front
Pharmacol. 6:2792015. View Article : Google Scholar
|
|
96
|
Ju L, Zhou C, Li W and Yan L: Integrin
beta1 over-expression associates with resistance to tyrosine kinase
inhibitor gefitinib in non-small cell lung cancer. J Cel Biochem.
111:1565–1574. 2010. View Article : Google Scholar
|
|
97
|
Kanda R, Kawahara A, Watari K, Murakami Y,
Sonoda K, Maeda M, Fujita H, Kage M, Uramoto H, Costa C, et al:
Erlotinib resistance in lung cancer cells mediated by integrin
β1/Src/Akt-driven bypass signaling. Cancer Res. 73:6243–6253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gachechiladze M, Skarda J, Janikova M,
Mgebrishvili G, Kharaishvili G, Kolek V, Grygarkova I, Klein J,
Poprachova A, Arabuli M and Kolar Z: Overexpression of filamin-A
protein is associated with aggressive phenotype and poor survival
outcomes in NSCLC patients treated with platinum-based combination
chemotherapy. Neoplasma. 63:274–281. 2016.PubMed/NCBI
|
|
99
|
Ji ZM, Yang LL, Ni J, Xu SP, Yang C, Duan
P, Lou LP and Ruan QR: Silencing filamin a inhibits the invasion
and migration of breast cancer cells by up-regulating 14-3-3σ. Curr
Med Sci. 38:461–466. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li L, Lu Y, Stemmer PM and Chen F: Filamin
A phosphorylation by Akt promotes cell migration in response to
arsenic. Oncotarget. 6:12009–12019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Guo Y, Li M, Bai G, Li X, Sun Z, Yang J,
Wang L and Sun J: Filamin a inhibits tumor progression through
regulating BRCA1 expression in human breast cancer. Oncol Lett.
16:6261–6266. 2018.PubMed/NCBI
|
|
102
|
Donadon M, Di Tommaso L, Soldani C,
Franceschini B, Terrone A, Mimmo A, Vitali E, Roncalli M, Lania A
and Torzilli G: Filamin A expression predicts early recurrence of
hepatocellular carcinoma after hepatectomy. Liver Int. 38:303–311.
2018. View Article : Google Scholar
|
|
103
|
Maynadier M, Farnoud R, Lamy PJ,
Laurent-Matha V, Garcia M and Rochefort H: Cathepsin D stimulates
the activities of secreted plasminogen activators in the breast
cancer acidic environment. Int J Oncol. 43:1683–1690. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hah YS, Noh HS, Ha JH, Ahn JS, Hahm JR,
Cho HY and Kim DR: Cathepsin D inhibits oxidative stress-induced
cell death via activation of autophagy in cancer cells. Cancer
Lett. 323:208–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhang C, Zhang M and Song S: Cathepsin D
enhances breast cancer invasion and metastasis through promoting
hepsin ubiquitin-proteasome degradation. Cancer Lett. 438:105–115.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kang SW: Superoxide dismutase 2 gene and
cancer risk: Evidence from an updated meta-analysis. Int J Clin Exp
Med. 8:14647–14655. 2015.PubMed/NCBI
|
|
107
|
Lee JH, Choi IY, Kil IS, Kim SY, Yang ES
and Park JW: Protective role of superoxide dismutases against
ionizing radiation in yeast. Biochim Biophys Acta. 1526:191–198.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Epperly MW, Gretton JE, Sikora CA,
Jefferson M, Bernarding M, Nie S and Greenberger JS: Mitochondrial
localization of superoxide dismutase is required for decreasing
radiation-induced cellular damage. Radiat Res. 160:568–578. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Takada Y, Hachiya M, Park SH, Osawa Y,
Ozawa T and Akashi M: Role of reactive oxygen species in cells
overexpressing manganese superoxide dismutase: Mechanism for
induction of radioresistance. Mol Cancer Res. 1:137–146.
2002.PubMed/NCBI
|
|
110
|
Hosoki A, Yonekura S, Zhao QL, Wei ZL,
Takasaki I, Tabuchi Y, Wang LL, Hasuike S, Nomura T, Tachibana A,
et al: Mitochondria-targeted superoxide dismutase (SOD2) regulates
radiation resistance and radiation stress response in HeLa cells. J
Radiat Res. 53:58–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Cruz-Bermúdez A, Laza-Briviesca R,
Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S,
Palacios-Zambrano S, Moreno-Villa MR, Ruiz-Valdepeñas AM, Lendinez
C, et al: Cisplatin resistance involves a metabolic reprogramming
through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS
inhibition. Free Radic Biol Med. 135:167–181. 2019. View Article : Google Scholar
|
|
112
|
Zuo J, Zhao M, Liu B, Han X, Li Y, Wang W,
Zhang Q, Lv P, Xing L, Shen H and Zhang X: TNF-α-mediated
upregulation of SOD-2 contributes to cell proliferation and
cisplatin resistance in esophageal squamous cell carcinoma. Oncol
Rep. 42:1497–1506. 2019.PubMed/NCBI
|
|
113
|
Yan X, Pan L, Yuan Y, Lang JH and Mao N:
Identification of platinum-resistance associated proteins through
proteomic analysis of human ovarian cancer cells and their
platinum-resistant sublines. J Proteome Res. 6:772–780. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Matsuzaki S, Enomoto T, Serada S, Yoshino
K, Nagamori S, Morimoto A, Yokoyama T, Kim A, Kimura T, Ueda Y, et
al: Annexin A4-conferred platinum resistance is mediated by the
copper transporter ATP7A. Int J Cancer. 134:1796–1809. 2014.
View Article : Google Scholar
|
|
115
|
Yao HS, Sun C, Li XX, Wang Y, Jin KZ,
Zhang XP and Hu ZQ: Annexin A4-nuclear factor-κB feedback circuit
regulates cell malignant behavior and tumor growth in gallbladder
cancer. Sci Rep. 6:310562016. View Article : Google Scholar
|
|
116
|
Yamashita T, Nagano K, Kanasaki S, Maeda
Y, Furuya T, Inoue M, Nabeshi H, Yoshikawa T, Yoshioka Y, Itoh N,
et al: Annexin A4 is a possible biomarker for cisplatin
susceptibility of malignant mesothelioma cells. Biochem Biophys Res
Commun. 421:140–144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Vizcaíno JA, Deutsch EW, Wang R, Csordas
A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, et
al: ProteomeXchange provides globally coordinated proteomics data
submission and dissemination. Nat Biotechnol. 32:223–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|