Introduction
Tuberous sclerosis complex (TSC) is a rare autosomal
dominant disease, caused by the heterozygous germline mutations in
TSC1 or TSC2 genes on chromosome 9 and 16, respectively. According
to the research, the overall mutation detection rate among patients
with TSC is 85-90% and 10-15% of patients were not identified
(1). Regarding the pathologic
mechanism for non-mutation patients with TSC, numerous theories
have been put forward, including somatic mosaicism or intronic
splicing variants affecting TSC1 or TSC2 (2). TSC affects multiple organs throughout
the body, although two-thirds of all cases are sporadic (1). According to research, the incidence
within newborn children is 1:6,000-10,000. Without disentangling
differences between ethnic groups or gender preferences (3), there are currently two million
individuals living with TSC worldwide (2). Treatments for TSC require
interdisciplinary cooperation and coordination from specialists
within various fields, as TSC is characterized by hamartomas within
multiple organs, including the brain, lungs, skin, kidneys, heart
and eyes (3).
Angiomyolipomas are the most common type of
TSC-related renal lesions, affecting 55-75% of all cases of TSC.
The abnormal vasculature (including aneurysms) may result in
spontaneous life-threatening bleeding, which is a leading cause of
early death in adult patients with TSC (4-6). The
pathological loss of heterozygosity resulting from tumor-suppressor
mutation of TSC genes disables the inhibitory role in controlling
mammalian target of rapamycin (mTOR). The mTOR pathway is a crucial
signaling pathway in regulating cellular growth and metabolization,
and overactive mTOR therefore leads to the formation of hamartoma
(3,7). Accordingly, mTOR inhibitors such as
everolimus have been approved for the treatment of TSC-associated
renal angiomyolipoma (TSC-RAML) and subependymal giant cell
astrocytoma (SEGA) (7). The
Exist-2 study (NCT00790400) is a double-blind, placebo-controlled,
phase III trial which confirmed the efficacy of everolimus in
reducing renal angiomyolipoma volume with an acceptable safety
profile (8). In addition, the
Exist-2 trial indicated that the plasma levels of VEGF-D diminished
after everolimus treatment, which correlated with the TSC-RAML
volume, indicating that VEGF-D may be used as a prognostic marker
(8).
Unlike sporadic angiomyolipoma (S-AML), TSC-RAML has
unique pathologic behaviors, including bilateral, early onset in
multiple sites (9). Therefore,
questions remain regarding how to differentiate TSC-RAML from S-AML
as a diagnostic marker, as well as the functional mechanism.
Undoubtedly, its low prevalence limits the availability of the
precious samples and feasibility of gaining insight into the
mechanisms involved. The aim of the present study was therefore to
identify potentially useful diagnostic markers and to assess their
relationship with the tumor and whole-body burden, as well as to
explore the tumor microenvironment.
Patients and methods
Patients and samples
A total of 25 consecutive patients with TSC
diagnosed according to the 2012 diagnostic criteria from the
International Tuberous Sclerosis Complex Consensus Conference
(10) at Peking Union Medical
College Hospital (Beijing, China) between November 2016 and
November 2017 were enrolled. All participants were ≥18 years old
and had at least 1 TSC-RAML with a size of >3 cm. All
participants were subjected to next-generation sequencing to
identify TSC1 or TSC2 mutation types. Blood samples from patients
with TSC at baseline and after 3-6 months of being administered
everolimus were collected. Those without plasma samples in our
sample bank or concurrent malignant tumors or metabolomic diseases,
such as diabetes or hyperlipidemia, were excluded.
Maximum renal tumor volumes and pulmonary
lymphangioleiomyomatosis (LAM) status were assessed independently
by radiologists and respiratory physicians using computed
tomography in a clinical setting. In addition, another 25 patients
with renal cyst and 25 patients with S-AML were simultaneously
enrolled to establish a control group. To further explore the
tissue proteomics and transcriptome, 15 paired TSC-RAML tumor and
non-tumor normal tissues (NATs) were collected from the surgery
room of Peking Union Medical College Hospital (Beijing, China)
between November 2016 and November 2021. After sample selection and
quality control, 8 paired TSC-RAML tumor and NATs were enrolled
into the following proteomic experiments, as well as 10 tumor and 8
NATs into the RNA sequencing experiments. In addition, proteomic
experiments were also conducted on another cohort of 8 paired S-AML
tumor and NATs during the same time period to establish control
groups.
