Introduction
Esophageal carcinoma is one of the most common
malignancies worldwide with a high mortality rate (1). China accounts for >50%
(324,422/604,100) of patients, and 90% of esophageal carcinoma
includes esophageal squamous cell carcinoma (ESCC) cases. Although
the diagnosis and treatment for ESCC have improved in recent years,
the overall 5-year survival rate of patients is 15 to 25% (2-4). The
leading cause of treatment failure and mortality among patients
with ESCC is tumor recurrence and metastasis (5,6).
Tumor metastasis is very complex and involves multiple steps
(7); thus, there is an urgent need
to discover the underlying molecular mechanisms responsible for
ESCC metastasis, as they may serve as targets to improve the
treatment of ESCC.
Numerous protein kinases have critical roles in
regulating cell growth and survival, and thus, have emerged as the
most important targets for drug discovery. Maternal embryonic
leucine zipper kinase (MELK) is a member of the
sucrose-non-fermenting/AMP-activated protein kinase family of
serine-threonine protein kinases and plays key functional roles in
multiple cellular processes, including the cell cycle, cell
proliferation, apoptosis, and cell migration (8-11).
MELK is overexpressed in a variety of human tumors, including
melanoma (12), breast (13), gastric (11), and high-grade prostate cancer
(14), as well as glioblastoma
multiforme (15). High levels of
MELK expression are associated with poor prognoses in patients
(14,16). Inhibition of MELK has been
demonstrated to suppress tumor growth in vitro and in
pre-clinical adult cancer models (17-19).
MELK is also elevated in cancer stem cells (CSCs) and can promote
CSC growth (20). Small molecule
inhibitors of MELK have anticancer activity in breast and other
cancers (20-22), indicating that MELK may be a target
for cancer therapy. A recent study reported that MELK enhances
tumorigenesis and metastasis of ESCC cells via activation of the
FOXM1 signaling pathway (23).
Epithelial-mesenchymal transition (EMT) is a
biological process that is known to be crucial for embryogenesis,
wound healing and malignant progression (24). In the context of cancer
pathogenesis, EMT confers on cancer cells increased
tumor-initiating and metastatic potential (25). EMT can be induced by several
intracellular signaling pathways, including the transforming growth
factor-β (TGFβ), WNT, PI3K/AKT and nuclear factor-κB (NF-κB)
pathways (24). NF-κB is a
ubiquitous transcription factor that plays a prominent role in
mediating EMT (26). NF-κB can be
activated in neoplastic cells by a series of cell intrinsic and
extrinsic signals (e.g., genetic alterations and microenvironmental
cytokines, respectively). Subsequently, nuclear NF-κB promotes EMT
by inducing the expression of EMT-inducing transcription factors
(EMT-TFs), including Snail, zinc finger E-box-binding homeobox
(ZEB)1, ZEB2 and TWIST1, and by directly inducing the expression of
mesenchymal proteins such as CD44, vimentin and N-cadherin
(27).
The prognostic role of MELK in ESCC and the precise
molecular mechanisms by which it promotes EMT and aggressive
progression in ESCC remain unknown. Whether there are other MELK
downstream signaling pathways besides FOXM1 in ESCC and whether
MELK promotes ESCC metastasis by activating NF-κB were investigated
in the present study.
Materials and methods
Patients and tissue samples
A total of 84 samples of ESCC tissue (including 84
cases of tumor tissues and 18 adjacent normal esophageal tissues)
were obtained from Meizhou People's Hospital (Meizhou, China)
between September 2004 and February 2008. None of the patients were
treated by radiotherapy or chemotherapy before surgical operation.
All samples were collected with written informed consent and
approved by the Research Ethics Committee of Meizhou People's
Hospital. The histopathological diagnosis was performed according
to the World Health Organization criteria. Tumor staging was
determined based on the 6th edition of the tumor-nodemetastasis
(TNM) classification of the International Union Against Cancer
(IUAC) (28). The characteristics
of the patients are detailed in Table
I.
 | Table IAssociation between MELK expression
and clinicopathological features in 84 patients with ESCC. |
Table I
Association between MELK expression
and clinicopathological features in 84 patients with ESCC.
| Variables | No. of cases | MELK expression
| P-value |
|---|
| Negative | Positive |
|---|
| Sex | | | | 0.034a |
| Male | 67 | 28 | 39 | |
| Female | 17 | 12 | 5 | |
| Age (years) | | | | 0.662 |
| ≤55 | 42 | 19 | 23 | |
| >55 | 42 | 21 | 21 | |
| Tumor size
(cm) | | | | 0.393 |
| ≤5.0 | 44 | 19 | 25 | |
| >5.0 | 40 | 21 | 19 | |
| Histological
grade | | | | 0.656 |
| I | 12 | 5 | 7 | |
| II-III | 72 | 35 | 37 | |
| T stage | | | | 0.447 |
| T1-T2 | 24 | 13 | 11 | |
| T3-T4 | 60 | 27 | 33 | |
| N stage | | | | 0.596 |
| N0 | 50 | 25 | 25 | |
| N1 | 34 | 15 | 19 | |
| Tumor stage | | | | 0.297 |
| I-II | 54 | 28 | 26 | |
| III-IV | 30 | 12 | 18 | |
Immunohistochemistry (IHC)
Tissues were fixed in 10% neutral buffered formalin
overnight at room temperature (RT) and embedded in paraffin. The
5-µm-thick sections of tissue paraffin samples were prepared
on pathological sections for immunohistochemical staining. Tissue
sections were heated in citrate buffer solution (pH 6.0) at 100°C
for 10 min to facilitate antigen retrieval. After tissue samples
cooled down to RT, the endogenous peroxidase activity was blocked
by incubating sections with 10% normal goat serum (product no.
C0265; Beyotime Institute of Biotechnology) for 1 h at RT followed
by incubation with an antibody against MELK (1:300; cat. no.
11403-1-AP; ProteinTech Group, Inc.) for 3 h at RT, and incubated
with a secondary antibody (Dako REAL EnVision; cat. no. K5007;
Agilent Technologies, Inc.) for 30 min at RT after rinsing with
PBS. Immunoreacted cells were visualized using diaminobenzidine,
and nuclei were counterstained with hematoxylin for 10 min at RT.
The results were independently assessed by two pathologists,
neither of whom knew the clinical data. The percentage of positive
cells in the sections was evaluated as 0-100%, while cases with
>20% of tumor cells exhibiting strong cytoplasmic staining were
considered positive MELK staining. A negative control tissue
section without primary MELK antibody was also included (Fig. S1). Leica Microsystems DM6000B
light microscope (Leica Microsystems GmbH) was used for evaluation
and image recording.
Cell lines and cell culture
Human normal esophageal epithelial cell line Het-1A
(cat. no. CC-Y1220) was purchased from Shanghai EK-Bioscience
Biotechnology Co., Ltd. (http://www.elisakits.cn/index.php). Human ESCC cell
lines KYSE140 (BNCC no. BNCC351870), KYSE450 (BNCC no. BNCC339896)
and KYSE510 (BNCC no. BNCC360126) were purchased from BeNa Culture
Collection; Beijing Beina Chunglian Institute of Biotechnology
(https://www.bncc.org.cn/). KYSE150 (TCHu236) was
obtained from the Shanghai Institute of Cell Biology, Chinese
Academy of Sciences. Cells were cultured in RPMI-1640 medium
(Biological Industries) with 10% fetal bovine serum (Biological
Industries) and 1% penicillin-streptomycin (Beyotime Institute of
Biotechnology) in a humidified atmosphere containing 5%
CO2 at 37°C. For pharmacological inhibition of MELK,
ESCC cells were treated with 4-16 nM of MELK inhibitor OTSSP167
(cat. no. S7159; Selleck Chemicals) for 24-32 h at 37°C.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total cellular RNA was obtained using TRIzol
(Invitrogen, Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions, and complementary DNA (cDNA) was
prepared from 1 µg of total RNA using PrimeScript RT reagent
Kit with gDNA Eraser (cat. no. RR047A; Takara Bio, Inc.) according
to the manufacturer's instructions. RT-qPCR was performed for the
gene fold change using SYBR Premix Ex Taq™ (cat. no. RR820A; Takara
Bio, Inc.). Thermocycling conditions were as follows: Initial
denaturation of 30 sec at 95°C; followed by 40 cycles of
denaturation at 95°C for 5 sec, and annealing and extension at 60°C
for 30 sec. The mRNA levels were analyzed by the comparative
2−ΔΔCq method after normalization to GAPDH (29). The primer sequences used in this
research were as follows: MELK forward, 5′-TGA TGA TTG CGT AAC AGA
AC-3′ and reverse, 5′-GAA GAT AGG TAG CCG TGA G-3′; human GAPDH
forward, 5′-ATC AAT GGA AAT CCC ATC ACC A-3′ and reverse, 5′-GAC
TCC ACG ACG TAC TCA GCG-3′.
Plasmid constructs and transfection
Corresponding short hairpin RNA (shRNA)
oligonucleotide sequences were cloned into the PLKO.1-TRC vector
(Sigma-Aldrich; Merck KGaA) to obtain an empty vector as the
control and the generation of lentiviral vectors encoding shRNA
targeting MELK. The targeting sequences of shMELK and shNC were as
follows: shMELK-1, 5′-GCC TGA AAG AAA CTC CAA TTA-3′; shMELK-2,
5′-CTG AGT TAA TAC AAG GCA AAT-3′; shMELK-3, 5′-CAG AAA CAA CAG GCA
AAC AAT-3′; sh negative control (NC), 5′-TTC TCC GAA CGT GTC ACG
T-3′. A total of 12 µg lentiviral expression vectors and
second-generation lentiviral packaging vectors pSPAX2 and pMD2.G
(Addgene, Inc.) were transfected into 293T (cat. no. SCSP-502;
Shanghai Cell Bank of the Chinese Academy of Sciences) cells at a
ratio of 4:3:2 and cultured at 37°C. The virus was collected three
times at an interval of 24 h between collections. ESCC cells were
infected with lentiviral particles (MOI=20) for 24 h in the
presence of polybrene (5 µg/ml; cat. no. TR-1003;
Sigma-Aldrich; Merck KGaA). Following selection by 2.5 mM puromycin
(Sigma-Aldrich; Merck KGaA) for 3 days, the cells with stable
knockdown of MELK were selected and maintained in 1 mM puromycin.
