Gliomas are the most common primary malignant tumors
in the brain. Glioblastoma multiforme (GBM), also known as World
Health Organization grade IV glioma, is a disease with a high
mortality rate worldwide, for which there is currently no effective
therapy (1). The current standard
treatments for GBM include maximal surgical resection, radiotherapy
and temozolomide chemotherapy. However, GBM tends to recur despite
therapy, with a recurrence rate as high as 90%. The median overall
survival is 15-18 months according to population-based studies, and
<10% of patients remain alive at 5 years post-diagnosis
(2).
GBM is a heterogeneous tumor that is characterized
by high resistance to therapy, which is promoted by GBM stem cells
(GSCs) (1). Cancer stem cells
constitute a small cell population that is highly involved in the
malignant behavior of numerous types of cancer (3), and they are defined by their
functionality, including maintenance of stemness, tumor formation,
tumor relapse and resistance to therapy (4). GBM follows a cellular hierarchy
model, with GSCs at the top and differentiated offspring cells at
the bottom of the model (5,6).
There are significant similarities between neural
stem/progenitor cells (NSCs) and GSCs, such as the expression of
stem cell markers (CD133, SOX2, oligodendrocyte transcription
factor 2 and nestin), and the ability to differentiate into
oligodendrocytes, astrocytes and neurons. However, GSCs are
characterized by the expression of multiple lineage markers in one
differentiated cell (7,8). GSCs harbor genetic abnormalities,
which contribute to tumor invasion (9), angiogenesis (10) and radio-resistance (11). The main characteristics of GSCs
are their capacity for self-renewal and differentiation (3). In a xenograft assay, GSCs displayed
greater tumorigenic capacity compared with non-stem tumor cells
(6). GSCs play a pivotal role in
the growth and therapeutic resistance of adult human GBM (7), suggesting that GSCs may lead to
tumor recurrence and, eventually, death. Thus, exploring the
signaling pathways regulating GSC self-renewal and the design of
therapies targeting these signaling pathways are important research
objectives. In recent years, numerous different signaling pathways
and potential therapeutic targets for GBM have been identified,
including TGF-β, Notch, Wnt, Hedgehog (HH) and STAT3, which also
play important roles in normal stem cell development and
differentiation. The focus of the present review was these five key
self-renewal GSC signaling pathways and the corresponding
differentiation therapies, with the aim of providing novel insight
and promoting advances in the clinical therapy of GBM.
The TGF-β family includes polypeptides that regulate
GSC maintenance and tumor differentiation (12,13). In addition, TGF-β signaling is
involved in carcinogenesis and tumor development (12).
The TGF-β family comprises TGF-βs, bone
morphogenetic proteins (BMPs) and other associated proteins
(14). The TGF-β signaling
cascade is a linear pathway from type II to type I receptor kinase
to SMAD activation, resulting in the transcription of target genes
in the cell nucleus. On the cell surface, TGF-β ligands bind to the
transmembrane receptor serine/threonine kinase (type I and II)
complex, and then type II receptor kinases [BMP receptor (BMPR)II,
activin receptor (ActR)II, ActRIIB, TGF-β receptor (TβR)II and
anti-Müllerian hormone receptor] and trans-phosphorylate type I
receptors [anaplastic lymphoma kinase (ALK); ALK5/1/2 for TβR and
ALK2/3/6 for BMPR]. The consequently activated type I receptors
trigger phosphorylation of SMAD and receptor-regulated SMAD
(R-SMAD) (TGF-β, R-SMAD2/3/1/5; BMP, R-SMAD1/5/8), which then form
a complex with common mediator SMAD4 (Co-SMAD). The activated
R-SMAD/Co-SMAD complex translocates to the nucleus to regulate gene
transcription. The activation of R-SMAD is inhibited by SMAD6 or
SMAD7 (14,15). It was previously demonstrated that
TGF-β signaling regulates cell fate (16), and that blockade of this signaling
pathway can inhibit proliferation of cancer cells and the GSC
subpopulation (17). Thus,
targeting the TGF-β pathway may be a meaningful treatment for GBM.
The current clinical trials on drugs targeting the TGF-β signaling
pathway are summarized in Table
I.
BMPs comprise a subfamily of the TGF-β superfamily,
and they are secreted signaling molecules that regulate embryonic
development (18). BMPRs, acting
as paracrine tumor suppressors, have a flexible oligomerization
pattern, which allows a greater variety of responses to ligands
(19). BMPs and growth
differentiation factors (GDFs) form a cystine-knot cytokine family,
which shares the characteristics of the TGF-β superfamily. GDFs are
extracellular factors containing a potential signaling sequence for
secretion and a proteolytic processing site (20,21). BMP/GDFs exist as homodimers and
heterodimers, and interact with complexes of type I and type II
receptor dimers, leading to the activation of one of two competing
sets of R-SMAD (22).
BMPs can cause a significant reduction in stem cell
numbers in GBM. BMPs induce GSC differentiation, attenuate the
expression of stemness markers, reduce self-renewal and block tumor
initiation (23-25). Thus, BMPs have been proposed as
potential differentiation therapies targeting GSCs, which may be
used to prevent GBM growth and recurrence (26).
BMPs can influence astrocyte fate and induce loss of
tumorigenicity, and they are considered as a GSC differentiation
targeted therapy; however, paradoxically, tumors express high
levels of BMPs (24,27). Gremlin, a protein of 184 amino
acids, contains a highly conserved cysteine knot domain shared by
the TGF-β superfamily (28). The
antagonist gremlin 1 has been demonstrated to be specifically
expressed by GSCs to protect against endogenous BMPs. Gremlin 1
blocks the differentiation effects of BMPs on GSCs and promotes the
maintenance of cancer cell stemness, thereby increasing tumor
formation ability. Targeting gremlin 1 results in impaired cell
proliferation and self-renewal. Mechanistically, gremlin 1 mediates
the downregulation of the cyclin-dependent kinase inhibitor
p21WAF1/CIP1, a key GSC signaling node (29). Thus, inhibition of gremlin 1 may
act synergistically with BMPs in GBM treatment. One therapy option
is to engineer a BMP variant that does not bind to gremlin 1
(30). Another option is combined
therapy of antibodies against gremlin 1 alongside BMP-based
therapy.
Previous studies have demonstrated that TGF-β
activity is present in aggressive and highly proliferative gliomas
(31). TGF-β has been shown to
induce self-renewal capacity and prevent differentiation in GSCs.
Furthermore, TGF-β may play a role in GSC-mediated oncogenesis via
leukemia inhibitory factor induction in vivo (32). Thus, blocking TGF-β signaling in
GBM may be of therapeutic value.
The expression of HMOX1, one of the cell surface
transmembrane proteins, is increased in GBM, and a high expression
level is associated with increased stemness and poor prognosis in
GBM (36). HMOX1 regulates
differentiation through the TGF-β signaling pathway (37). Specifically, TGF-β regulates HMOX1
expression on the cell surface, and endogenous activators (such as
EGFR) and inhibitors (such as PTEN) of TGF-β signaling may also
interfere with the expression of HMOX1. These findings indicate
that targeting HMOX1 may be a novel therapeutic approach to
GBM.
Notch proteins (Notch 1-4) are transmembrane
receptors that mediate cell-cell signaling. Notch signaling can
amplify and consolidate molecular differences, eventually dictating
cell growth, proliferation, survival and differentiation. Notch
activity affects cell differentiation, proliferation and apoptotic
programs (41). Both the
receptors and ligands of the Notch family are cell surface type I
transmembrane proteins. Notch ligands include delta (Dl) and
Serrate. Upon ligand binding, Notch receptors undergo three
proteolytic cleavages. The first cleavage, S1, generates fragments
and forms a heterodimeric receptor, which is inserted in the cell
membrane (42). S2 occurs after
the heterodimers bind to the ligand (Dl-like-1, -3 and -4, and
Jagged-1 and -2). S3 is mediated by the γ-secretase complex,
leading to the release of Notch intracellular domain (NICD) into
the nucleus (43).