The present study was approved by the Institutional
Review Board in Peking Union Medical College Hospital and the
Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences (Beijing, China; approval no. KS2020127). Informed consent
was requested and obtained from each patient prior to being
accepted to participate.
Genotype and phenotype
TSC gene mutations were further divided into five
types, namely frameshift mutation, missense mutation, nonsense
mutation, other mutations e.g. splicing abnormality and gene
deletion, and no mutation. TSC-RAML was graded according to the
Renal Angiomyolipoma Staging Criteria Proposed by the University
Medical Center Utrecht: Stage 1 to 6 (11). The LAM status was determined by
radiologists and respiratory specialists in this field and graded
as either 0 (without LAM) or 1 (with LAM). The whole-body disease
burden was calculated for each participant as the sum of AML
grading and LAM status, with scores ranging from 1 to 7.
Plasma and tissue proteomics according to
ultra-performance liquid chromatography mass spectrometry (UPLC-MS)
Plasma sample collection and pre-experiment processing
For blood samples, 4 ml EDTA tubes, containing whole
blood, were collected in the morning between 07:00-09:00 am after
overnight fasting to reduce the effect of the diet. Plasma and
peripheral blood mononuclear cells were isolated through density
gradient centrifugation over Ficoll-Hypaque, at room temperature,
within 1 h of initial collection. The plasma layer was extracted
and then stored at -80°C until the formal experiment.
High Select™ Top14 Abundant Protein Depletion Mini
Spin Columns (Thermo Fisher Scientific, Inc.) were applied to
eliminate extraneous proteins in the plasma following the
manufacturer's instructions and 30-µl samples were obtained
after the above process. From each sample, 10 µl were
removed to measure protein concentrations using the Pierce™ BCA
assay protein assay kit (cat. no. 23225; Thermo Fisher Scientific,
Inc.).
Tissue sample preparation for
proteomics
Tumor tissues and NATs were removed in the operation
room and were transferred into liquid nitrogen within 0.5 h and
preserved until the experiments took place. Approximately 25-120 mg
tissue was homogenized separately in an appropriate volume of lysis
buffer [i.e. 2% SDS, 20 mM Tris, cocktail (1:100 dilution), DNAse
(1:100 dilution), RNAse (1:1,000 dilution)] by repeated vortexing.
The protein concentration was determined using the Pierce™ BCA
assay protein assay kit.
Protein digestion
Dithiothreitol (20 mM for every 100 mg of protein
within each sample) was applied for protein reduction for 5 min at
95°C and subsequently alkylated with 50 mM iodoacetamide for 45 min
at room temperature in the dark. The filter-aided sample
preparation technique was performed to digest proteins and 30 kDa
filter devices (Pall Filtersystems GmbH) were used. Trypsin
(Trypsin Gold; mass spec grade; Promega Corporation) was added
(enzyme to protein ratio, 1:50) and incubated at 37°C overnight.
After centrifugation at 14,000 × g (4°C, 10 min), ~30 µl
liquid was retrieved from each sample.
Quality control samples were taken from randomly
selected representative samples and injected with formal samples.
All of the samples were loaded on the autosamplers with a mixture
of indexed retention time (iRT).
Electron spray ionization (ESI)-LC-MS/MS
for proteome library generation
Pooled peptide samples of each group were separated
by high-pH reverse phase LC columns (4.6×250 mm, C18, 3 µm;
Waters Corporation) and then loaded onto the column in buffer A1
(H2O; pH 10) using a Waters H-class UPLC (Waters
Corporation). The elution gradient was 5-30% buffer B1 [90%
acetonitrile (ACN); pH 10; flow rate, 1 ml/min] over 30 min.