Western blotting was then performed to identify the knockdown of
MELK. For overexpression of inhibitor of nuclear factor-κB kinase
subunit β (IKKβ; MELK-depleted ESCC cells were transfected with 2
µg IKKβ expression plasmid (pcDNA3.1-IKKβ) or the empty
vector (pcDNA 3.1-EV) as the control. Following selection using 400
µg/ml G418 (MedChemExpress) for 2 weeks, the cells with
stable overexpression of IKKβ were selected and maintained in 200
µg/ml G418.
Proliferation assays with Cell Counting
Kit-8 (CCK-8)
CCK-8 assays were performed according to the
manufacturer's instructions, to detect cell proliferation. KYSE150
and KYSE510 cells were seeded into 96-well plates with a density of
1,000 cells/well in 100 µl cell medium. Each group was
assessed every 24 h. The plates were placed in a 37°C incubator for
2 h after adding 10 µl of CCK-8 working solution (Bimake),
and then the absorbance was measured at 450 nm using a microplate
reader. The average values of the five replicates were calculated
and used to draw the growth curve. This experiment was repeatedly
performed three times. The OD450 value was statistically
evaluated by one-way ANOVA analysis followed by Tukey's post hoc
test using SPSS 19.0 statistical software.
Colony formation assay
Colony formation assays were performed to detect
cell proliferation. KYSE150 and KYSE510 cells were inoculated into
6-well plates with a concentration of 100 cells/well. The plates
were placed in a cell incubator at 37°C for 2 weeks, and the medium
with 10% FBS was renewed every 4 days. When visible colonies had
formed, each well was fixed with 1 ml of 4% paraformaldehyde for 30
min at RT and stained with 1 ml of 0.1% crystal violet (Beyotime
Insitute of Biotechnology) for 30 min at RT. The number of visible
colonies (consisting of >50 cells) of three replicates was
counted manually after the plates dried up. The experiment was
independently performed three times.
Transwell assays
Transwell assays were performed to detect cell
vertical migration and invasion. A total of 5×104
KYSE150 and KYSE510 cells in 150 µl serum-free medium were
seeded in the upper compartment of an 8-µm pore size
Transwell chamber (Corning, Inc.) after pre-treatment with
serum-free medium for 12 h. The upper chambers had been pre-coated
with 50 µl of 2.5 mg/ml solution of Matrigel at 37°C for 1 h
(Corning, Inc.) for the invasion assays or uncoated for the
migration assays. The lower chambers were filled with 600 µl
RPMI-1640 medium containing 10% FBS. After 24 h (migration
experiment) or 36 h (invasion experiment), the cells on the upper
surface of the chamber membranes were wiped off. The membranes were
then fixed with 4% polymethanol at RT for 30 min and stained with
0.5% crystal violet at RT for 30 min. Five fields randomly selected
under a light microscope were captured to count the cells on the
lower surface of the filter at a magnification of ×100. This
experiment was performed in triplicate independently.
Wound healing assays
Wound healing assays were performed to detect cell
lateral migration. A total of 5×105 KYSE150 and KYSE510
cells were seeded in a 6-well plate and exposed to serum-free
RPMI-1640 medium for 12 h to inhibit cell proliferation. When the
confluency reached >80%, a horizontal scratch was made using a
sterile 200-µl microliter pipette tip. After the wound was
induced, the cells were washed twice with PBS and cultured in a
serum-free medium. Images of migration were obtained at 0, 16, and
20 h after inducing under a light microscope. The assays were
repeatedly performed three times.
Western blotting
Western blotting was performed to detect the
expression level of related proteins. The whole KYSE150 and KYSE510
cell lysates were prepared in SDS buffer with 1% protease inhibitor
cocktail and 1% PMSF. The nuclear proteins were obtained using
Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Institute
of Biotechnology) according to the manufacturer's instructions. The
concentration of protein samples was quantified using BCA Protein
Assay Kit (Beyotime Institute of Biotechnology). A total of 50
µg of proteins were separated by 10% SDS-PAGE and then
transferred onto PVDF membranes. Incubated with TBST buffer
(Tris-buffered saline with 0.1% Tween-20) including 5% dried
skimmed milk at RT for 1 h, the corresponding PVDF membranes were
incubated with a primary antibody at 4°C overnight. The membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (1:2,000; product no. 7074; Cell Signaling
Technology, Inc.) at RT for 1 h. Finally, protein bands were
detected using enhanced chemiluminescence (ECL) detection reagent
(MilliporeSigma) according to the manufacturer's instructions. The
grayscale value of the immunoreactive bands was measured by ImageJ
(version 1.53; National Institutes of Health). Antibodies used in
this study were as follows: Antibodies against zonula occludens-1
(ZO-1; 1:1,000; product no. 8193), E-cadherin (1:1,000; product no.
14472), N-cadherin (1:1,000; product no. 13116), matrix
metalloproteinase (MMP)-2 (1:1,000; product no. 40994), MMP-9
(1:1,000; product no. 13667), ZEB2 (1:1,000; product no. 97885),
Slug (1:1,000; product no. 9585), Snail (1:1,000; product no.
3879), phosphorylated (p)-IKKα/β (Ser176/180; 1:1,000; product no.
2697) were purchased from Cell Signaling Technology, Inc.
Antibodies against GAPDH (1:1,000; cat. no. 60004-1-Ig), MELK
(1:2,000; cat. no. 11403-1-AP), p65 (1:1,000; cat. no. 10745-1-AP)
were obtained from ProteinTech Group, Inc. Antibodies against
NF-κB1 (1:500; cat. no. BM3946) and NF-κB2 (1:500; cat. no. BA1298)
were purchased from Boster Biological Technology. Antibodies
against p-p65 (Ser536; 1:1,000; cat. no. WL02169) and p-IKB-α
(Ser32/36; 1:500; cat. no. WL02495) were purchased from Wanleibio
Co., Ltd. Antibody against p62 (SQSTM1) (1:1,000; cat. no. PM045)
were obtained from MBL Beijing Biotech Co., Ltd.
Immunofluorescence staining
A total of 4×104 KYSE150 and KYSE510
cells were singly seeded on coverslips in 24-well plates and fixed
with 4% paraformaldehyde at RT for 20 min. After being washed
thrice with PBS for 5 min each time, cells were permeabilized with
0.3% Triton X-100 (Sigma-Aldrich; Merck KGaA) in PBS for 10 min at
4°C and blocked with 10% goat serum in PBS at RT for 60 min. Cells
were incubated with a primary antibody overnight at 4°C and then
were incubated with Alexa Fluor 488/647-conjugated secondary
antibodies (1:500; product nos. 4412 and 4414; Cell Signaling
Technology, Inc.) at RT for 1 h. Following washing three times with
PBS for 10 min each time, cells were counterstained with DAPI for
15 min at RT. Fluorescence images were captured by Zeiss LSM 780 or
LSM 800 laser scanning microscope (Carl Zeiss AG). Antibodies used
in this study were as follows: MELK (1:100; cat. no. 11403-1-AP;
ProteinTech Group, Inc.), Slug (1:100; product no. 9585; Cell
Signaling Technology, Inc.), NF-κB1 (1:25; cat. no. BM3946; Boster
Biological Technology), and NF-κB2 (1:50; cat. no. BA1298; Boster
Biological Technology).
Co-immunoprecipitation (Co-IP)
assays
Protein lysate of KYSE510 cells was collected using
IP lysis buffer (cat. no. P0013; Beyotime Insitute of
Biotechnology) and mixed with the indicated antibody or negative
control IgG overnight at 4°C. Subsequently, 200 µl lysate
was mixed with 20 µl of protein G magnetic beads (cat. no.
37478; Cell Signaling Technology, Inc.) for 4 h at 4°C; the binding
complex was obtained using a magnetic stand. After being washed
thrice with IP buffer, the complex was boiled for 5 min at 100°C
with SDS buffer and centrifuged at 4°C and 12,000 × g for 2 min to
gain bound protein samples, which were used for western blotting.
Antibodies used in this study were as follows: MELK (1:50; cat. no.
11403-1-AP; ProteinTech Group, Inc.), IKKα/β (1:50; cat. no.
WL01900; Wanleibio Co., Ltd.), and IgG (1:50; cat. no. A7016;
Beyotime Institute of Biotechnology).
Bioinformatics analysis
Two human microarray datasets, namely, GSE20347
(30) and GSE23400 (31), were obtained from the public Gene
Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). The mRNA sequencing
data of patients with ESCC were downloaded from The Cancer Genome
Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Gene expression of
MELK in the GEO database and TCGA ESCC database were assessed using
GraphPad Prism 7 software (GraphPad Software, Inc.). Gene set
enrichment analysis (GSEA) was performed by generating the Gene
MatriX file (.gmx) using published signatures for the Rickman et
al 'metastasis' (32), the
Sarrio et al 'epithelial mesenchymal transition' (33), and GO biological process gene sets
from the Molecular Signatures Database (MSigDB; version 6.2)
(34). The gene cluster text file
(.gct) was generated from the TCGA ESCC database. The number of
permutations for GSEA was set to 1,000, and the TCGA gene list was
used as the chip platform.
Experimental in vivo metastasis
assays
A total of 14 male BALB/c-nu/nu mice (aged 4 to 5
weeks; weighing 18 to 25 g) were purchased from the Shanghai
Laboratory Animal Center of the Chinese Academy of Sciences and
raised in a pathogen-free laminar flow cabinet throughout the
experiments under the following conditions: Controlled humidity,
50±10%; temperature, 23±2°C; 12-h light/dark cycle and ad
libitum access to food and water. KYSE150 cells stably
transfected with shNC or shMELK were collected and re-suspended in
PBS at a concentration of 1×108 cells/ml. Prepared cells
were injected into the tail veins of 14 mice (100 µl per
mouse). The health and behavior of the nude mice was monitored
every 2 days. The mice would be humanely euthanized if they
experienced unrelieved pain or distress, based on the euthanasia
criteria. Following 5 weeks of injection, the mice were euthanized
by cervical dislocation. The lungs and livers of the mice were
removed and photographed. The visible tumors on the surface of the
lung tissue were then counted. The largest diameter of the tumor
nodules was 2 mm, and fusion nodules exceeding 2 mm were calculated
as the diameter/2 mm. The experiment was strictly carried out
following the Guide for the Care and the Use of Laboratory Animals
of the National Institutes of Health and was approved by the Animal
Care and Experimental Ethics Committee of Jinan University (approva
l no. IACUC-20190211-04; Guangzhou, China).