The Notch signaling pathway, including NICD,
hairy/enhancer-of-split (Hes)1 and Hes related family basic
helix-loop-helix (bHLH) transcription factor with YRPW motif 1
(Hey1), regulates cell stemness and differentiation. Activation of
the Notch receptor rapidly inhibits the death of NSCs (44). Inhibitors and activators targeting
Notch receptors and ligands that exert antitumor effects have been
developed. Notch stimulation results in poorly infiltrative but
highly vascularized grafts, in contrast to the highly infiltrative
and poorly vascularized characteristics of GBM stem cells. This
indicates that the Notch pathway is crucial for regulating GSC fate
(45).
During the early stages of embryogenesis, Notch
signaling serves as a critical quality control pathway to prevent
premature neurogenesis and maintain pools of progenitor cells in
the developing central nervous system. In the perinatal stages,
Notch signaling increases progenitor cell proliferation and drives
astrocyte differentiation, thereby serving a critical function in
human brain development. The Notch pathway is involved in
maintaining adult neural stem cells bivalently by promoting
self-renewal and repressing differentiation (46,47). The activity of the Notch signaling
pathway plays an instrumental role in regulating self-renewal and
determining cell fate of normal NSCs (48). The Notch pathway is active in NSCs
during neurogenesis, gliogenesis and tumorigenesis (49,50). It has been demonstrated that the
Notch target genes Hes1 and Hes5 are strongly associated with the
regulation of neurogenesis and gliomagenesis in the brain (51).
The self-renewal capacity of GSCs relies on the
activity of the Notch signaling pathway. The expression level of
the Notch receptor gene and its downstream activation cascade of
events are associated with the phenotypic plasticity and intratumor
heterogeneity of GBM cells (49).
A previous study that used computational modeling methods
demonstrated that the stem cell renewal induced by the Notch
pathway and the antagonistic effects exerted on the p53 pathway are
highly involved in maintaining the regenerative properties of the
NSCs (49,52). In agreement with this, previous
in vitro and in vivo studies on glioma cell lines
have indicated that CD133-positive GSCs are particularly sensitive
to γ-secretase inhibitors or Notch1/2 knockdown compared with
CD133-negative glioma cells (53). Blocking Notch signaling or
recombination signal-binding protein for immunoglobulin κJ region
(RBP-κJ), which is a major transcriptional effector of this
pathway, reduced clonogenicity potential in tumor sphere assays and
engraftment capacity in glioma xenograft models (48). Notch activity may contribute to
intratumor heterogeneity by promoting stem cell behavior in poorly
differentiated subpopulations of glioma cells. Notch signaling
potentially regulates multiple steps of gliomagenesis, including
tumor initiation, progression and recurrence.
However, the actual sequence of regulating events
and the exact mechanisms through which Notch activity controls
stemness and tumorigenicity remain to be elucidated. Since Notch
can promote and maintain the stem cell characteristics of brain
tumors, it may represent a promising target for developing more
effective therapies against glioma. A phase I clinical trial
investigating the use of γ-secretase/Notch inhibition in
combination with temozolomide and radiotherapy in newly diagnosed
GBM or anaplastic astrocytoma demonstrated that the addition of
Notch inhibition to standard treatment was associated with certain
benefits (Table II) (54), although it also clearly
demonstrated that Notch inhibition, alone or combined with
radiation and chemotherapy, may be insufficient for fully
controlling tumor progression. However, those findings indicated
that Notch may serve as a targeted biological tool that counteracts
tumor stem cell-like behavior by preventing self-renewal and,
possibly, angiogenesis (55).
NICD regulates transcription in the cell nucleus,
and is directly involved in transcriptional control by associating
with the DNA-binding protein CBF1, Suppressor of Hairless, Lag-1
(also known as RBP-κJ) (48). It
was previously reported that Notch1 is overexpressed in GSCs
(41,45). Enhancing the protein expression
and nuclear transport of NICD may upregulate Notch signaling. The
canonical importin α/β pathway, which targets proteins to the
nuclear pore complex and facilitates their translocation across the
nuclear envelope (56), can
regulate the transport of NICD into the nucleus, thus being
directly involved in the Notch signaling pathway (57).
CXCR4 is a cell surface chemokine receptor that is
closely associated with glioma growth. It is overexpressed in GSCs
and plays a critical role in regulating carcinogenesis (60). CXCL12, which is a
CXCR4-stimulating factor, was highly expressed in glioma cells.
Blockade of the CXCL12/CXCR4 signaling axis induces apoptosis and
inhibits cell cycle progression, thus promoting the survival of GBM
cells (61). In GBM, Notch1 and
CXCR4 are enriched in GSCs, and are co-expressed with stemness
markers (41,45,60). Blocking the Notch1 signaling
pathway may suppress the proliferation of GSCs, and this effect may
be reversed by upregulation of CXCL12. In addition, Notch1 could
directly enhance the transcription of CXCR4 (62). Decreasing Notch1 expression levels
may downregulate CXCR4 expression, leading to the inhibition of the
PI3K/AKT/mTOR signaling pathway, and attenuation of the ability of
GSC self-renewal and GBM growth (62). Therefore, investigating the
crosstalk between Notch1 and the CXCL12/CXCR4 axis may uncover more
effective therapies for Notch1-targeted treatment of GBM.
The Hes family comprises bHLH-type transcriptional
repressors that negatively regulate the expression of downstream
target genes (such as tissue-specific transcription factors). In
the nucleus, NICD associates with the nuclear proteins of the
RBP-κJ family and activates the transcription of primary target
genes of the Notch signaling pathway, such as Hes1-7 (63). Members of the Hes family are the
best characterized transcriptional targets of Notch signaling, and
negatively regulate downstream target gene expression. Thus, Hes
directly affects cell differentiation (63).
CPEB1 is a highly conserved RNA-binding protein that
specifically binds to CPE, which is indirectly involved in
translational repression and activation. Previous studies
demonstrated that CPEB1 could reduce sphere formation ability,
downregulate the expression of stemness markers and control cell
differentiation in GSCs, and it was positively associated with the
overall survival of patients with glioma (64,65). The detailed molecular mechanism of
action of CPEB1 is by specifically suppressing the translation of
Hes1, inducing the differentiation of GSCs at the
post-transcriptional level. Thus, CPEB1 is as a critical factor
involved in the Notch signaling pathway and may provide novel
approaches to GSC differentiation therapy.
Hey [also known as Hes-related repressor protein
(Herp), and Hey/Hesr/Hrt/CHF/gridlock], is a member of the bHLH
protein family and is associated with Hes. Hey expression is
directly upregulated by Notch ligand binding and has intrinsic
transcriptional repression activity (63). Hes and Hey form a stable
heterodimer that has DNA-binding and transcription-suppressive
activities, thus regulating Notch signaling and target gene
expression (66,67).
The canonical Wnt/β-catenin pathway is a highly
evolutionarily conserved signaling pathway that regulates
pluripotency in stem cells (68).
Frizzled proteins are receptors involved in Wnt signaling (69) that interact as co-receptors with
Arrow, a low-density lipoprotein receptor-related protein that
forms part of a receptor complex with Frizzled protein (70). When the Wnt ligand is activated by
binding to the co-receptors, the AKT/glycogen synthase kinase
(GSK)3β/adenomatous polyposis coli (APC) complex separates. The
AKT/GSK3β/APC complex promotes the degradation of β-catenin, an
intracellular signaling molecule. APC antagonizes the Wnt signaling
pathway directly at the β-catenin effector level in several
different ways: It acts as an adaptor between β-catenin and
C-terminal binding protein, removes β-catenin, abrogates
transcriptional transactivation, and inhibits the binding of the
lymphoid enhancer-binding factor (LEF)/T-cell factor (TCF) proteins
to β-catenin (71,72). Therefore, through the degradation
of the AKT/GSK3β/APC complex, intracytoplasmic β-catenin becomes
stable, and non-phosphorylated β-catenin accumulates in the
cytoplasm and translocates to the nucleus to facilitate the
transcription of target genes by interacting with the TCF and LEF
transcription factors (71). The
XTC-3 transcription factor mediates β-catenin-induced axis
formation in Xenopus embryos. Functional interaction of β-catenin
with the transcription factor LEF-1 leads to the nuclear
localization of β-catenin (73-75). Wnt plays a key role in maintaining
stemness in GBM cells (76,77). Thus, abnormal activation of the
Wnt pathway may promote GSC self-renewal.