The eluted peptides were collected at one fraction
per minute. After lyophilization using a CENTRIVAP (ChristRVC 2-25
CD plus; Martin Christ GmbH), the 30 fractions were resuspended in
0.1% formic acid and then concatenated into 10 fractions by
combining fractions 1, 11, 21, etc. To generate the spectral
library, the fractions from RPLC were analyzed in data-dependent
acquisition (DDA) mode using an EXPLORIS 480 (Thermo Fisher
Scientific, Inc.). The parameters were set as follows: the MS was
recorded at 350-1,500 m/z at a resolution of 60,000 m/z; the
maximum injection time was 50 msec, the auto gain control (AGC) was
1×106, and the cycle time was 3 sec. MS/MS scans were
performed at a resolution of 15,000 with an isolation window of 1.6
Da and high-collision dissociation (HCD) collision energy of 32%;
the AGC target was 50,000 and the maximum injection time was 30
msec.
ESI-LC-MS/MS for proteome
data-independent acquisition analysis
Digested peptides were dissolved in 0.1% formic acid
and separated on an RP C18 self-packing capillary LC column (75
µm x 150 mm, 3 µm; Dr Masch GmbH). The eluted
gradient was 5-30% buffer B2 (0.1% formic acid and 99.9% ACN; flow
rate, 0.3 µl/min) for 60 min. For MS acquisition, the
variable isolation window data-independent acquisition (DIA) method
with 38 windows was developed. The specific window lists were
constructed based on the DDA experiment of the pooled sample. The
full scan was set at a resolution of 120,000 over the m/z range of
400 to 900, followed by DIA scans with a resolution of 30,000; the
HCD collision energy was 32%, the AGC target was 1E6 and the
maximal injection time was 50 msec.
Spectral library generation
To generate a comprehensive spectral library, a
pooled sample from each group was processed. DDA data were
processed using Proteome Discoverer software (Thermo Fisher
Scientific, Inc.) and searched against the human UniProt database
(https://sparql.uniprot.org/) appended
with the iRT fusion protein sequence (Biognosys AG).
A maximum of two missed cleavages for trypsin were
used, cysteine carbamidomethylation was set as a fixed modification
and methionine oxidation deamination and +43 on Kn (Carbamyl) were
used as variable modifications. Parent and fragment ion mass
tolerances were set to 10 ppm and 0.02 Da, respectively. The
applied false discovery rate (FDR) cut-off was 0.01 at the protein
level. The results were then imported to Spectronaut Pulsar
software (Biognosys AG) to generate the spectral library.
In addition, DIA data were imported into Spectronaut
Pulsar software and searched against the human UniProt database to
generate the DIA library. The final library was generated by
combining the DDA and DIA libraries of all samples.
Data analysis
DIA-MS data were analyzed using Spectronaut Pulsar
(Biognosys AG) with default settings. All results were filtered
with a Q-value cutoff of 0.01 which corresponded to an FDR of 1%.
Proteins identified in >50% of the samples in each group were
retained for further analysis. Missing values were imputed based on
the k-nearest neighbor method.
Raw proteomics data were transformed with log2 and
then centralized. Supervised orthogonal partial least squares
discriminant analysis (O2PLS-DA) was applied to view the
distribution tendency of all samples with SIMCA version 14.1
(Umetrics). The unpaired, two-sided t-test was implemented to
calculate the differentially expressed proteins with the software R
v4.1.1 (https://www.r-project.org/).
Bulk RNA sequencing
RNA quantification and
qualification
A total of 10 TSC-RAML tumor tissues and 8 paired
non-tumor normal tissues samples were resected from the operation
room and stored at −80°C until final analysis. Subsequently, total
RNA was isolated from the above tissue using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.). RNA integrity was assessed using
the RNA Nano 6000 Assay Kit from the Bioanalyzer 2100 system
(Agilent Technologies, Inc.). The following RNA-seq experiments
were performed by Novogene (Beijing, China).