Statistical analysis
All data were analyzed with SPSS 19.0 software (IBM
Corp.). The association between MELK expression and
clinicopathological feathers of patients with ESCC was analyzed
using the chi-squared test. The Kaplan-Meier method and log-rank
tests were used to compare the overall survival. The numerical data
are reported as the mean ± standard deviation (SD). The difference
between two groups was analyzed by a two-tailed unpaired Student's
t-test. A one-way ANOVA with Tukey's post hoc test were used to
analyze three or more groups. P-values <0.05 were considered to
indicate a statistically significant difference.
Results
MELK is highly expressed in human ESCC
tissues and is associated with poor overall survival of patients
with ESCC
MELK is highly expressed in cancer and plays key
roles in tumor progression in some cancer types (11,12);
however, the role of MELK in ESCC remains unknown. To assess the
roles of MELK in ESCC, the expression levels of MELK in 84 ESCC
tissues and 18 adjacent normal esophageal tissues were first
assessed by immunohistochemical staining. The MELK protein was
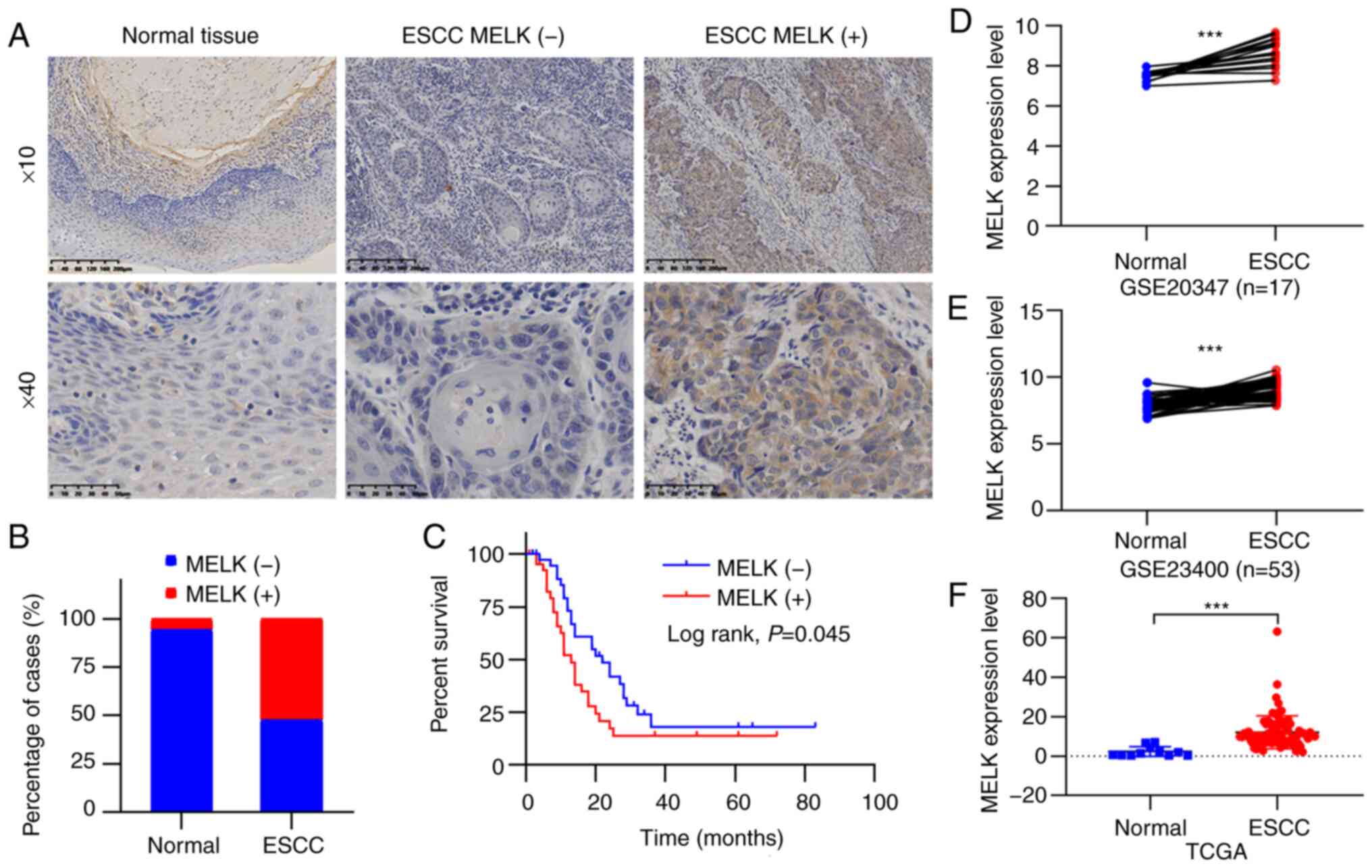
found to be localized in the cytoplasm (Fig. 1A). The positive rates of MELK
protein detection were 5.6% (1/18) in non-tumor tissues and 52.4%
(44/84) in tumor tissues, and the expression of MELK was
significantly upregulated in ESCC compared with normal tissues
(P<0.001; Fig. 1B). The
associations between the expression status of MELK and
clinicopathological characteristics of patients were also analyzed,
although the results revealed that positive MELK expression was not
significantly associated with characteristics, including age,
histological grade, and tumor stage (P>0.05; Table I). By using Kaplan-Meier analysis,
it was determined that positive MELK expression indicated the poor
overall survival of patients with ESCC (P<0.05; Fig. 1C). Furthermore, the GEO and TCGA
databases were searched to analyze MELK levels in patients with
ESCC, and the increased expression of MELK was also observed in
ESCC (Fig. 1D-F). These results
indicated that the expression of MELK protein was elevated in ESCC
and predicted a poor prognosis in patients.
MELK inhibition reduces proliferation and
colony formation in ESCC cells
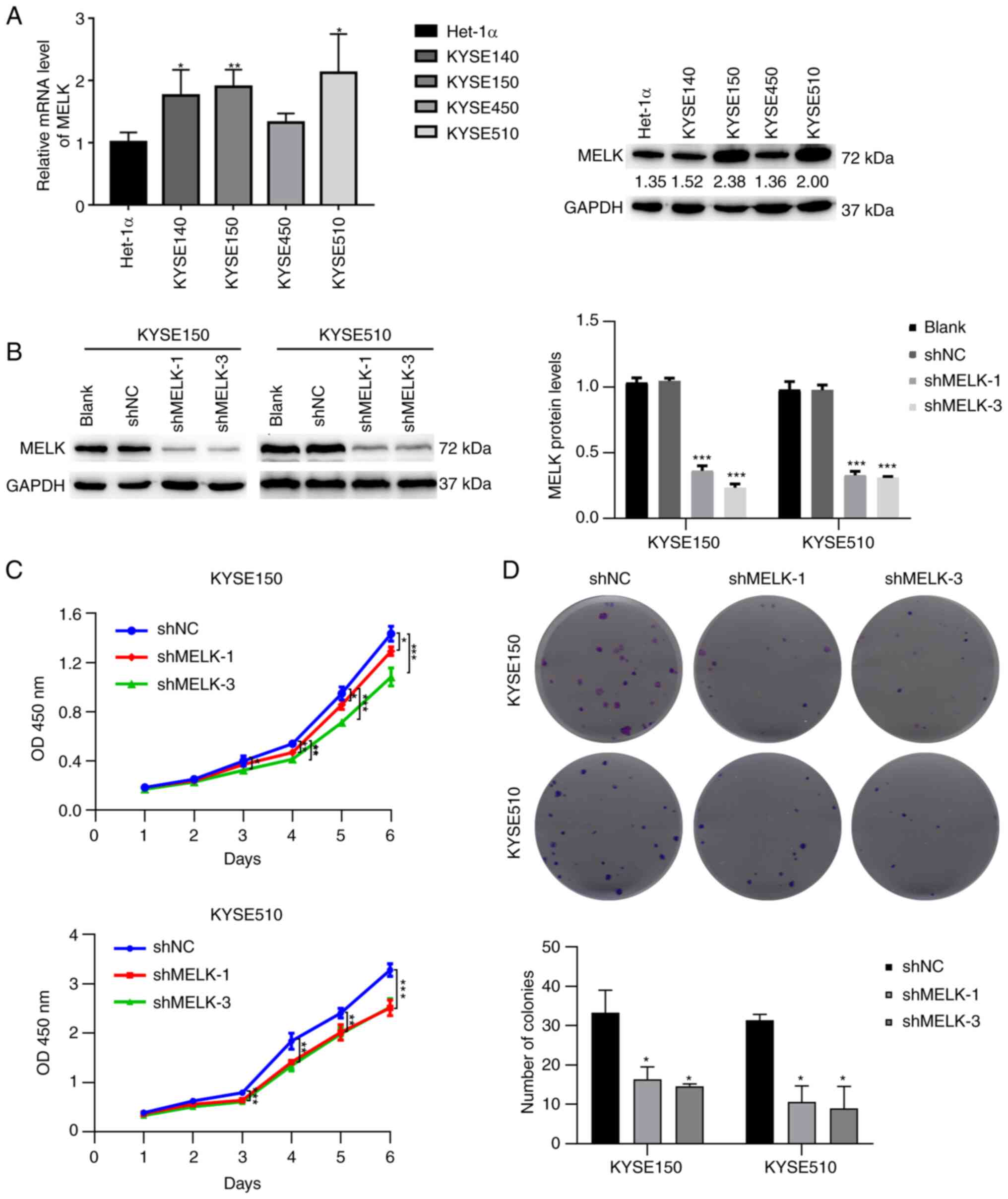
The aforementioned results revealed that MELK was
more highly expressed in ESCC and positively associated with the
poor survival of patients. To determine the biological function of
MELK in ESCC, the protein level of MELK in ESCC cell lines was
first analyzed, and then KYSE150 and KYSE510 cell lines, which had
higher expression levels of MELK and represented poorly
differentiated and well differentiated ESCC respectively, were
selected (35), to be used for
further study (Fig. 2A). Stable
knockdown of MELK was generated by transfecting KYSE150 and KYSE510
cells with shRNA of MELK. Following transfection and selection with
puromycin, the expression of MELK in KYSE150 and KYSE510 cells was
significantly decreased (Fig. 2B).
The effect of MELK on the proliferation of ESCC cells was then
investigated using CCK-8 assays. The results revealed that
knockdown of MELK significantly reduced the growth of KYSE150 and
KYSE510 cells compared with the control cells (Fig. 2C). Colony formation assays
demonstrated that knock-down of MELK efficiently decreased the
ability of ESCC cells to proliferate (Fig. 2D). These results indicated that
MELK regulated the proliferative ability of ESCC cells.