The architectural transcription factor LEF-1
interacts with β-catenin (thus forming a localized complex in the
nucleus), and regulates transcriptional activation and tumor
growth. The complex forms a ternary complex with DNA that displays
an altered DNA bend (74,79,80). AEG-1 is an oncogene that is
upregulated in GBM, which plays a key role in cancer cell
metastasis and regulates tumorigenesis (81). In GBM cells, the internal domain
of AEG-1 directly interacts with the pleckstrin homology domain of
AKT2, thus contributing to tumor cell survival and proliferation
(82). It has been reported that
the expression level of AEG-1 is strongly associated with the
presence of stemness markers in GBM. AEG-1 promotes the
translocation of β-catenin into the nucleus by forming a complex
with LEF1 and β-catenin, and then activating Wnt signaling in GSCs
via the AEG-1/AKT/GSK3β signaling axis (83). Thus, AEG-1 acts as a critical
regulator of Wnt/β-catenin signaling to control GSC stemness and
differentiation.
CD163/casein kinase (CK)2. CD163 has been reported
to act as a receptor that scavenges hemoglobin by regulating the
endocytosis of haptoglobin-hemoglobin complexes (84). CD163 is considered to be a marker
of the tumor-associated macrophage (TAM) M2 phenotype (85). TAMs are reported to secrete
pleiotrophin to stimulate GSCs and promote GBM growth through its
receptor protein tyrosine phosphatase receptor type Z1 (86). A high expression level of CD163 in
glioma has been demonstrated to be correlated with poor prognosis
(85).
CK2, whose constitutive phosphorylation is required
for AKT activation, can interact with CD163 and plays an essential
role in CD163 signaling (87,88). CD163 is necessary for maintaining
GSC stemness, and downregulation of CD163 decreases stemness marker
expression in GBM by interacting directly with CK2 and then
inhibiting the CK2/AKT/GSK3β/β-catenin pathway. A previous study
found that anti-CD163 antibodies induce cytotoxicity against glioma
cells, indicating that CD163 may serve as a therapeutic target for
glioma cells, specifically GSCs (89).
The ING family of epigenetic regulators (ING1-5) can
target histone acetyltransferase and histone deacetylase complexes
to alter histone acetylation and gene expression. The ectopic
expression of ING5 increases stemness, promotes self-renewal and
prevents differentiation of GSCs by enhancing PI3K/AKT activity.
This suggests that ING5 may represent a valuable target for
therapeutic strategies in GBM (90).
β-catenin, a cytoplasmic protein, has two functions:
Linking cadherin-mediated cell-adhesion molecules with the
cytoskeleton and participating in the Wnt signaling pathway
(74). A previous study has shown
that the content of β-catenin affects the Wnt signaling pathway
(74). When the AKT/GSK3β/APC
complex is degraded, β-catenin becomes stable and translocates into
the nucleus to facilitate the transcription of target genes
(91).
CypA belongs to the peptidyl-prolyl isomerase
family. CypA is a specific cytosolic protein and can form a complex
with cyclosporin A to induce immunosuppression (92). It was previously demonstrated that
CypA is associated with GBM growth (93). CypA has been found to promote GSC
stemness, self-renewal and proliferation (94). Mechanistically, CypA binds to
β-catenin and increases the interaction between β-catenin and TCF4
to regulate gene transcription (94). Thus, CypA is a potential target
for glioma therapy.
Classical HH signaling is required to maintain stem
cell niches in the adult brain (95). HH has three gene homologs: Sonic
HH (SHH), Desert HH and Indian HH. Upon inhibiting SHH signaling,
the number of neural progenitors is reduced. Activation of the HH
protein requires Rasp-dependent acylation (96). HH ligands initiate signaling
pathways by binding to the transmembrane receptor protein patched
homolog (PTCH). The HH-PTCH complex is internalized, and the
inhibition of the receptor Smoothened (Smo) is abolished, thus
allowing Smo activation, which induces the activation of the
glioma-associated oncogene homolog (Gli) family. As a result, Gli
translocates to the nucleus to regulate the transcription of target
genes (97).
The Gli family consists of zinc-finger transcription
factors, including Gli1, Gli2 and Gli3. Gli3 and SHH repress each
other, while Gli2 and Gli1 are the SHH signaling targets. However,
only Gli1 can mediate SHH-induced cell differentiation. Gli3 mostly
acts as a repressor, whereas Gli2 has both activator and repressor
functions (98).
The STAT3 signaling pathway is involved in multiple
biological processes, including cell proliferation, differentiation
and self-renewal of GSCs. Cytokines and growth factors bind to
their receptor, which, once dimerized, activates Janus kinase
(JAK). JAK induces STATs phosphorylation, and activated STATs
translocate into the nucleus to regulate target gene expression.
Previous studies found that phosphorylated STAT3 interacts with the
switch/sucrose non-fermentable complex in the nucleus (91,103). TRIM8, the expression of which is
highly correlated with stem cell markers, is reported to activate
STAT3 signaling to maintain the stemness and self-renewal of GSCs.
TRIM8 activates STAT3 by suppressing the expression of the protein
inhibitor of activated STAT3, and STAT3 activation can upregulate
TRIM8, demonstrating that bidirectional TRIM8/STAT3 signaling is
involved in the regulation of the stemness of GSCs (104).
Tetraspanin CD9, a regulator of cell adhesion,
stabilizes the IL-6 receptor glycoprotein 130 (gp130) by preventing
its ubiquitin-dependent lysosomal degradation, thus promoting bone
marrow tyrosine kinase gene on chromosome X/STAT3 signaling in
GSCs. Disrupting CD9 or gp130 can inhibit the self-renewal of GSCs
and promote their differentiation (105). Currently, there are various
ongoing clinical trials in USA investigating the targeting of STAT3
with the small molecule inhibitor WP1066 (Table III).
ID1 is highly expressed in GSCs and is involved in
the TGF-β, Wnt and SHH signaling pathways. ID proteins are
transcriptional regulators that are implicated in cell fate
determination and differentiation of stem-like cells (106). Ubiquitination-specific proteases
and cyclooxygenase-2-derived prostaglandin E2 have been reported to
positively regulate the stability of ID1, and to promote GSC
maintenance and treatment resistance (107,108). ID1 induces cell proliferation
and promotes self-renewal through increasing cyclin E, the target
molecule of cullin 3. Cullin 3 interacts with Gli2 and dishevelled
segment polarity protein 2, and induces their degradation through
ubiquitination. Loss of cullin 3 is the common signaling node in
the Wnt and SHH signaling pathways through ID1 (109).
ID1 was previously found to inhibit BMP-mediated GSC
differentiation through BMPRII and to maintain GSC traits (110). BMPs bind to a cognate
high-affinity type II receptor (BMPRII) to phosphorylate the type I
receptor (BMPRI). Activated BMPRI initiates downstream signaling by
phosphorylating R-SMAD. ID1 could decrease BMPRII expression and
the phosphorylation of its downstream signaling molecules SMAD1,
SMAD5 and SMAD8 in cells (15,16). These results indicate that
targeting ID1-driven intrinsic stemness signaling may be an
effective therapeutic strategy for GBM.