Library preparation for transcriptome
sequencing
NEBNext® Ultra™ RNA Library Prep Kit for
Illumina® (New England Biolabs, Inc.) was used to
construct sequencing libraries following the manufacturer's
recommendations. Total RNA was used as input material for RNA
sample preparations. mRNA was purified using poly-T oligo-attached
magnetic beads. Fragmentation was performed using divalent cations
at an elevated temperature in First Strand Synthesis Reaction
Buffer (5X). First strand cDNA was synthesized using random hexamer
primer and M-MuLV Reverse Transcriptase, then RNaseH was used to
degrade the RNA. Second strand cDNA synthesis was subsequently
performed using DNA Polymerase I and dNTP. Remanent overhangs were
converted into blunt ends through exonuclease/polymerase
activities. After adenylation of 3' ends of DNA fragments, adaptors
with hairpin loop structures were ligated to prepare for
hybridization. In order to select cDNA fragments of preferentially
370-420 bp in length, the library fragments were purified with an
AMPure XP system (Beckman Coulter, Inc.). Subsequently, PCR was
performed using Phusion High-Fidelity DNA polymerase, Universal PCR
primers and an Index (X) Primer according to a previously published
protocol (12). PCR products were
purified (AMPure XP system; Beckman Coulter, Inc.) and the library
quality was assessed on the Agilent Bioanalyzer 2100 system
(Agilent Technologies, Inc.).
Clustering and sequencing
This experiment was performed at the Novogene
Experimental Department. The clustering of the index-coded samples
was performed on a cBot Cluster Generation System using TruSeq PE
Cluster Kit v3-cBot-HS (Illumia, Inc.) according to the
manufacturer's instructions. After cluster generation, the library
preparations were sequenced on an Illumina Novaseq platform and 150
bp paired-end reads were generated.
Data processing and differential
analysis
Raw data (raw reads) in the fastq format were first
processed through in-house perl scripts. Reads mapping to the
reference genome (GRCh38) and gene model annotation files were
downloaded from the genome website directly (http://www.ensembl.org/Homo_sapiens/Info/Index).
Paired-end clean reads were aligned to the reference genome using
Hisat2 v2.0.5 (http://daehwankimlab.github.io/hisat2/). For the
quantification of gene expression levels, featureCounts v1.5.0-p3
(http://subread.sourceforge.net/) was
used to count the number of reads mapped to each gene.
Subsequently, the FPKM of each gene was calculated based on the
length of the gene and the reads count was mapped to this gene.
Differential expression analysis was performed using
the DESeq2 R package v1.20.0. The resulting P-values were adjusted
using the Benjamini-Hochberg approach for controlling the FDR.
Genes with an adjusted P<0.05 were assigned as being
differentially expressed.
Single-cell RNA sequencing
The single-cell RNA sequencing data (10X data) of a
patient with TSC1-mutated AML were downloaded from the Gene
Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/; GEO accession no.
GSM4035469) and used to simulate the microenvironment of TSC-RAML,
which has been analyzed and presented in the original study by Guo
et al (13). In addition,
the raw experimental procedures are available in the original
article and the following steps describe the quality control and
analytic process.
The quality control and secondary analysis were
conducted with R v4.1.1 and the Seurate package (14,15).
Cells with a mitochondrial gene percentage of >30% and with
<200 genes and >2,500 genes were filtered. After this
selection, 1,675 high-quality cells were obtained for further
analysis. After normalization scaling of the raw data, all highly
variable genes were included into the downstream primary component
analysis. Elbow-plot analysis was used to identify significant
principal components. With the resolution of 0.5, all the cells
were classified into 10 groups. The differentially expressed genes
(DEGs) within groups were found with the FindAllMarkers function
and the cells were firstly annotated by the R package 'SingleR'
(16) automatically and then
annotated manually through searching published articles. The top
200 ranked genes from each cell were then selected to perform gene
ontology analysis with the R package 'clusterProfiler' (17,18).
The R package 'monocle' (version 2.24.1) was used to construct the
pseudo-time trajectories (19).
Functional analysis
The Gene Ontology (GO) functional enrichment was
performed with the R package 'clusterProfiler' (17,18).
The characteristic modules of each subgroup were completed by the
weighted gene correlation network analysis '(WGCNA)' package
(20,21) and the correlation between modules
and clinical information was assessed by Pearson correlation
analysis. The Gene Set Enrichment Analysis (GSEA) application
(version 4.1.0) was applied to perform GSEA hallmark analysis. The
cell composition within the tumor microenvironment was calculated
with the R package 'MCPcounter' (22).