MELK inhibition decreases the migration
and invasion of ESCC cells
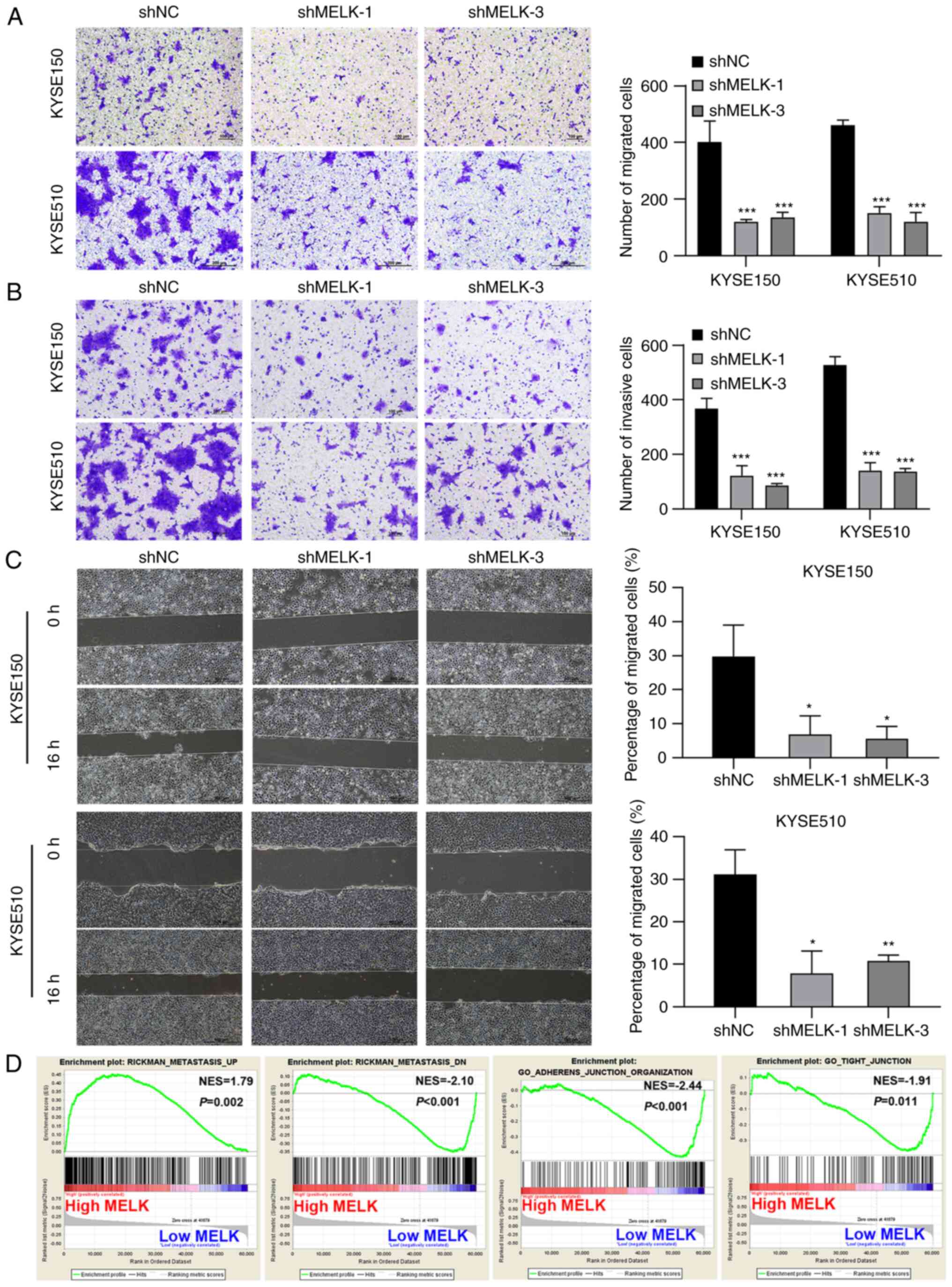
Next, the cell migration and invasive abilities of
cells were examined using Transwell migration and invasion assays,
as well as wound healing assays. The number of migrated and
invasive cells in MELK-knockdown KYSE150 and KYSE510 cells was
significantly decreased compared with the control cells (Fig. 3A and B). In the wound healing
assays, the control cells markedly closed the wound 16 h after
scratching, but MELK-knockdown cells were unable to heal the wound,
and the wound areas of the experimental groups and controls were
significantly different. MELK knockdown significantly inhibited
cell migration (Fig. 3C). To
further ascertain whether MELK accelerates the motility capacity of
ESCC cells, GSEA was used to determine the degree of MELK
expression with metastasis-related signatures. The results showed
that ESCC with high MELK expression had significant enrichment with
an aggressive signature. To further confirm this observation, MELK
expression was examined with metastasis-related markers and it was
determined that GSEA highlighted the positive association of
increased MELK expression with metastasis and the negative
association of increased MELK expression with adherens junction and
tight junction (Fig. 3D).
Collectively, these findings indicated that high MELK expression
may be associated with the highly infiltrative and aggressive
characteristics of ESCC cells.
MELK facilitates the EMT in ESCC
cells
EMT is an early event in tumor metastasis, and tumor
cells show enhanced migration and invasion abilities during the EMT
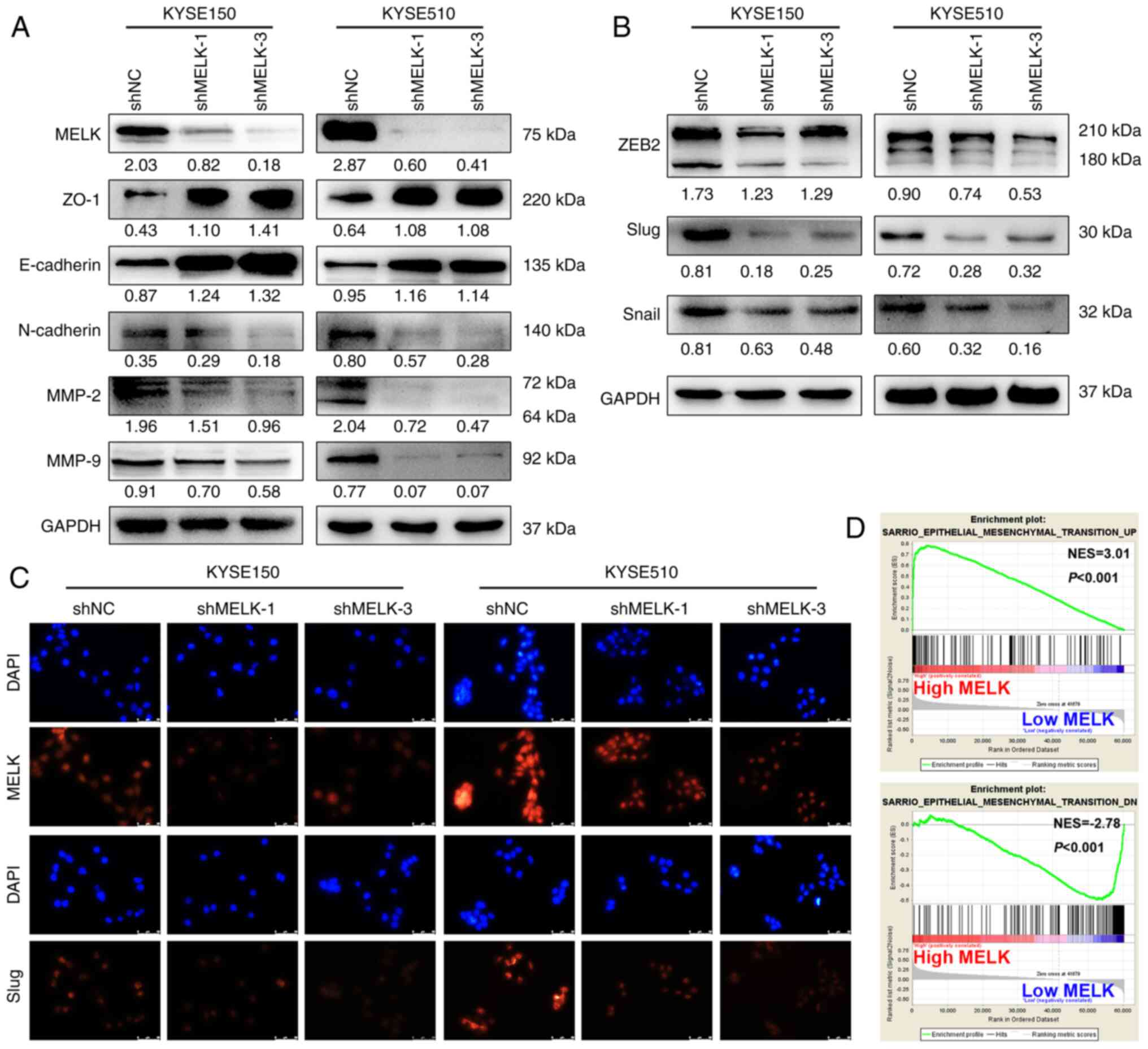
(24,25). Therefore, the expression of markers
associated with EMT were examined using western blotting. MELK
inhibition upregulated the expression of epithelial-related
proteins, including E-cadherin and ZO-1, and downregulated
mesenchymal-related protein N-cadherin in both KYSE150 and KYSE510
cells (Fig. 4A). Moreover,
EMT-TFs, ZEB2, Slug and Snail, were also inhibited after MELK
knockdown (Fig. 4B). In
particular, the protein expression of Slug was demonstrated to be
markedly reduced by immunofluorescence staining (Fig. 4C). In addition, MELK knockdown
decreased MMP-9 and MMP-2 protein levels, which are involved in
facilitating cell metastasis by extracellular matrix (ECM)
degradation (Fig. 4A).
Furthermore, GSEA highlighted the positive association of increased
MELK expression with the EMT signature (Fig. 4D). Thus, the results demonstrated
that MELK may promote cell migration and invasion by promoting EMT
in ESCC cells.
MELK positively regulates NF-κB
signaling
To decipher the mechanisms by which MELK regulates
EMT, GSEA was used to compare the expression profiles between high-
and low-MELK expression cases. The NF-κB-activated gene signature
was enriched in high-MELK expression cases (Fig. S2A). Aberrant NF-κB pathway
activation is a significant contributor to the EMT (36,37).