GBM is a primary brain tumor with a high mortality
rate, for which there is currently no effective therapy. Previous
studies have found that GSCs promote the heterogeneity and
treatment resistance of GBM. The main characteristics of GSCs are
their capacity for self-renewal and differentiation. Therefore,
elucidating the mechanism through which GSCs regulate the
self-renewal response is meaningful in order to design therapeutic
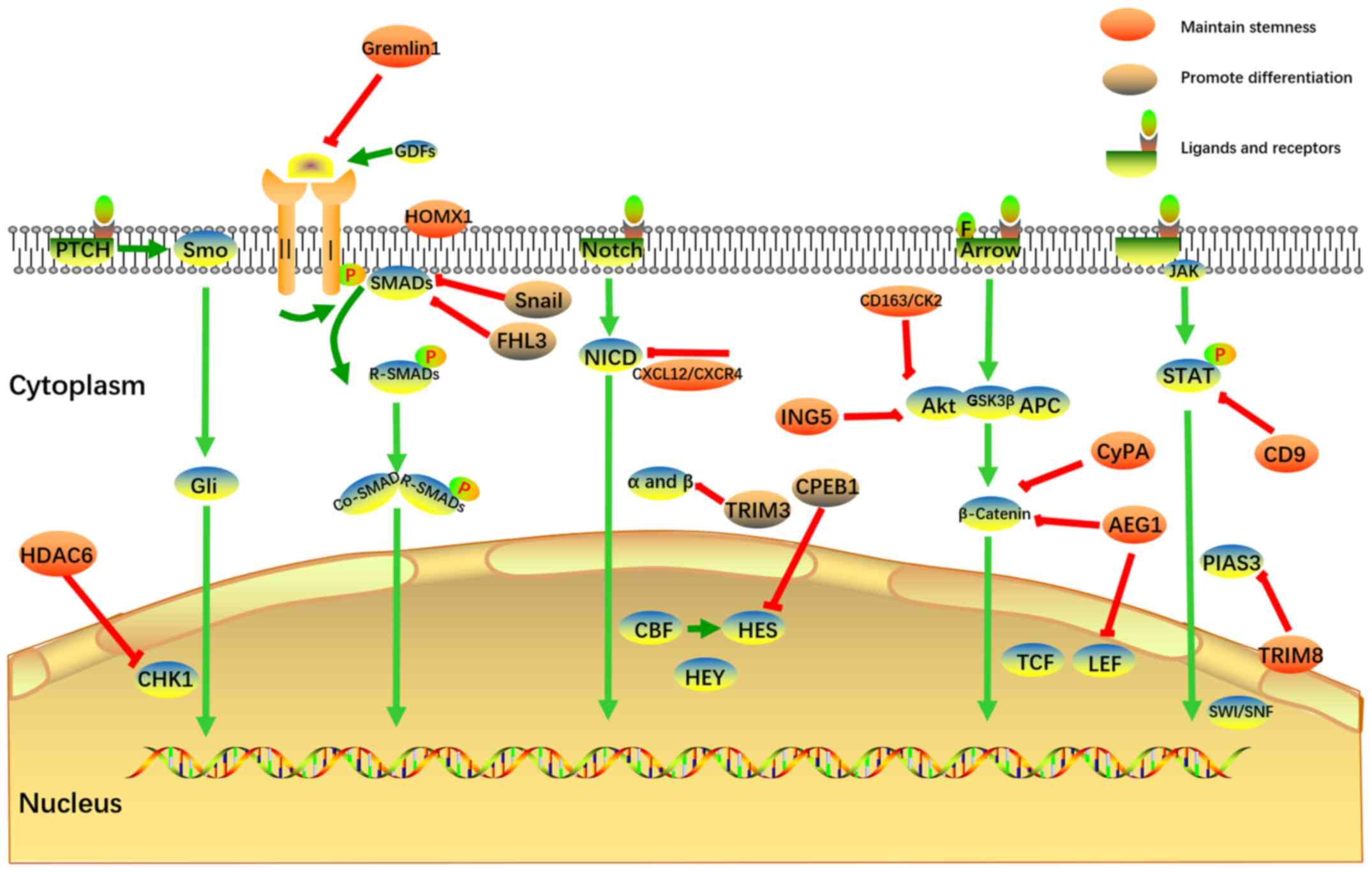
approaches targeting the self-renewal signaling pathways. The focus
of the present review was five key self-renewal GSC signaling
pathways, including TGF-β, Notch, Wnt, HH and STAT3, and the
corresponding therapeutic targets, and the aim was to provide novel
insight to enable advances in clinical therapy. Among these
signaling pathways, gremlin 1, HMOX1, CXCL12/CXCR4, AEG1,
CD163/CK2, ING5, CypA, HDAC6, CD9 and TRIM8 can increase stemness,
promote self-renewal and prevent differentiation of GSCs.
Therefore, their corresponding inhibitors may represent a novel
type of therapeutic approach to glioma. On the other hand, Snail,
FHL3, TRIM3 and CPEB1 may promote differentiation of GSCs. Thus,
their corresponding agonists should be further investigated in this
context. The aforementioned self-renewal pathways and corresponding
differentiation-targeting treatments of GSCs are summarized in
Fig. 1. Considering its
complexity, the crosstalk between these pathways is not shown in
Fig. 1. The current clinical
trials targeting the TGF-β, Notch and STAT3 pathways are summarized
in Tables I-III.
Based on the aforementioned findings, there are
numerous potential treatments that are currently being explored.
There are also emerging signaling pathways under investigation that
may uncover potential treatment targets for GBM. In GSCs, lysine
demethylase (KDM)1A (111), the
transcription factors forkhead box G1 and transducin-like enhancer
of split 1 (40,112), hypoxia-inducible factors
(113), proliferating cell
nuclear antigen (PCNA)-associated factor (PAF) (114), MEK partner-1 (MP1) (115), erythropoietin-producing
hepatocellular receptors (116),
progranulin (PGRN) (117) and
DNA polymerase delta subunit 2 (POLD2) (118) are all overexpressed, and
maintain GSC self-renewal capacity and stemness. In addition to the
aforementioned classical pathways, the molecular mechanisms through
which these factors maintain stemness require deeper and more
comprehensive investigation. For example, PAF promotes the
maintenance of self-renewal ability and stemness by interacting
with PCNA, and regulates PCNA-associated DNA translesion synthesis
(114), while MP1 contributes to
GSC stemness by driving ERK activity (115).
Other factors play a unique role in the damage and
repair of DNA, such as POLD2 and PGRN (117,118). Previous studies have
demonstrated that PGRN promotes DNA repair through activator
protein 1 transcription factor, cFos and JunB (117). In terms of their relevance to
treatment, the knockdown of these molecules can reduce GSC stemness
and induce their differentiation. Based on the identification of
these factors that maintain stemness, corresponding inhibitors may
be developed to target GSCs. For example, a series of inhibitors
have already been developed and evaluated. Two novel KDM1A-specific
inhibitors (NCL-1 and NCD-38) were found to significantly reduce
GSCs-driven tumor progression by inducing the activation of the
unfolded protein response pathway (111). GLPG1790, a small-molecule ephrin
receptor inhibitor, completely blocks ephrin type-A receptor 2
signaling and exerts antitumor effects (116). Similarly, GSC gap junctions also
have pro-tumorigenic effects depending on connexin expression
(119). However, to the best of
our knowledge, the detailed mechanisms remain elusive and further
research is needed in this field.
In addition to the aforementioned factors that
maintain stemness, other factors promote differentiation, and
regulating their activity may be of value in the context of
differentiation therapy. For example, MAPK phosphatase 1 (MKP1), a
dual-specificity phosphatase, acts as a negative inhibitor of JNK,
ERK1/2 and p38 MAPK. High levels of MKP1 expression impair
self-renewal and induce differentiation in GSCs (120). The let-7 miRNA family has also
been shown to induce GSC differentiation. The mechanism is as
follows: Its recognition elements may be bound by insulin-like
growth factor 2 mRNA-binding protein 2, which prevents let-7 target
gene silencing and impairs the maintenance of GSC stemness
(121).
In summary, inhibitors of the factors found to
maintain stemness may be developed in the future to provide
possible differentiation therapies. For the factors that can
promote differentiation, increasing their expression levels is an
important method for targeting GSCs. It is expected that more
clinically feasible differentiation treatments will be developed in
the future in order to improve GBM treatment efficacy and
prognosis.
Not applicable.
JJ, FG and ML performed the literature search. JJ,
FG, CCC and ML wrote the manuscript. All the authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the National Natural Sciences
Foundation of China (grant nos. 82072802 and 81572480).
|
1
|
Prager BC, Bhargava S, Mahadev V, Hubert
CG and Rich JN: Glioblastoma Stem Cells: Driving Resilience through
Chaos. Trends Cancer. 6:223–235. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weller M, Cloughesy T, Perry JR and Wick
W: Standards of care for treatment of recurrent glioblastoma - are
we there yet? Neuro Oncol. 15:4–27. 2013. View Article : Google Scholar
|
|
3
|
Jordan CT: Cancer stem cells:
Controversial or just misunderstood? Cell Stem Cell. 4:203–205.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choi SA, Lee JY, Phi JH, Wang KC, Park CK,
Park SH and Kim SK: Identification of brain tumour initiating cells
using the stem cell marker aldehyde dehydrogenase. Eur J Cancer.