Statistical analysis
Unless specially mentioned above, all the data
analyses were performed and figures were generated using R (version
4.1.1) with required packages described in the sections above.
χ2 and Kruskal-Wallis tests were used to assess the
gender and age distribution within different subgroups. All of the
tests were two-sided and P≤0.05 was used as the threshold for
statistical significance.
Results
Baseline information of enrolled
patients
A total of 75 participants with 100 plasma samples
were divided into four subgroups, namely the pre-treatment TSC-RAML
(n=25), post-treatment TSC-RAML (n=25), S-AML (n=25) and renal cyst
(CY, n=25) groups. The demographics and clinical information of the
patients are summarized in Table
I. Among the 25 patients with TSC-RAML, 36% (n=9) were male and
the rates for S-AML and CY (renal cyst) were 20% (n=5) and 44%
(n=11), respectively (P=0.186). In terms of age, the subjects in
the TSC-RAML group were younger with a median age of 30 years old,
compared with 39 for S-AML and 45 for CY (P<0.001).
 | Table IBaseline information of all the
enrolled patients. |
Table I
Baseline information of all the
enrolled patients.
| Item | TSC-AML (n=25) | S-AML (n=25) | CY (n=25) |
|---|
| Sex | | | |
| Male | 9 | 5 | 11 |
| Female | 16 | 20 | 14 |
| Age, years | 30 (18-42) | 39 (15-54) | 45 (13-57) |
| Gene mutation
type | | | |
| TSC2 | 19 | - | - |
| None | 6 | - | - |
| AML grading | | | |
| 1 | 3 | - | - |
| 2 | 2 | - | - |
| 3 | 3 | - | - |
| 4 | 1 | - | - |
| 5 | 7 | - | - |
| 6 | 9 | - | - |
| LAM | | | |
| Yes | 10 | - | - |
| No | 15 | - | - |
| Whole body disease
burdena | 5 (1-7) | - | - |
In the genotype group (n=25), 76% (n=19) were
indicated to have TSC2 gene mutations, and among these, nonsense
mutations were the most frequent with 28% (n=7). This was followed
by frameshift mutations in 20% (n=5), other mutations (16%; n=4)
and then missense mutations (12%; n=3), as presented in Fig. S1A.
In the clinical phenotype group, most TSC-RAML
grades were 5 and 6, and were therefore considered severe,
suggesting a heavy renal tumor burden. Among the 25 participants,
10 had LAM (40%) and the median whole body disease burden was 5
(Table I).
Unique plasma proteomics of patients with
TSC-RAML distinguished from cases of S-AML and renal cyst
After censoring and filling missing values, 903
proteins were selected for further analysis. First, O2PLS-DA
analysis was performed with SIMCA (version 14.1) and a
three-dimensional distribution of all the samples was established.