The NF-κB pathway can directly and indirectly affect Snail, Slug,
and ZEB1 expression to facilitate the EMT. The
expression/phosphorylation of proteins involved in the NF-κB
signaling were examined by western blotting. The knockdown effect
of MELK on various pathways involved in EMT, including
WNT/β-catenin, TGFβ and AKT signaling pathways, were also assessed
to search for the downstream pathway of MELK. The results
demonstrated that MELK inhibition significantly downregulated the
expression of main NF-κB subunits, including NF-κB p50, NF-κB p52,
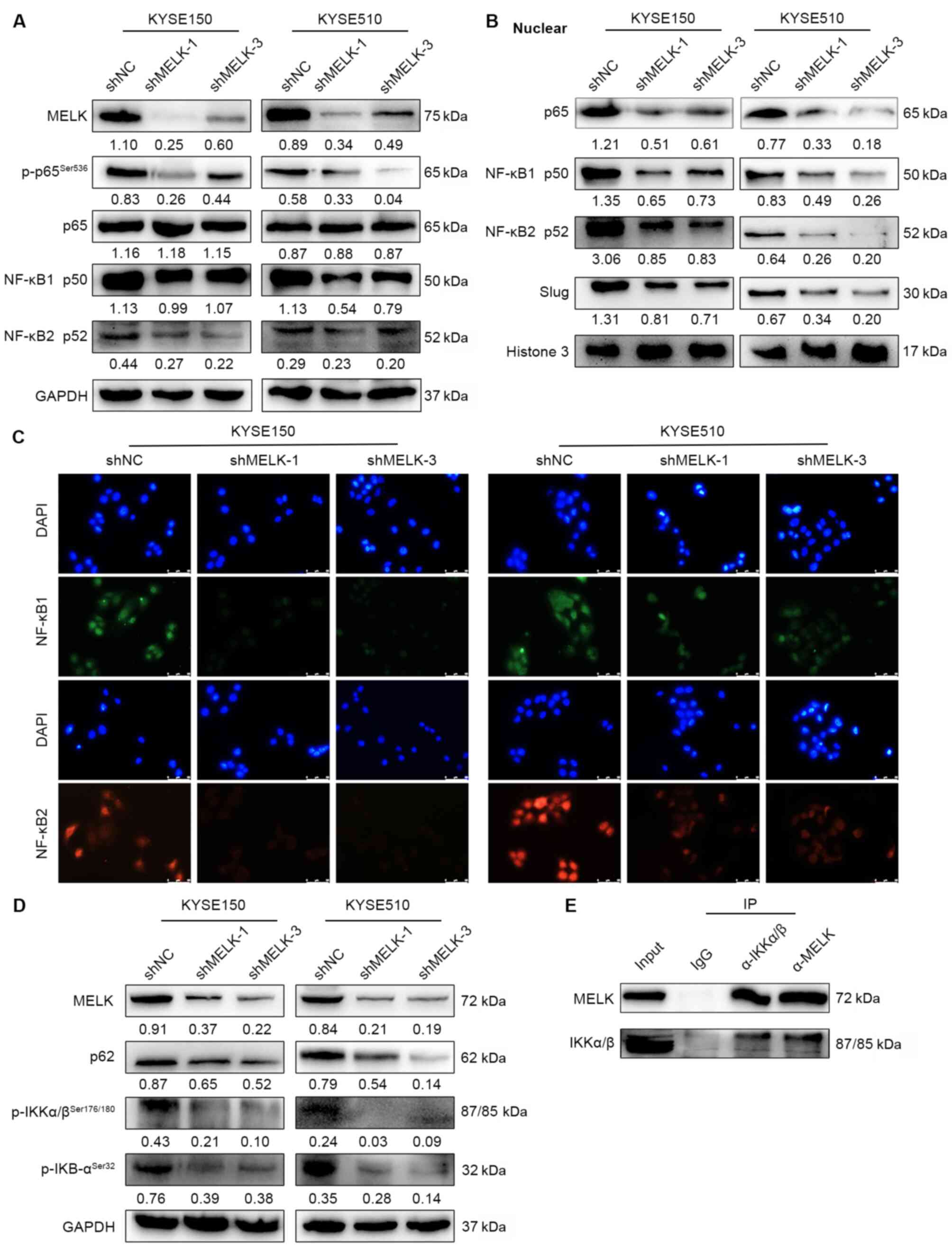
NF-κB p65, and phosphorylated NF-κB (p-NF-κB) p65 (Fig. 5A), but it did not affect the
expression of β-catenin, p-SMAD2 and p-AKT (Fig. S2B). The nuclear translocation of
NF-κB p65, NF-κB p50, and NF-κB p52 was also examined, and MELK
inhibition led to decreased translocation of NF-κB p65, NF-κB p50,
and NF-κB p52 to the nucleus (Fig.
5B-C), which suggested downregulation of this signaling
pathway. Additionally, Slug, the downstream target of the NF-κB
pathway was evaluated, and its expression was shown to be reduced
in the nucleus (Fig. 5B). These
results suggested that MELK knockdown inhibited activation of the
NF-κB pathway. NF-κB complex is usually inactive and located in the
cytoplasm while bound to IκB inhibitor proteins (27). When IκB protein is phosphorylated
by the IκB kinase (IKK) complex, which leads to IκB ubiquitination
and subsequent degradation, NF-κB subunits may translocate to the
nucleus and activate downstream gene transcription (27). Thus, the expression levels of NF-κB
upstream kinases were examined; the results revealed that MELK
inhibition reduced the protein levels of p-IκB, p-IKK, and p62 by
which the activation of NF-κB was decreased (Fig. 5D). Furthermore,
co-immunoprecipitation in ESCC cells was performed to detect the
interaction between MELK and the upstream kinases of the NF-κB
signaling and it was determined that MELK could bind to IKK and
regulated phosphorylation of IKK (Fig.
5E). Hence, the results suggested that MELK may activate the
canonical NF-κB signaling by interacting with IKK.
Overexpression of IKKβ rescues NF-κB
signaling and the migration of ESCC cells after MELK
inhibition
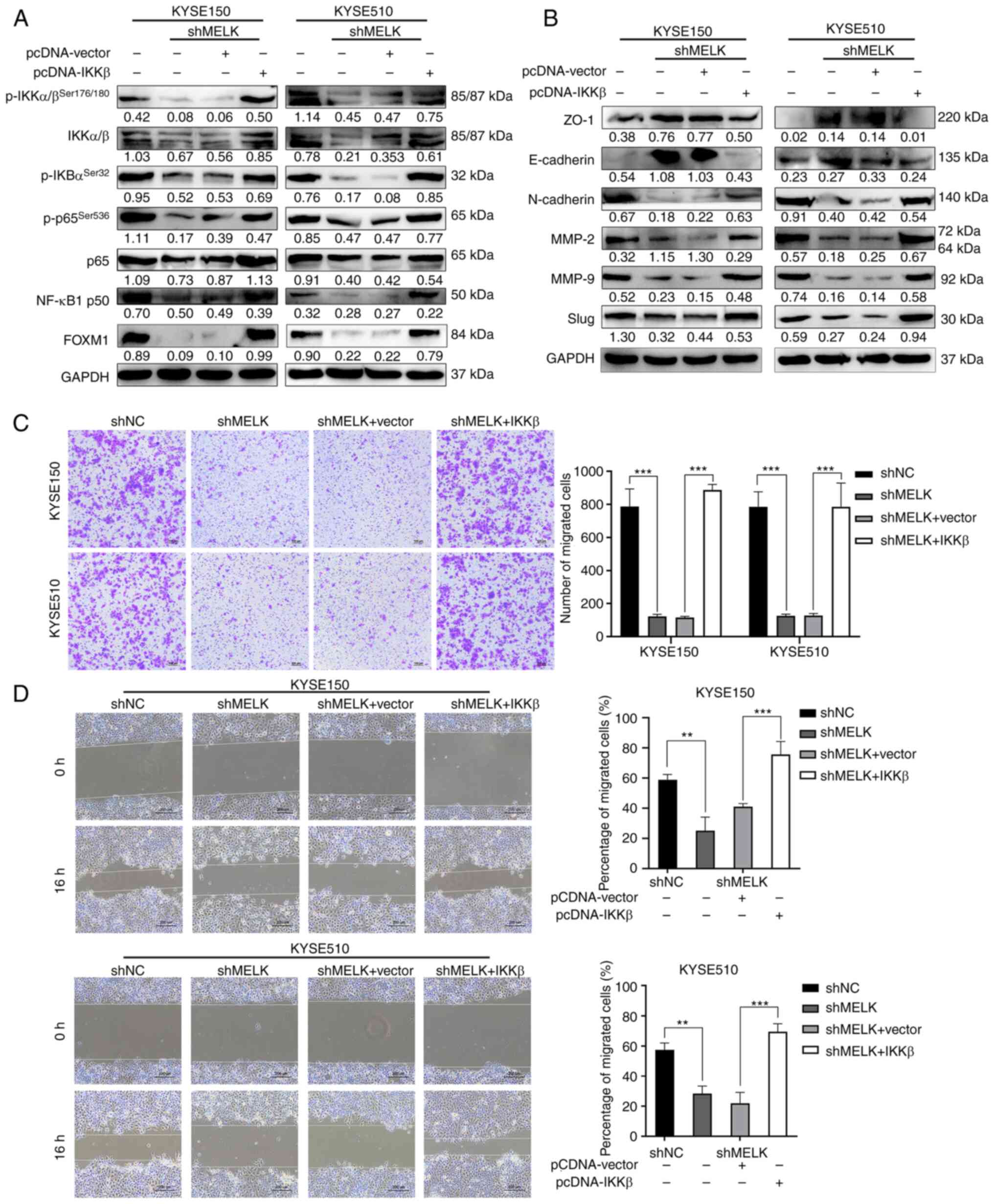
Because IKKβ acts downstream of MELK, it was
investigated whether ectopic expression of IKKβ could rescue the
inhibition of NF-κB signaling caused by MELK inhibition.
IKKβ-overexpressing plasmid was stably transduced into
MELK-depleted ESCC cells, and then the expression of IKKβ was
detected by western blotting. As revealed in Fig. 6A, the protein levels of p-IKKβ,
IKKβ and its downstream targets including p-IκBα, p-p65 and NF-κB
p50 were markedly upregulated by IKKβ in ESCC cells with MELK
depletion. A previous study reported that MELK promoted tumor
growth and metastasis via stimulating FOXM1 signaling in ESCC
(23). In view of the reciprocal
regulation between NK-κB and FOXM1 (38), the expression of FOXM1 in ESCC
cells with MELK knockdown and IKKβ overexpression was also
assessed. As revealed in Fig. 6A,
MELK knockdown inhibited FOXM1 expression, which was rescued by
IKKβ overexpression. Immunoblotting analysis also revealed that
ectopic expression of IKKβ markedly restored Slug, E-cadherin,
N-cadherin, ZO-1, MMP-2 and MMP-9 expression in MELK-depleted ESCC
cells (Fig. 6B). Moreover,
overexpression of IKKβ in MELK-depleted ESCC cells also attenuated
the inhibitory effects on the migration in both Transwell and wound
healing assays (Fig. 6C and D). By
contrast, the expression of an empty vector did not rescue the
migration of ESCC cells (Fig. 6C and
D). Collectively, these results demonstrated that attenuation
of NF-κB signaling was partly responsible for blocking ESCC
migration inhibition after MELK inhibition.