50:137–149. 2014. View Article : Google Scholar
|
|
6
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galli R, Binda E, Orfanelli U, Cipelletti
B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F and Vescovi
A: Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Z, Wang H, Eyler CE, Hjelmeland AB and
Rich JN: Turning cancer stem cells inside out: An exploration of
glioma stem cell signaling pathways. J Biol Chem. 284:16705–16709.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng L, Wu Q, Guryanova OA, Huang Z,
Huang Q, Rich JN and Bao S: Elevated invasive potential of
glioblastoma stem cells. Biochem Biophys Res Commun. 406:643–648.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Folkins C, Shaked Y, Man S, Tang T, Lee
CR, Zhu Z, Hoffman RM and Kerbel RS: Glioma tumor stem-like cells
promote tumor angiogenesis and vasculogenesis via vascular
endothelial growth factor and stromal-derived factor 1. Cancer Res.
69:7243–7251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Z, Bandyopadhyay A, Nichols RW, Wang
L, Hinck AP, Wang S and Sun LZ: Blockade of Autocrine TGF-β
signaling inhibits stem cell phenotype, survival, and metastasis of
murine breast cancer cells. J Stem Cell Res Ther. 2:1–8. 2012.
View Article : Google Scholar
|
|
13
|
Xi Q, Wang Z, Zaromytidou AI, Zhang XH,
Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K,
Kaartinen V, et al: A poised chromatin platform for TGF-β access to
master regulators. Cell. 147:1511–1524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Massagué J: How cells read TGF-beta
signals. Nat Rev Mol Cell Biol. 1:169–178. 2000. View Article : Google Scholar
|
|
16
|
Oshimori N and Fuchs E: The harmonies
played by TGF-β in stem cell biology. Cell Stem Cell. 11:751–764.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang Y, Zhu F, Zhang H, Chen D, Zhang X,
Gao Q and Li Y: Conditional ablation of TGF-β signaling inhibits
tumor progression and invasion in an induced mouse bladder cancer
model. Sci Rep. 6:294792016. View Article : Google Scholar
|
|
18
|
Furuta Y, Piston DW and Hogan BL: Bone
morphogenetic proteins (BMPs) as regulators of dorsal forebrain
development. Development. 124:2203–2212. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gilboa L, Nohe A, Geissendörfer T, Sebald
W, Henis YI and Knaus P: Bone morphogenetic protein receptor
complexes on the surface of live cells: A new oligomerization mode
for serine/threonine kinase receptors. Mol Biol Cell. 11:1023–1035.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee SJ: Identification of a novel member
(GDF-1) of the transforming growth factor-beta superfamily. Mol
Endocrinol. 4:1034–1040. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McPherron AC and Lee SJ: GDF-3 and GDF-9:
Two new members of the transforming growth factor-beta superfamily
containing a novel pattern of cysteines. J Biol Chem.
268:3444–3449. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rider CC and Mulloy B: Bone morphogenetic
protein and growth differentiation factor cytokine families and
their protein antagonists. Biochem J. 429:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chirasani SR, Sternjak A, Wend P, Momma S,
Campos B, Herrmann IM, Graf D, Mitsiadis T, Herold-Mende C, Besser
D, et al: Bone morphogenetic protein-7 release from endogenous
neural precursor cells suppresses the tumourigenicity of stem-like
glioblastoma cells. Brain. 133:1961–1972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Piccirillo SG, Reynolds BA, Zanetti N,
Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F and Vescovi
AL: Bone morphogenetic proteins inhibit the tumorigenic potential
of human brain tumour-initiating cells. Nature. 444:761–765. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raja E, Komuro A, Tanabe R, Sakai S, Ino
Y, Saito N, Todo T, Morikawa M, Aburatani H, Koinuma D, et al: Bone
morphogenetic protein signaling mediated by ALK-2 and DLX2
regulates apoptosis in glioma-initiating cells. Oncogene.
36:4963–4974. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tso JL, Yang S, Menjivar JC, Yamada K,
Zhang Y, Hong I, Bui Y, Stream A, McBride WH, Liau LM, et al: Bone
morphogenetic protein 7 sensitizes O6-methylguanine
methyltransferase expressing-glioblastoma stem cells to clinically
relevant dose of temozolomide. Mol Cancer. 14:1892015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee J, Son MJ, Woolard K, Donin NM, Li A,
Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al:
Epigenetic-mediated dysfunction of the bone morphogenetic protein
pathway inhibits differentiation of glioblastoma-initiating cells.
Cancer Cell. 13:69–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Namkoong H, Shin SM, Kim HK, Ha SA, Cho
GW, Hur SY, Kim TE and Kim JW: The bone morphogenetic protein
antagonist gremlin 1 is overexpressed in human cancers and
interacts with YWHAH protein. BMC Cancer. 6:742006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan K, Wu Q, Yan DH, Lee CH, Rahim N,
Tritschler I, DeVecchio J, Kalady MF, Hjelmeland AB and Rich JN:
Glioma cancer stem cells secrete Gremlin1 to promote their
maintenance within the tumor hierarchy. Genes Dev. 28:1085–1100.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tate CM, Pallini R, Ricci-Vitiani L,
Dowless M, Shiyanova T, D'Alessandris GQ, Morgante L, Giannetti S,
Larocca LM, di Martino S, et al: A BMP7 variant inhibits the
tumorigenic potential of glioblastoma stem-like cells. Cell Death
Differ. 19:1644–1654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bruna A, Darken RS, Rojo F, Ocaña A,
Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, et al:
High TGFbeta-Smad activity confers poor prognosis in glioma
patients and promotes cell proliferation depending on the
methylation of the PDGF-B gene. Cancer Cell. 11:147–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peñuelas S, Anido J, Prieto-Sánchez RM,
Folch G, Barba I, Cuartas I, García-Dorado D, Poca MA, Sahuquillo
J, Baselga J, et al: TGF-beta increases glioma-initiating cell
self-renewal through the induction of LIF in human glioblastoma.
Cancer Cell. 15:315–327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Savary K, Caglayan D, Caja L, Tzavlaki K,
Bin Nayeem S, Bergström T, Jiang Y, Uhrbom L, Forsberg-Nilsson K,
Westermark B, et al: Snail depletes the tumorigenic potential of
glioblastoma. Oncogene. 32:5409–5420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caja L, Tzavlaki K, Dadras MS, Tan EJ,
Hatem G, Maturi NP, Morén A, Wik L, Watanabe Y, Savary K, et al:
Snail regulates BMP and TGFβ pathways to control the
differentiation status of glioma-initiating cells. Oncogene.
37:2515–2531. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Teh JL and Chen S: Glutamatergic signaling
in cellular transformation. Pigment Cell Melanoma Res. 25:331–342.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ghosh D, Ulasov IV, Chen L, Harkins LE,
Wallenborg K, Hothi P, Rostad S, Hood L and Cobbs CS:
TGFβ-responsive HMOX1 expression is associated with stemness and
invasion in glioblastoma multiforme. Stem Cells. 34:2276–2289.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han W, Xin Z, Zhao Z, Bao W, Lin X, Yin B,
Zhao J, Yuan J, Qiang B and Peng X: RNA-binding protein PCBP2
modulates glioma growth by regulating FHL3. J Clin Invest.
123:2103–2118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han W, Hu P, Wu F, Wang S, Hu Y, Li S,
Jiang T, Qiang B and Peng X: FHL3 links cell growth and
self-renewal by modulating SOX4 in glioma. Cell Death Differ.