MELK inhibitor blocks the EMT via NF-κB
pathway
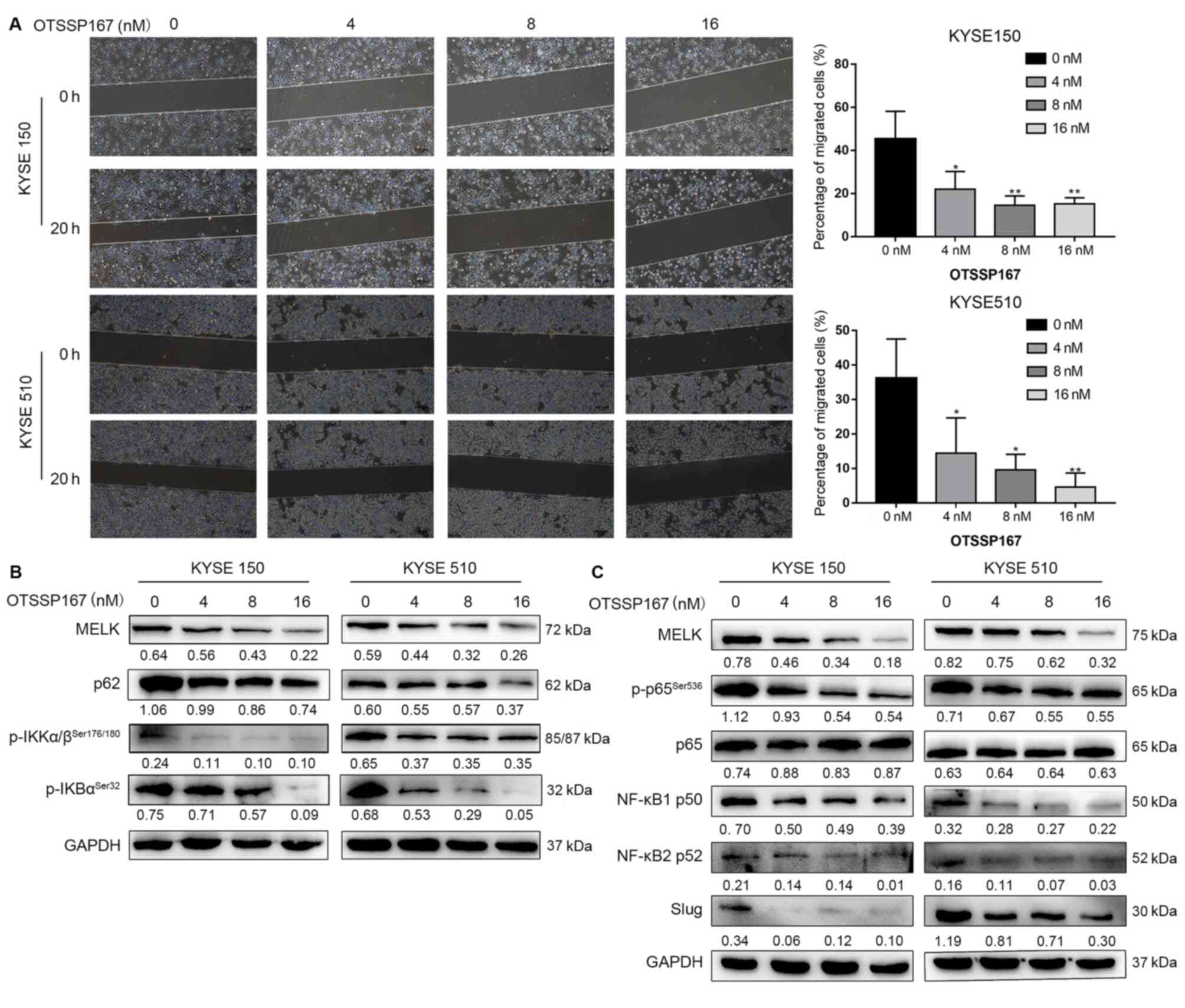
To further confirm whether MELK inhibition, but not
an off-target effect, would reduce the activation of the NF-κB
pathway, ESCC cell lines were treated with the MELK inhibitor
OTSSP167 (16,39,40).
OTSSP167 treatment significantly inhibited ESCC cell migration
(P<0.05; Fig. 7A). OTSSP167
reduced the protein levels of p-IKK, p-IκB, NF-κB1 p50, NF-κB2 p52,
p62, and p-NF-κB p65 and the downstream protein Slug (Fig. 7B-C). Thus, MELK inhibition was
confirmed to block the EMT by decreasing the activation of NF-κB
signaling in ESCC cells.
Knockdown of MELK inhibits metastatic
capacities of ESCC cells in vivo
To determine whether MELK regulates ESCC cell
migration and invasion potential in vivo, KYSE150-shMELK and
control KYSE150 vector cells were injected into the tail vein of
nude mice (seven mice per group). At 5 weeks after cell injection,
mice were sacrificed, and metastatic tumors formed in the lung and
liver were examined. The results showed that no tumor nodule was
formed in the liver of mice. However, metastatic tumor nodules were
frequently observed in the lung of mice, and knockdown of MELK
significantly decreased the number of metastatic nodules in the
lung (P<0.05; Fig. 8A-C). These
results demonstrated that MELK inhibition significantly reduced
metastasis of ESCC.
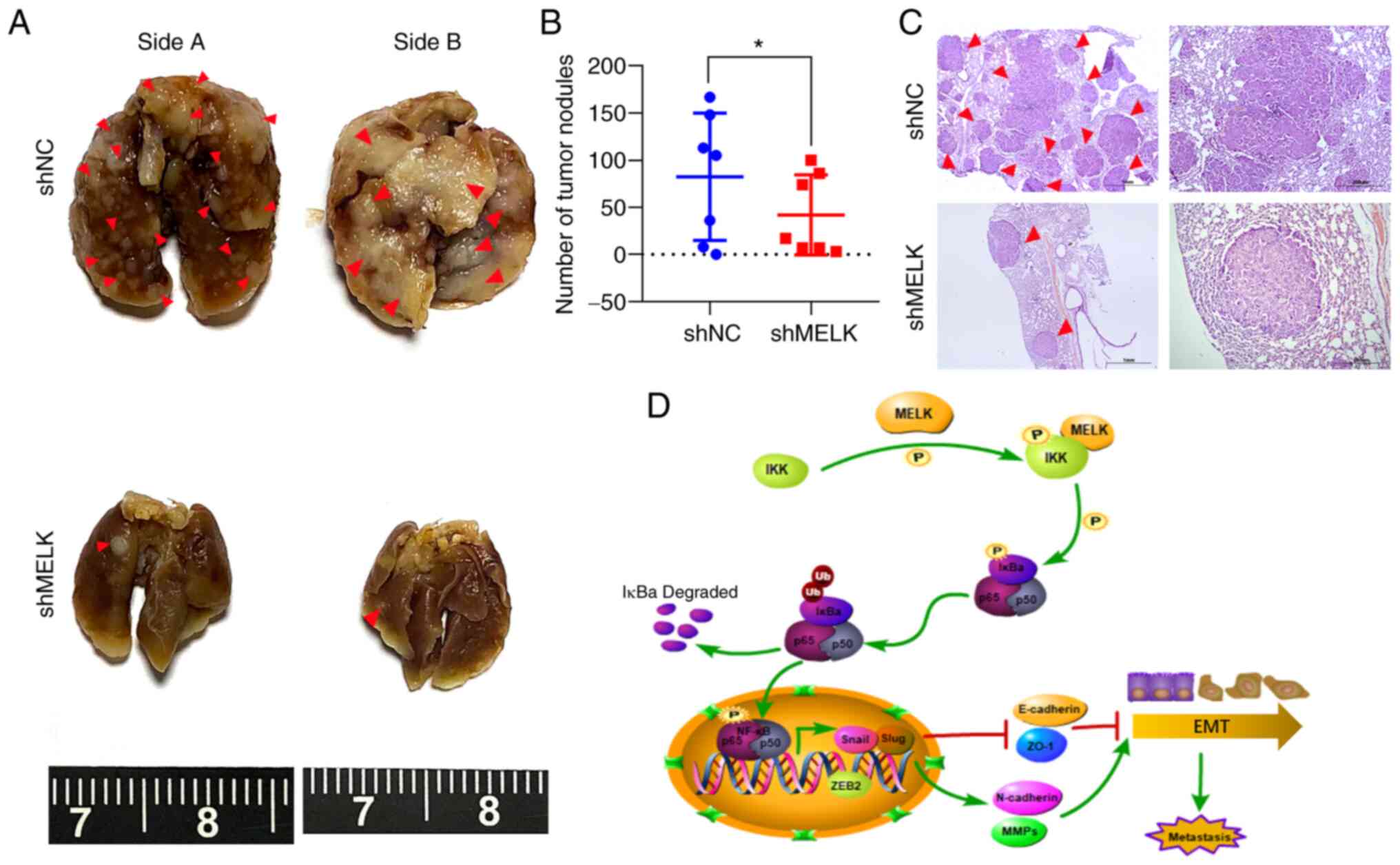 | Figure 8Knockdown of MELK inhibits the
metastasis of ESCC cells in vivo. (A) KYSE150 cells with
MELK knockdown were intravenously injected into nude mice (n=7 per
group) through the tail vein. Representative images of excised
lungs after 5 weeks of injection are shown (arrows indicate the
metastatic foci). (B) The number of tumor nodules on lung surfaces.
(C) Lung metastases in each mouse were stained with H&E. Arrows
indicate the metastatic colonization of tumor cells in the lung
tissues. Scale bars, 1 mm (left), 200 µm (right). (D)
Schematic illustrating the role of MELK in stimulating metastasis
of ESCC cells. *P<0.05. MELK, maternal embryonic
leucine zipper kinase; ESCC, esophageal squamous cell carcinoma;
H&E, hematoxylin and eosin; sh, short hairpin RNA; NC, negative
control; IKK, IκB kinase; NF-κB, nuclear factor-κB; ZEB2, zinc
finger E-box-binding homeobox 2; E-cadherin, epithelial cadherin;
ZO-1, zonula occludens-1; N-caherin, neural cadherin; MMPs, matrix
metalloproteinases; EMT, epithelial-mesenchymal transition. |
Discussion
Recent studies have shown that MELK plays an
important role in tumor progression of numerous types of tumors
(11,14,16,41).
However, little is known about the function and related mechanisms
of MELK in ESCC (23). In the
present study, it was demonstrated that MELK expression was
upregulated in ESCC tissues and was important for the acquisition
of an aggressive and poor prognostic phenotype. It was also
demonstrated that MELK inhibition decreased cell proliferation,
migration, and invasion of ESCC cells both in vitro and
in vivo. Furthermore, it was observed that MELK induced EMT
and promoted cell migration and invasion via the NF-κB pathway
(Fig. 8D).
ESCC is one of the most common malignant tumors, and
the vast majority of tumor-related deaths are the result of tumor
metastasis, but the precise molecular mechanisms that drive this
metastasis process are largely unknown. Therefore, it is important
to understand the molecular events governing tumor proliferation
and metastasis to develop novel therapeutic targets in ESCC.
Numerous studies have shown that MELK is highly expressed in
several tumors, and its expression is correlated with tumor grade
and prognosis (42,43). In the present study, the results
showed that MELK expression was significantly upregulated in ESCC
compared with normal esophagus epithelium, which is consistent with
previous studies in other types of tumors (16,21).
Survival analysis revealed that high MELK expression predicted a
poor prognosis in patients with ESCC. To further confirm the
findings, large sample data from TCGA and GEO databases confirmed
the results of MELK expression in the tissue samples of the present
study. It was also revealed that positive MELK expression was
significantly associated with males. In a previous study, Oliva
et al revealed that the effects of sex on gene expression
are ubiquitous (13,294 sex-biased genes across all tissues)
(44). They also discovered that
promoters of sex-biased genes are enriched for hormone-related and
other transcription factor binding sites (TFBSs) (44). The strongest difference between
male- and female-biased enrichment profiles was observed for TFBSs
of SP2, SP4, NFYB, TWIST1, and STAT5B (female-biased) and of HNF4G,
NFKB1, E2F6, HNF4A, and ETS1 (male-biased), respectively (44). The potential transcription factor
of MELK was also analyzed using UCSC Genome Browser, and it was
found that HNF4G, E2F6 and HNF4A are the potential transcription
factors of MELK. Thus, MELK may be transcriptionally regulated by
HNF4G, E2F6 and HNF4A, and function as a male-biased gene. However,
this hypothesis warrants further demonstration.
As a serine/threonine protein kinase, MELK was
reported to regulate the cell cycle, stem cell renewal, and
apoptosis (21,45,46).
MELK was also identified to contribute to tumor metastasis and
recurrence in breast and gastric cancers (11,47).
In the present study, a series of assays were employed to
investigate the role of MELK in regulating the characteristic
aggressive phenotype of MELK. Knockdown of the expression of MELK
ESCC cells in vitro was established due to the high
expression in ESCC cell lines, and the cell growth, colony
formation, and invasion of these cells were investigated. Most
previous studies on the function of MELK in tumors have focused on
cell growth and apoptosis. The results of the present study
suggested that MELK inhibition decreased cell proliferation and
colony formation in ESCC cells, consistent with previous studies
(16,22,48).
However, the cell cycle and apoptosis were not found to be
significantly influenced by MELK inhibition in ESCC cells. Of note,
the cell migration and invasion abilities were inhibited in
MELK-knockdown ESCC cells. The metastasis of ESCC cells was also
reduced by MELK inhibition in vivo. Collectively, the
results demonstrated that MELK is an important oncogenic factor and
plays critical roles in the cell growth and metastasis of ESCC.
The migration and invasion of cancer cells involve
multiple changes in tumor cells (49). EMT is considered one of the major
mechanisms involved in solid tumor metastasis (25). It has been reported that EMT
progression is accompanied by several crucial changes of epithelial
cells, including loss of epithelial adherence, tight junction
proteins, loss of cell polarity, and acquisition of a mesenchymal
phenotype, leading to invasive and migratory behaviors (7,33).
In the present study, the expression levels of EMT-related proteins
were examined and it was found that the epithelial markers
E-cadherin and ZO-1 were upregulated, while mesenchymal markers
N-cadherin, ZEB1, Snail and Slug were downregulated by MELK
inhibition in ESCC cells. In addition, MMPs, which are capable of
degrading the ECM, were upregulated. Furthermore, GSEA analysis
confirmed the association between MELK and aggressive
characteristics in ESCC. Collectively, these results revealed that
MELK is an important factor for the support of EMT and
aggressiveness in ESCC cells.
To date, the precise molecular mechanisms of MELK
that promote ESCC cell EMT and aggressive potential remain unknown.
MELK reportedly regulates tumor progression by several signaling
pathways (16,50,51).
The NF-κB pathway is a major tumor-promoting and EMT-related
pathway (36,52). A previous study showed that MELK
regulates the NF-κB pathway by phosphorylating SQSTM1/p62 and
promoting melanoma growth (12).
The results of the present study revealed that MELK inhibition
decreased the expression of NF-κB transcriptional targets, and
reduced translocation to the nucleus of p65, p50, and p52. The
expression of p62 was also decreased in MELK-knockdown cells, which
was consistent with a previous study (12). The activation of the NF-κB complex
relies on the IκB protein being phosphorylated by the IKK complex,
which leads to IκB ubiquitination and subsequent degradation
(27). In the present study it was
determined that the phosphorylation of IκB and IKK were decreased
in MELK-knockdown ESCC cells. The findings of the present study
were also confirmed by treatment of ESCC cells with the MELK
inhibitor OTSSP167 (21,22,40).
In addition, co-immunoprecipitation experiments showed that MELK
interacted with IKK. The inhibitory effect of MELK knockdown on
migration ability and NF-κB signaling could be markedly rescued by
overexpression of IKKβ. Therefore, it is proposed that MELK
activates the NF-κB pathway by phosphorylating IKKβ. It was
determined that MELK interacts with IKK, upstream of IκB, and
regulates the expression of p-IκB. Although MELK may interact with
p-IκB, it is considered that the interaction between MELK and IKK
is more critical for its molecular function. A previous study
claimed that MELK enhanced tumorigenesis, migration, invasion and
metastasis of ESCC cells via activation of FOXM1 signaling pathway
(23). FOXM1 promoter region
contains a functional NF-κB element and is transcriptionally
activated upon NF-κB binding in chronic myelogenous leukemia cells
(53). There are also FOX binding
motifs within the FOXM1 promoter, whose activity is markedly
induced after overexpression of p65 (38). In the present study, ectopic
expression of IKKβ markedly rescued FOXM1 expression in
MELK-silenced ESCC cells, which indicated that FOXM1 is a
downstream effector of IKKβ function. Collectively, these results
identified MELK as a regulator of the NF-κB pathway and
demonstrated that MELK at least partly promotes ESCC metastasis by
activating this pathway.
In summary, the results of the present study
revealed that MELK regulated the activation of the NF-κB pathway
via its inhibitor IKK. In addition, MELK promoted tumor metastasis
of ESCC and predicted a poor prognosis in patients with ESCC.
Therefore, MELK may be a potential focus of future therapeutic
options for ESCC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LW and XZ conceived and designed the study. JY, WD,
YZ, HL, BG, ZQ and PL performed the experiments and acquired the
data. JY and WD performed data analysis and drafted the manuscript.
JY and LW revised the manuscript for important intellectual
content. JY and WD confirmed the authenticity of all the raw data.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All patients and healthy donors signed the written
informed consents. The collection and use of tissue samples were
approved by the Ethics Committee of the Meizhou People's Hospital
(Meizhou, China). The animal experiments were reviewed and approved
by the Animal Care and Experimental Committee of Jinan University
(approval no. IACUC-2019021 1-04; Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Guangdong Province (grant nos. 2017A030313117 and
2018A030313301), the Guangzhou Science Technology and Innovation
Commission (grant no. 201804010075), and the China Postdoctoral
Science Foundation (grant no. 2019M663304).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen MF, Yang YH, Lai CH, Chen PC and Chen
WC: Outcome of patients with esophageal cancer: A nationwide
analysis. Ann Surg Oncol. 20:3023–3030. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeng H, Zheng R, Guo Y, Zhang S, Zou X,
Wang N, Zhang L, Tang J, Chen J, Wei K, et al: Cancer survival in
China, 2003-2005: A population-based study. Int J Cancer.
136:1921–1930. 2015. View Article : Google Scholar
|
|
4
|
Zeng H, Chen W, Zheng R, Zhang S, Ji JS,
Zou X, Xia C, Sun K, Yang Z, Li H, et al: Changing cancer survival
in China during 2003-15: A pooled analysis of 17 population-based
cancer registries. Lancet Glob Health. 6:e555–e567. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pennathur A, Farkas A, Krasinskas AM,
Ferson PF, Gooding WE, Gibson MK, Schuchert MJ, Landreneau RJ and
Luketich JD: Esophagectomy for T1 esophageal cancer: Outcomes in
100 patients and implications for endoscopic therapy. Ann Thorac
Surg. 87:1048–1054. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rustgi A and El-Serag HB: Esophageal
carcinoma. N Engl J Med. 372:1472–1473. 2015.PubMed/NCBI
|
|
7
|
Diepenbruck M and Christofori G:
Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no,
maybe? Curr Opin Cell Biol. 43:7–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heyer BS, Warsowe J, Solter D, Knowles BB
and Ackerman SL: New member of the Snf1/AMPK kinase family, Melk,
is expressed in the mouse egg and preimplantation embryo. Mol
Reprod Dev. 47:148–156. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakano I, Paucar AA, Bajpai R, Dougherty
JD, Zewail A, Kelly TK, Kim KJ, Ou J, Groszer M, Imura T, et al:
Maternal embryonic leucine zipper kinase (MELK) regulates
multipotent neural progenitor proliferation. J Cell Biol.
170:413–427. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Y, Begley M, Li Q, Huang HT, Lako A,
Eck MJ, Gray NS, Mitchison TJ, Cantley LC and Zhao JJ: Mitotic
MELK-eIF4B signaling controls protein synthesis and tumor cell
survival. Proc Natl Acad Sci USA. 113:9810–9815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Du T, Qu Y, Li J, Li H, Su L, Zhou Q, Yan
M, Li C, Zhu Z and Liu B: Maternal embryonic leucine zipper kinase
enhances gastric cancer progression via the FAK/Paxillin pathway.