26:796–811. 2019. View Article : Google Scholar :
|
|
40
|
Bulstrode H, Johnstone E, Marques-Torrejon
MA, Ferguson KM, Bressan RB, Blin C, Grant V, Gogolok S, Gangoso E,
Gagrica S, et al: Elevated FOXG1 and SOX2 in glioblastoma enforces
neural stem cell identity through transcriptional control of cell
cycle and epigenetic regulators. Genes Dev. 31:757–773. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Blaumueller CM, Qi H, Zagouras P and
Artavanis-Tsakonas S: Intracellular cleavage of Notch leads to a
heterodimeric receptor on the plasma membrane. Cell. 90:281–291.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stockhausen MT, Kristoffersen K and
Poulsen HS: The functional role of Notch signaling in human
gliomas. Neuro Oncol. 12:199–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Androutsellis-Theotokis A, Leker RR,
Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK,
Kittappa R and McKay RD: Notch signalling regulates stem cell
numbers in vitro and in vivo. Nature. 442:823–826. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guichet PO, Guelfi S, Teigell M, Hoppe L,
Bakalara N, Bauchet L, Duffau H, Lamszus K, Rothhut B and Hugnot
JP: Notch1 stimulation induces a vascularization switch with
pericyte-like cell differentiation of glioblastoma stem cells. Stem
Cells. 33:21–34. 2015. View Article : Google Scholar
|
|
46
|
Kanamori M, Kawaguchi T, Nigro JM,
Feuerstein BG, Berger MS, Miele L and Pieper RO: Contribution of
Notch signaling activation to human glioblastoma multiforme. J
Neurosurg. 106:417–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chowdhury S and Sarkar RR: Exploring Notch
pathway to elucidate phenotypic plasticity and intra-tumor
heterogeneity in fliomas. Sci Rep. 9:94882019. View Article : Google Scholar
|
|
48
|
Basak O, Giachino C, Fiorini E, Macdonald
HR and Taylor V: Neurogenic subventricular zone stem/progenitor
cells are Notch1-dependent in their active but not quiescent state.
J Neurosci. 32:5654–5666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ge W, Martinowich K, Wu X, He F, Miyamoto
A, Fan G, Weinmaster G and Sun YE: Notch signaling promotes
astrogliogenesis via direct CSL-mediated glial gene activation. J
Neurosci Res. 69:848–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bansod S, Kageyama R and Ohtsuka T: Hes5
regulates the transition timing of neurogenesis and gliogenesis in
mammalian neocortical development. Development. 144:3156–3167.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Armesilla-Diaz A, Bragado P, Del Valle I,
Cuevas E, Lazaro I, Martin C, Cigudosa JC and Silva A: p53
regulates the self-renewal and differentiation of neural
precursors. Neuroscience. 158:1378–1389. 2009. View Article : Google Scholar
|
|
53
|
Fan X, Khaki L, Zhu TS, Soules ME, Talsma
CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al: NOTCH pathway
blockade depletes CD133-positive glioblastoma cells and inhibits
growth of tumor neurospheres and xenografts. Stem Cells. 28:5–16.
2010.
|
|
54
|
Xu R, Shimizu F, Hovinga K, Beal K, Karimi
S, Droms L, Peck KK, Gutin P, Iorgulescu JB, Kaley T, et al:
Molecular and clinical effects of Notch inhibition in glioma
patients: A phase 0/I trial. Clin Cancer Res. 22:4786–4796. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tanaka S, Nakada M, Yamada D, Nakano I,
Todo T, Ino Y, Hoshii T, Tadokoro Y, Ohta K, Ali MA, et al: Strong
therapeutic potential of γ-secretase inhibitor MRK003 for CD44-high
and CD133-low glioblastoma initiating cells. J Neurooncol.
121:239–250. 2015. View Article : Google Scholar
|
|
56
|
Goldfarb DS, Corbett AH, Mason DA,
Harreman MT and Adam SA: Importin alpha: A multipurpose
nuclear-transport receptor. Trends Cell Biol. 14:505–514. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Huenniger K, Krämer A, Soom M, Chang I,
Köhler M, Depping R, Kehlenbach RH and Kaether C: Notch1 signaling
is mediated by importins alpha 3, 4, and 7. Cell Mol Life Sci.
67:3187–3196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen G, Kong J, Tucker-Burden C, Anand M,
Rong Y, Rahman F, Moreno CS, Van Meir EG, Hadjipanayis CG and Brat
DJ: Human Brat ortholog TRIM3 is a tumor suppressor that regulates
asymmetric cell division in glioblastoma. Cancer Res. 74:4536–4548.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mukherjee S, Tucker-Burden C, Zhang C,
Moberg K, Read R, Hadjipanayis C and Brat DJ: Drosophila brat and
human ortholog TRIM3 maintain stem cell equilibrium and suppress
brain tumorigenesis by attenuating Notch nuclear transport. Cancer
Res. 76:2443–2452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gagliardi F, Narayanan A, Reni M, Franzin
A, Mazza E, Boari N, Bailo M, Zordan P and Mortini P: The role of
CXCR4 in highly malignant human gliomas biology: Current knowledge
and future directions. Glia. 62:1015–1023. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Calinescu AA, Yadav VN, Carballo E,
Kadiyala P, Tran D, Zamler DB, Doherty R, Srikanth M, Lowenstein PR
and Castro MG: Survival and proliferation of neural
progenitor-derived glioblastomas under hypoxic stress is controlled
by a CXCL12/CXCR4 autocrine-positive feedback mechanism. Clin
Cancer Res. 23:1250–1262. 2017. View Article : Google Scholar
|
|
62
|
Yi L, Zhou X, Li T, Liu P, Hai L, Tong L,
Ma H, Tao Z, Xie Y, Zhang C, et al: Notch1 signaling pathway
promotes invasion, self-renewal and growth of glioma initiating
cells via modulating chemokine system CXCL12/CXCR4. J Exp Clin
Cancer Res. 38:3392019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Iso T, Kedes L and Hamamori Y: HES and
HERP families: Multiple effectors of the Notch signaling pathway. J
Cell Physiol. 194:237–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tay J and Richter JD: Germ cell
differentiation and synaptonemal complex formation are disrupted in
CPEB knockout mice. Dev Cell. 1:201–213. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yin J, Park G, Lee JE, Park JY, Kim TH,
Kim YJ, Lee SH, Yoo H, Kim JH and Park JB: CPEB1 modulates
differentiation of glioma stem cells via downregulation of HES1 and
SIRT1 expression. Oncotarget. 5:6756–6769. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Iso T, Sartorelli V, Poizat C, Iezzi S, Wu
HY, Chung G, Kedes L and Hamamori Y: HERP, a novel heterodimer
partner of HES/E(spl) in Notch signaling. Mol Cell Biol.
21:6080–6089. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Iso T, Sartorelli V, Chung G, Shichinohe
T, Kedes L and Hamamori Y: HERP, a new primary target of Notch
regulated by ligand binding. Mol Cell Biol. 21:6071–6079. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sokol SY: Maintaining embryonic stem cell
pluripotency with Wnt signaling. Development. 138:4341–4350. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bhanot P, Brink M, Samos CH, Hsieh JC,
Wang Y, Macke JP, Andrew D, Nathans J and Nusse R: A new member of
the frizzled family from Drosophila functions as a Wingless
receptor. Nature. 382:225–230. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wehrli M, Dougan ST, Caldwell K, O'Keefe
L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A and DiNardo
S: arrow encodes an LDL-receptor-related protein essential for
Wingless signalling. Nature. 407:527–530. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Korinek V, Barker N, Morin PJ, van Wichen
D, de Weger R, Kinzler KW, Vogelstein B and Clevers H: Constitutive
transcriptional activation by a beta-catenin-Tcf complex in
APC−/− colon carcinoma. Science. 275:1784–1787. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hamada F and Bienz M: The APC tumor
suppressor binds to C-terminal binding protein to divert nuclear
beta-catenin from TCF. Dev Cell. 7:677–685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Molenaar M, van de Wetering M, Oosterwegel
M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O and
Clevers H: XTcf-3 transcription factor mediates
beta-catenin-induced axis formation in Xenopus embryos. Cell.
86:391–399. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Behrens J, von Kries JP, Kühl M, Bruhn L,
Wedlich D, Grosschedl R and Birchmeier W: Functional interaction of
beta-catenin with the transcription factor LEF-1. Nature.
382:638–642. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huber O, Korn R, McLaughlin J, Ohsugi M,
Herrmann BG and Kemler R: Nuclear localization of beta-catenin by
interaction with transcription factor LEF-1. Mech Dev. 59:3–10.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Takahashi-Yanaga F and Kahn M: Targeting
Wnt signaling: Can we safely eradicate cancer stem cells? Clin
Cancer Res. 16:3153–3162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rajakulendran N, Rowland KJ, Selvadurai
HJ, Ahmadi M, Park NI, Naumenko S, Dolma S, Ward RJ, So M, Lee L,
et al: Wnt and Notch signaling govern self-renewal and
differentiation in a subset of human glioblastoma stem cells. Genes
Dev. 33:498–510. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sonoda Y, Ozawa T, Aldape KD, Deen DF,
Berger MS and Pieper RO: Akt pathway activation converts anaplastic
astrocytoma to glioblastoma multiforme in a human astrocyte model
of glioma. Cancer Res. 61:6674–6678. 2001.PubMed/NCBI
|
|
79
|
Morgan RG, Ridsdale J, Payne M, Heesom KJ,
Wilson MC, Davidson A, Greenhough A, Davies S, Williams AC, Blair
A, et al: LEF-1 drives aberrant β-catenin nuclear localization in
myeloid leukemia cells. Haematologica. 104:1365–1377. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen J, Liu G, Wu Y, Ma J, Wu H, Xie Z,
Chen S, Yang Y, Wang S, Shen P, et al: CircMYO10 promotes
osteosarcoma progression by regulating miR-370-3p/RUVBL1 axis to
enhance the transcriptional activity of β-catenin/LEF1 complex via
effects on chromatin remodeling. Mol Cancer. 18:1502019. View Article : Google Scholar
|
|
81
|
Brown DM and Ruoslahti E: Metadherin, a
cell surface protein in breast tumors that mediates lung
metastasis. Cancer Cell. 5:365–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hu B, Emdad L, Bacolod MD, Kegelman TP,
Shen XN, Alzubi MA, Das SK, Sarkar D and Fisher PB: Astrocyte
elevated gene-1 interacts with Akt isoform 2 to control glioma
growth, survival, and pathogenesis. Cancer Res. 74:7321–7332. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hu B, Emdad L, Kegelman TP, Shen XN, Das
SK, Sarkar D and Fisher PB: Astrocyte elevated Gene-1 regulates
β-catenin signaling to maintain glioma stem-like stemness and
self-renewal. Mol Cancer Res. 15:225–233. 2017. View Article : Google Scholar
|
|
84
|
Kristiansen M, Graversen JH, Jacobsen C,
Sonne O, Hoffman HJ, Law SK and Moestrup SK: Identification of the
haemoglobin scavenger receptor. Nature. 409:198–201. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ostuni R, Kratochvill F, Murray PJ and
Natoli G: Macrophages and cancer: From mechanisms to therapeutic
implications. Trends Immunol. 36:229–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Shi Y, Ping YF, Zhou W, He ZC, Chen C,
Bian BS, Zhang L, Chen L, Lan X, Zhang XC, et al: Tumour-associated
macro-phages secrete pleiotrophin to promote PTPRZ1 signalling in
glioblastoma stem cells for tumour growth. Nat Commun. 8:150802017.
View Article : Google Scholar
|
|
87
|
Ritter M, Buechler C, Kapinsky M and
Schmitz G: Interaction of CD163 with the regulatory subunit of
casein kinase II (CKII) and dependence of CD163 signaling on CKII
and protein kinase C. Eur J Immunol. 31:999–1009. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Di Maira G, Salvi M, Arrigoni G, Marin O,
Sarno S, Brustolon F, Pinna LA and Ruzzene M: Protein kinase CK2
phosphorylates and upregulates Akt/PKB. Cell Death Differ.
12:668–677. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen T, Chen J, Zhu Y, Li Y, Wang Y, Chen
H, Wang J, Li X, Liu Y, Li B, et al: CD163, a novel therapeutic
target, regulates the proliferation and stemness of glioma cells
via casein kinase 2. Oncogene. 38:1183–1199. 2019. View Article : Google Scholar
|
|
90
|
Wang F, Wang AY, Chesnelong C, Yang Y,
Nabbi A, Thalappilly S, Alekseev V and Riabowol K: ING5 activity in
self-renewal of glioblastoma stem cells via calcium and follicle
stimulating hormone pathways. Oncogene. 37:286–301. 2018.
View Article : Google Scholar :
|
|
91
|
Zhu Q, Shen Y, Chen X, He J, Liu J and Zu
X: Self-renewal signalling pathway inhibitors: Perspectives on
therapeutic approaches for cancer stem cells. OncoTargets Ther.
13:525–540. 2020. View Article : Google Scholar
|
|
92
|
Handschumacher RE, Harding MW, Rice J,
Drugge RJ and Speicher DW: Cyclophilin: A specific cytosolic
binding protein for cyclosporin A. Science. 226:544–547. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Sun S, Wang Q, Giang A, Cheng C, Soo C,
Wang C, Liu L and Chiu R: Knockdown of CypA inhibits interleukin-8
(IL-8) and IL-8-mediated proliferation and tumor growth of
glioblastoma cells through down-regulated NF-κB. J Neurooncol.
101:1–14. 2011. View Article : Google Scholar
|
|
94
|
Wang G, Shen J, Sun J, Jiang Z, Fan J,
Wang H, Yu S, Long Y, Liu Y, Bao H, et al: Cyclophilin A maintains
glioma-initiating cell stemness by regulating Wnt/β-catenin
signaling. Clin Cancer Res. 23:6640–6649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Machold R, Hayashi S, Rutlin M, Muzumdar
MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA,
Rubin LL, et al: Sonic hedgehog is required for progenitor cell
maintenance in telencephalic stem cell niches. Neuron. 39:937–950.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Micchelli CA, The I, Selva E, Mogila V and
Perrimon N: Rasp, a putative transmembrane acyltransferase, is
required for Hedgehog signaling. Development. 129:843–851. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Takebe N, Harris PJ, Warren RQ and Ivy SP:
Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog
pathways. Nat Rev Clin Oncol. 8:97–106. 2011. View Article : Google Scholar
|
|
98
|
Ruiz i Altaba A and Altaba A:
Combinatorial Gli gene function in floor plate and neuronal
inductions by Sonic hedgehog. Development. 125:2203–2212. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Baylin SB and Jones PA: Epigenetic
determinants of cancer. Cold Spring Harb Perspect Biol.
8:a0195052016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Marampon F, Megiorni F, Camero S,
Crescioli C, McDowell HP, Sferra R, Vetuschi A, Pompili S, Ventura
L, De Felice F, et al: HDAC4 and HDAC6 sustain DNA double strand
break repair and stem-like phenotype by promoting radioresistance
in glioblastoma cells. Cancer Lett. 397:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yang W, Liu Y, Gao R, Yu H and Sun T:
HDAC6 inhibition induces glioma stem cells differentiation and
enhances cellular radiation sensitivity through the SHH/Gli1
signaling pathway. Cancer Lett. 415:164–176. 2018. View Article : Google Scholar
|
|
102
|
Auzmendi-Iriarte J, Saenz-Antoñanzas A,
Mikelez-Alonso I, Carrasco-Garcia E, Tellaetxe-Abete M, Lawrie CH,
Sampron N, Cortajarena AL and Matheu A: Characterization of a new
small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis.
11:4172020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Dolatabadi S, Jonasson E, Lindén M,
Fereydouni B, Bäcksten K, Nilsson M, Martner A, Forootan A, Fagman
H, Landberg G, et al: JAK-STAT signalling controls cancer stem cell
properties including chemotherapy resistance in myxoid liposarcoma.
Int J Cancer. 145:435–449. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang C, Mukherjee S, Tucker-Burden C,
Ross JL, Chau MJ, Kong J and Brat DJ: TRIM8 regulates stemness in
glioblastoma through PIAS3-STAT3. Mol Oncol. 11:280–294. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Shi Y, Zhou W, Cheng L, Chen C, Huang Z,
Fang X, Wu Q, He Z, Xu S, Lathia JD, et al: Tetraspanin CD9
stabilizes gp130 by preventing its ubiquitin-dependent lysosomal
degradation to promote STAT3 activation in glioma stem cells. Cell
Death Differ. 24:167–180. 2017. View Article : Google Scholar :
|
|
106
|
Lasorella A, Benezra R and Iavarone A: The
ID proteins: Master regulators of cancer stem cells and tumour
aggressiveness. Nat Rev Cancer. 14:77–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lee JK, Chang N, Yoon Y, Yang H, Cho H,
Kim E, Shin Y, Kang W, Oh YT, Mun GI, et al: USP1 targeting impedes
GBM growth by inhibiting stem cell maintenance and radioresistance.
Neuro Oncol. 18:37–47. 2016. View Article : Google Scholar
|
|
108
|
Cook PJ, Thomas R, Kingsley PJ, Shimizu F,
Montrose DC, Marnett LJ, Tabar VS, Dannenberg AJ and Benezra R:
Cox-2-derived PGE2 induces Id1-dependent radiation resistance and
self-renewal in experimental glioblastoma. Neuro Oncol.