Mol Cancer. 13:1002014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Janostiak R, Rauniyar N, Lam TT, Ou J, Zhu
LJ, Green MR and Wajapeyee N: MELK promotes melanoma growth by
stimulating the NF-κB pathway. Cell Rep. 21:2829–2841. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Lee YM, Baitsch L, Huang A, Xiang
Y, Tong H, Lako A, Von T, Choi C, Lim E, et al: MELK is an
oncogenic kinase essential for mitotic progression in basal-like
breast cancer cells. Elife. 3:e017632014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuner R, Falth M, Pressinotti NC, Brase
JC, Puig SB, Metzger J, Gade S, Schäfer G, Bartsch G, Steiner E, et
al: The maternal embryonic leucine zipper kinase (MELK) is
upregulated in high-grade prostate cancer. J Mol Med (Berl).
91:237–248. 2013. View Article : Google Scholar
|
|
15
|
Kig C, Beullens M, Beke L, Van Eynde A,
Linders JT, Brehmer D and Bollen M: Maternal embryonic leucine
zipper kinase (MELK) reduces replication stress in glioblastoma
cells. J Biol Chem. 292:127862017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Q, Ge Q, Zhou Y, Yang B, Yang Q, Jiang
S, Jiang R, Ai Z, Zhang Z and Teng Y: MELK promotes endometrial
carcinoma progression via activating mTOR signaling pathway.
EBioMedicine. 51:1026092020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pitner MK, Taliaferro JM, Dalby KN and
Bartholomeusz C: MELK: A potential novel therapeutic target for
TNBC and other aggressive malignancies. Expert Opin Ther Targets.
21:849–859. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guan S, Lu J, Zhao Y, Yu Y, Li H, Chen Z,
Shi Z, Liang H, Wang M, Guo K, et al: MELK is a novel therapeutic
target in high-risk neuroblastoma. Oncotarget. 9:2591–2602. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ikeda Y, Sato S, Yabuno A, Shintani D,
Ogasawara A, Miwa M, Zewde M, Miyamoto T, Fujiwara K, Nakamura Y
and Hasegawa K: High expression of maternal embryonic
leucine-zipper kinase (MELK) impacts clinical outcomes in patients
with ovarian cancer and its inhibition suppresses ovarian cancer
cells growth ex vivo. J Gynecol Oncol. 31:e932020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu H, Sun Q, Sun Y, Zhang J, Yuan H, Pang
S, Qi X, Wang H, Zhang M, Zhang H, et al: MELK and EZH2 cooperate
to regulate medulloblastoma cancer stem-like cell proliferation and
differentiation. Mol Cancer Res. 15:1275–1286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen S, Zhou Q, Guo Z, Wang Y, Wang L, Liu
X, Lu M, Ju L, Xiao Y and Wang X: Inhibition of MELK produces
potential anti-tumour effects in bladder cancer by inducing G1/S
cell cycle arrest via the ATM/CHK2/p53 pathway. J Cell Mol Med.
24:1804–1821. 2020. View Article : Google Scholar
|
|
22
|
Kohler RS, Kettelhack H,
Knipprath-Meszaros AM, Fedier A, Schoetzau A, Jacob F and
Heinzelmann-Schwarz V: MELK expression in ovarian cancer correlates
with poor outcome and its inhibition by OTSSP167 abrogates
proliferation and viability of ovarian cancer cells. Gynecol Oncol.
145:159–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen L, Wei Q, Bi S and Xie S: Maternal
embryonic leucine zipper kinase promotes tumor growth and
metastasis via stimulating FOXM1 signaling in esophageal squamous
cell carcinoma. Front Oncol. 10:102020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dongre A and Weinberg RA: New insights
into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. Nat Rev Mol Cell Biol. 20:69–84. 2019.
View Article : Google Scholar
|
|
25
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar
|
|
26
|
Yamini B: NF-κB, mesenchymal
differentiation and glioblastoma. Cells. 7:1252018. View Article : Google Scholar
|
|
27
|
Zhang Q, Lenardo MJ and Baltimore D: 30
years of NF-κB: A blossoming of relevance to human pathobiology.
Cell. 168:37–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greene FL, Page DL, Fleming ID, Fritz AG,
Balch CM and Haller DG: AJCC cancer staging manual. Springer; New
York, NY: pp. 91–98. 2002
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Hu N, Clifford RJ, Yang HH, Wang C,
Goldstein AM, Ding T, Taylor PR and Lee MP: Genome wide analysis of
DNA copy number neutral loss of heterozygosity (CNNLOH) and its
relation to gene expression in esophageal squamous cell carcinoma.
BMC Genomics. 11:5762010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su H, Hu N, Yang HH, Wang C, Takikita M,
Wang QH, Giffen C, Clifford R, Hewitt SM, Shou JZ, et al: Global
gene expression profiling and validation in esophageal squamous
cell carcinoma and its association with clinical phenotypes. Clin
Cancer Res. 17:2955–2966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rickman DS, Millon R, De Reynies A, Thomas
E, Wasylyk C, Muller D, Abecassis J and Wasylyk B: Prediction of
future metastasis and molecular characterization of head and neck
squamous-cell carcinoma based on transcriptome and genome analysis
by microarrays. Oncogene. 27:6607–6622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarrio D, Rodriguez-Pinilla SM, Hardisson
D, Cano A, Moreno-Bueno G and Palacios J: Epithelial-mesenchymal
transition in breast cancer relates to the basal-like phenotype.
Cancer Res. 68:989–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liberzon A, Birger C, Thorvaldsdottir H,
Ghandi M, Mesirov JP and Tamayo P: The molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar
|
|
35
|
Shimada Y, Imamura M, Wagata T, Yamaguchi
N and Tobe T: Characterization of 21 newly established esophageal
cancer cell lines. Cancer. 69:277–284. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pires BR, Mencalha AL, Ferreira GM, de
Souza WF, Morgado-Díaz JA, Maia AM, Corrêa S and Abdelhay ESF:
NF-kappaB is involved in the regulation of EMT genes in breast
cancer cells. PLoS One. 12:e01696222017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
38
|
Li Y, Lu L, Tu J, Zhang J, Xiong T, Fan W,
Wang J, Li M, Chen Y, Steggerda J, et al: Reciprocal regulation
between forkhead box M1/NF-κB and methionine adenosyltransferase 1A
drives liver cancer. Hepatology. 72:1682–1700. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Zhou X, Li Y, Xu Y, Lu K, Li P
and Wang X: Inhibition of maternal embryonic leucine zipper kinase
with OTSSP167 displays potent anti-leukemic effects in chronic
lymphocytic leukemia. Oncogene. 37:5520–5533. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cho YS, Kang Y, Kim K, Cha YJ and Cho HS:
The crystal structure of MPK38 in complex with OTSSP167, an orally
administrative MELK selective inhibitor. Biochem Biophys Res
Commun. 447:7–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li G, Yang M, Zuo L and Wang MX: MELK as a
potential target to control cell proliferation in triple-negative
breast cancer MDA-MB-231 cells. Oncol Lett. 15:9934–9940.
2018.PubMed/NCBI
|
|
42
|
Li Y, Li Y, Chen Y, Xie Q, Dong N, Gao Y,
Deng H, Lu C and Wang S: Correction to: MicroRNA-214-3p inhibits
proliferation and cell cycle progression by targeting MELK in
hepatocellular carcinoma and correlates cancer prognosis. Cancer
Cell Int. 18:552018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bollu LR, Shepherd J, Zhao D, Ma Y,
Tahaney W, Speers C, Mazumdar A, Mills GB and Brown PH: Mutant P53
induces MELK expression by release of wild-type P53-dependent
suppression of FOXM1. NPJ Breast Cancer. 6:22020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oliva M, Munoz-Aguirre M, Kim-Hellmuth S,
Wucher V, Gewirtz ADH, Cotter DJ, Parsana P, Kasela S, Balliu B,
Viñuela A, et al: The impact of sex on gene expression across human
tissues. Science. 369:eaba30662020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ren L, Deng B, Saloura V, Park JH and
Nakamura Y: MELK inhibition targets cancer stem cells through
downregulation of SOX2 expression in head and neck cancer cells.
Oncol Rep. 41:2540–2548. 2019.PubMed/NCBI
|
|
46
|
Wang K, Zhu X, Yao Y, Yang M, Zhou F and
Zhu L: Corosolic acid induces cell cycle arrest and cell apoptosis
in human retinoblastoma Y-79 cells via disruption of MELK-FoxM1
signaling. Oncol Rep. 39:2777–2786. 2018.PubMed/NCBI
|
|
47
|
Speers C, Zhao SG, Kothari V, Santola A,
Liu M, Wilder-Romans K, Evans J, Batra N, Bartelink H, Hayes DF, et
al: Maternal embryonic leucine zipper kinase (MELK) as a novel
mediator and biomarker of radioresistance in human breast cancer.
Clin Cancer Res. 22:5864–5875. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tian JH, Mu LJ, Wang MY, Zeng J, Long QZ,
Guan B, Wang W, Jiang YM, Bai XJ and Du YF: BUB1B promotes
proliferation of prostate cancer via transcriptional regulation of
MELK. Anticancer Agents Med Chem. 20:1140–1146. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mierke CT: The matrix environmental and
cell mechanical properties regulate cell migration and contribute
to the invasive phenotype of cancer cells. Rep Prog Phys.
82:0646022019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Seong HA, Manoharan R and Ha H: Zinc
finger protein ZPR9 functions as an activator of AMPK-related
serine/threonine kinase MPK38/MELK involved in ASK1/TGF-beta/p53
signaling pathways. Sci Rep. 7:425022017. View Article : Google Scholar
|
|
51
|
Gu C, Banasavadi-Siddegowda YK, Joshi K,
Nakamura Y, Kurt H, Gupta S and Nakano I: Tumor-specific activation
of the C-JUN/MELK pathway regulates glioma stem cell growth in a
p53-dependent manner. Stem Cells. 31:870–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ren D, Yang Q, Dai Y, Guo W, Du H, Song L
and Peng X: Oncogenic miR-210-3p promotes prostate cancer cell EMT
and bone metastasis via NF-κB signaling pathway. Mol Cancer.
16:1172017. View Article : Google Scholar
|
|
53
|
Jin B, Wang C, Li J, Du X, Ding K and Pan
J: Anthelmintic niclosamide disrupts the interplay of p65 and
FOXM1/β-catenin and eradicates leukemia stem cells in chronic
myelogenous leukemia. Clin Cancer Res. 23:789–803. 2017. View Article : Google Scholar
|