18:1379–1389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jin X, Jeon HM, Jin X, Kim EJ, Yin J, Jeon
HY, Sohn YW, Oh SY, Kim JK, Kim SH, et al: The ID1-CULLIN3 axis
regulates intracellular SHH and WNT signaling in glioblastoma stem
cells. Cell Rep. 16:1629–1641. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Jin X, Jin X, Kim LJY, Dixit D, Jeon HY,
Kim EJ, Kim JK, Lee SY, Yin J, Rich JN, et al: Inhibition of
ID1-BMPR2 Intrinsic Signaling Sensitizes Glioma Stem Cells to
Differentiation Therapy. Clin Cancer Res. 24:383–394. 2018.
View Article : Google Scholar
|
|
111
|
Sareddy GR, Viswanadhapalli S, Surapaneni
P, Suzuki T, Brenner A and Vadlamudi RK: Novel KDM1A inhibitors
induce differentiation and apoptosis of glioma stem cells via
unfolded protein response pathway. Oncogene. 36:2423–2434. 2017.
View Article : Google Scholar :
|
|
112
|
Dali R, Verginelli F, Pramatarova A,
Sladek R and Stifani S: Characterization of a FOXG1:TLE1
transcriptional network in glioblastoma-initiating cells. Mol
Oncol. 12:775–787. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Semenza GL: Dynamic regulation of stem
cell specification and maintenance by hypoxia-inducible factors.
Mol Aspects Med. 47-48:15–23. 2016. View Article : Google Scholar
|
|
114
|
Ong DST, Hu B, Ho YW, Sauvé CG, Bristow
CA, Wang Q, Multani AS, Chen P, Nezi L, Jiang S, et al: PAF
promotes stemness and radioresistance of glioma stem cells. Proc
Natl Acad Sci USA. 114:E9086–E9095. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kwon SJ, Kwon OS, Kim KT, Go YH, Yu SI,
Lee BH, Miyoshi H, Oh E, Cho SJ and Cha HJ: Role of MEK partner-1
in cancer stemness through MEK/ERK pathway in cancerous neural stem
cells, expressing EGFRviii. Mol Cancer. 16:1402017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Gravina GL, Mancini A, Colapietro A, Delle
Monache S, Sferra R, Vitale F, Cristiano L, Martellucci S, Marampon
F, Mattei V, et al: The small molecule Ephrin receptor inhibitor,
glpg1790, reduces renewal capabilities of cancer stem cells,
showing anti-tumour efficacy on preclinical glioblastoma models.
Cancers (Basel). 11:3592019. View Article : Google Scholar
|
|
117
|
Bandey I, Chiou SH, Huang AP, Tsai JC and
Tu PH: Progranulin promotes Temozolomide resistance of glioblastoma
by orchestrating DNA repair and tumor stemness. Oncogene.
34:1853–1864. 2015. View Article : Google Scholar
|
|
118
|
Xu Q, Hu C, Zhu Y, Wang K, Lal B, Li L,
Tang J, Wei S, Huang G, Xia S, et al: ShRNA-based POLD2 expression
knockdown sensitizes glioblastoma to DNA-Damaging therapeutics.
Cancer Lett. 482:126–135. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hitomi M, Deleyrolle LP, Mulkearns-Hubert
EE, Jarrar A, Li M, Sinyuk M, Otvos B, Brunet S, Flavahan WA,
Hubert CG, et al: Differential connexin function enhances
self-renewal in glioblastoma. Cell Rep. 11:1031–1042. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Arrizabalaga O, Moreno-Cugnon L,
Auzmendi-Iriarte J, Aldaz P, Ibanez de Caceres I, Garros-Regulez L,
Moncho-Amor V, Torres-Bayona S, Pernía O, Pintado-Berninches L, et
al: High expression of MKP1/DUSP1 counteracts glioma stem cell
activity and mediates HDAC inhibitor response. Oncogenesis.
6:4012017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Degrauwe N, Schlumpf TB, Janiszewska M,
Martin P, Cauderay A, Provero P, Riggi N, Suvà ML, Paro R and
Stamenkovic I: The RNA binding protein IMP2 preserves glioblastoma
stem cells by preventing let-7 target gene silencing. Cell Rep.
15:1634–1647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Iwamaru A, Szymanski S, Iwado E, Aoki H,
Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, et al: A
novel inhibitor of the STAT3 pathway induces apoptosis in malignant
glioma cells both in vitro and in vivo. Oncogene. 26:2435–2444.
2007. View Article : Google Scholar
|
|
123
|
Ott M, Kassab C, Marisetty A, Hashimoto Y,
Wei J, Zamler D, Leu JS, Tomaszowski KH, Sabbagh A, Fang D, et al:
Radiation with STAT3 blockade triggers dendritic cell-T cell
interactions in the glioma microenvironment and therapeutic
efficacy. Clin Cancer Res. 26:4983–4994. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lim D, Kim KS, Kim H, Ko KC, Song JJ, Choi
JH, Shin M, Min JJ, Jeong JH and Choy HE: Anti-tumor activity of an
immunotoxin (TGFα-PE38) delivered by attenuated Salmonella
typhimurium. Oncotarget. 8:37550–37560. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sampson JH, Akabani G, Archer GE, Berger
MS, Coleman RE, Friedman AH, Friedman HS, Greer K, Herndon JE II,
Kunwar S, et al: Intracerebral infusion of an EGFR-targeted toxin
in recurrent malignant brain tumors. Neuro Oncol. 10:320–329. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hau P, Jachimczak P, Schlingensiepen R,
Schulmeyer F, Jauch T, Steinbrecher A, Brawanski A, Proescholdt M,
Schlaier J, Buchroithner J, et al: Inhibition of TGF-beta2 with AP
12009 in recurrent malignant gliomas: from preclinical to phase
I/II studies. Oligonucleotides. 17:201–12. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Rodon J, Carducci MA, Sepulveda-Sánchez
JM, Azaro A, Calvo E, Seoane J, Braña I, Sicart E, Gueorguieva I,
Cleverly AL, et al: First-in-human dose study of the novel
transforming growth factor-β receptor I kinase inhibitor LY2157299
monohydrate in patients with advanced cancer and glioma. Clin
Cancer Res. 21:553–560. 2015. View Article : Google Scholar
|
|
128
|
Zhang M, Lahn M and Huber PE: Translating
the combination of TGFβ blockade and radiotherapy into clinical
development in glioblastoma. OncoImmunology. 1:943–945. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhang M, Kleber S, Röhrich M, Timke C, Han
N, Tuettenberg J, Martin-Villalba A, Debus J, Peschke P, Wirkner U,
et al: Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor
LY2109761 enhances radiation response and prolongs survival in
glioblastoma. Cancer Res. 71:7155–7167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Pan E, Supko JG, Kaley TJ, Butowski NA,
Cloughesy T, Jung J, Desideri S, Grossman S, Ye X and Park DM:
Phase I study of RO4929097 with bevacizumab in patients with
recurrent malignant glioma. J Neurooncol. 130:571–579. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Yahyanejad S, King H, Iglesias VS, Granton
PV, Barbeau LM, van Hoof SJ, Groot AJ, Habets R, Prickaerts J,
Chalmers AJ, et al: NOTCH blockade combined with radiation therapy
and temozolomide prolongs survival of orthotopic glioblastoma.
Oncotarget. 7:41251–41264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tolcher AW, Messersmith WA, Mikulski SM,
Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari
A, Shapiro GI, et al: Phase I study of RO4929097, a gamma secretase
inhibitor of Notch signaling, in patients with refractory
metastatic or locally advanced solid tumors. J Clin Oncol.
30:2348–2353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
den Hollander MW, Bensch F, Glaudemans AW,
Oude Munnink TH, Enting RH, den Dunnen WF, Heesters MA, Kruyt FA,
Lub-de Hooge MN, Cees de Groot J, et al: TGF-β Antibody Uptake in
Recurrent High-Grade Glioma Imaged with 89Zr-Fresolimumab PET. J
Nucl Med. 56:1310–1314. 2015. View Article : Google Scholar : PubMed/NCBI
|