TSC-RAML plasma proteomic profiling was distinguished from S-AML
and renal cyst (Fig. S1B). From
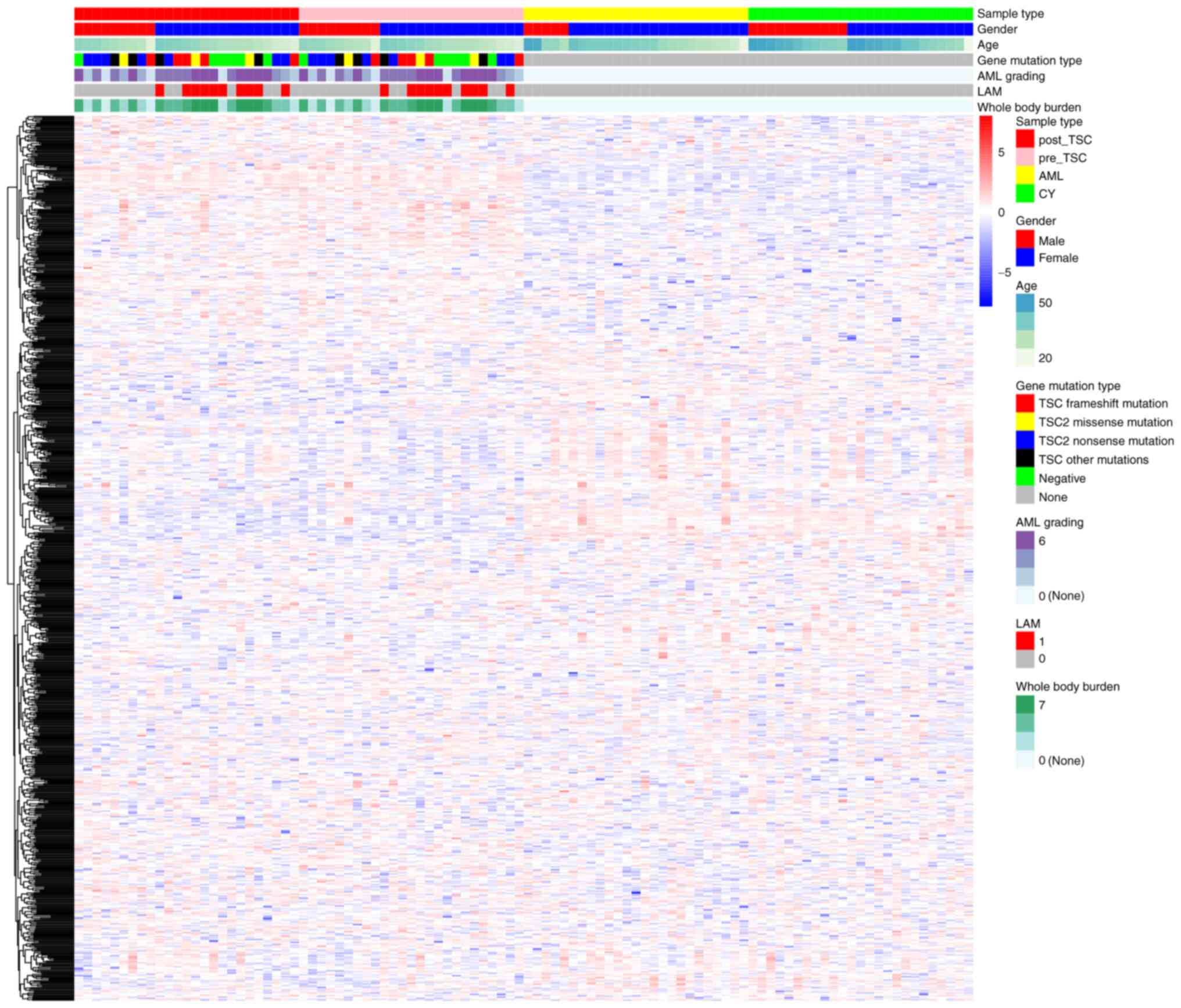
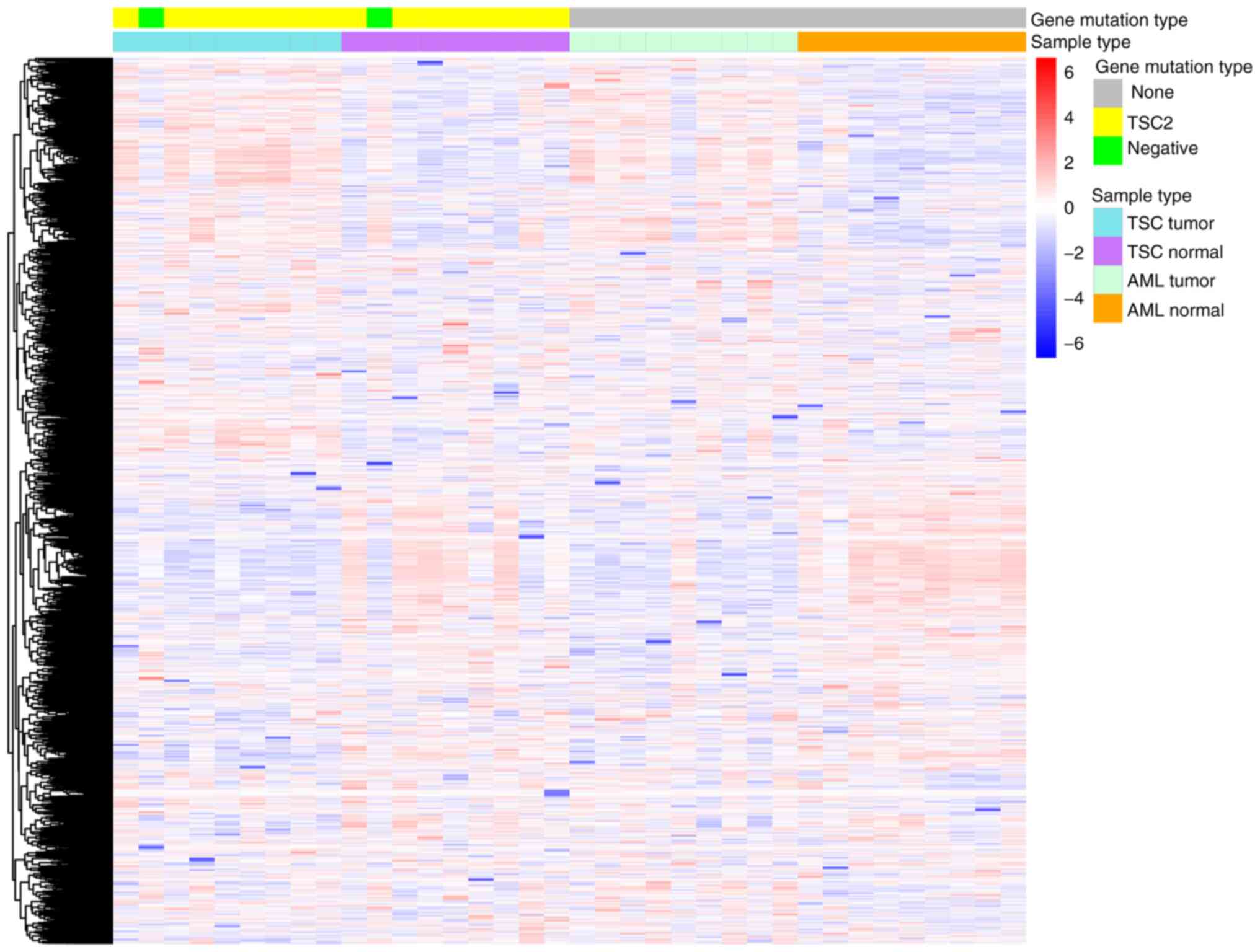
the heatmap, the characteristic plasma proteomics of TSC-RAML as
well as the similarity within everolimus treatment were directly
observed (Fig. 1).
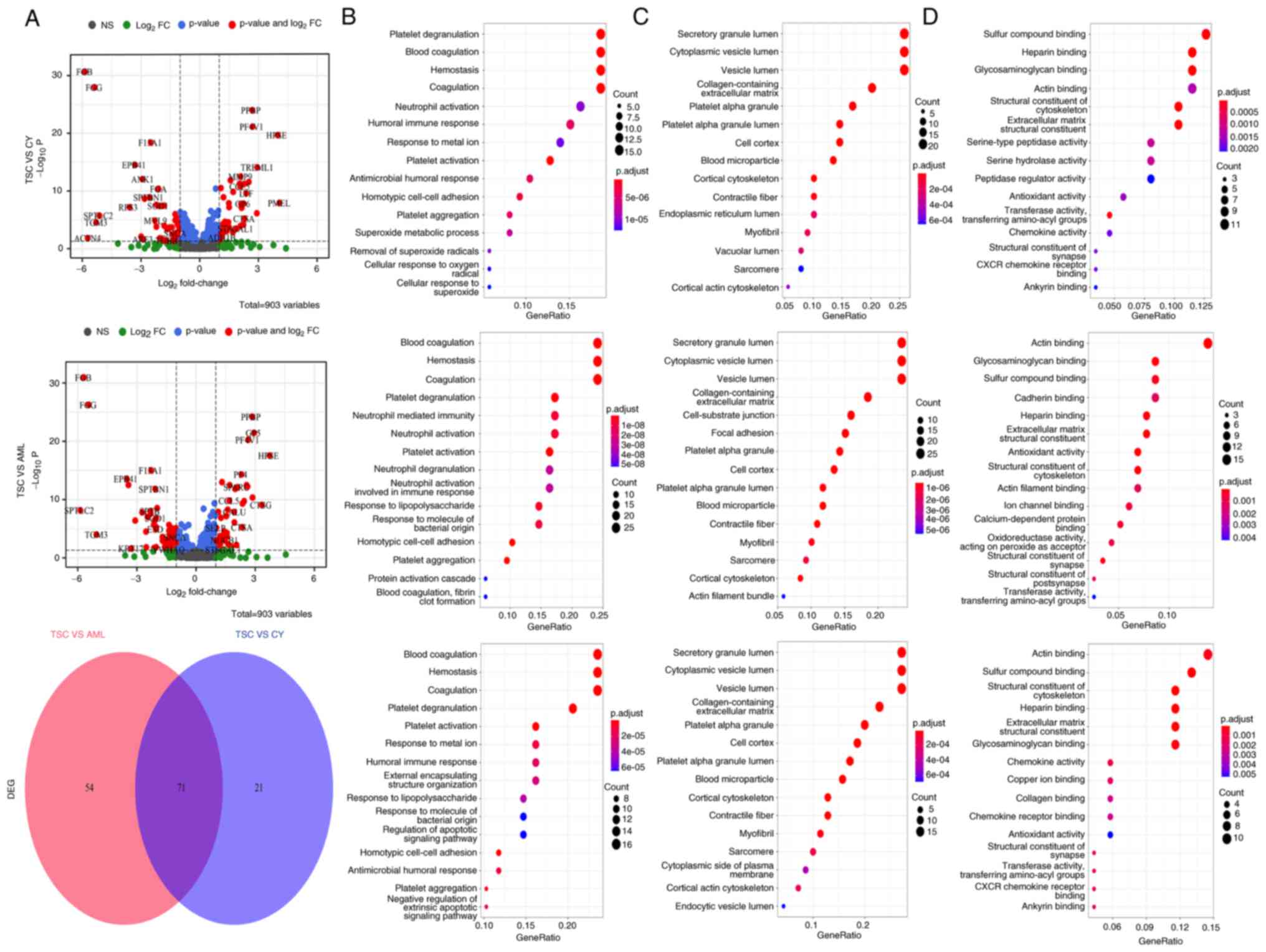
Differential analysis was applied to search for
TSC-RAML-specific plasma proteins, regardless of the everolimus
treatment status. In the volcano plots in Fig. 2A, it is apparent that numerous
proteins, such as the upregulated MMP9, C-C motif chemokine ligand
5 (CCL5), premelanosome protein (PMEL) and the downregulated FGB,
FGG, EPB41, appear to have differential roles, not only in TSC-RAML
as opposed to renal cysts, but also in TSC-RAML vs. S-AML. PMEL
(also known as HMB45) has already been regarded as a diagnostic
marker for angiomyolipoma and MMP9 has been indicated to be
elevated in TSC-related cortical tubes and subependymal giant cell
astrocytoma at both the protein and transcriptome level (23,24).
The Venn diagram revealed that there existed 71 proteins
demonstrating similar differential functions like PMEL and CCL5
(Fig. 2A, lower panel).
Proinflammatory chemokine CCL5, responsible for
recruiting immune cells such as monocytes and T cells to the site
of inflammation (25), is
significantly upregulated in TSC-RAML, suggesting a high
inflammatory status. The functional enrichment analysis of
differentially expressed proteins (DEPs) indicated that they were
mostly enriched in the platelet degranulation, blood coagulation,
hemostasis (category biological process), secretory granule lumen,
cytoplasmic vesicle lumen, vesicle lumen (category cellular
component) and actin binding, sulfur compound binding and
glycosaminoglycan binding (category molecular function). The DEPs
mainly participated in the process of blood cell function,
secretory lumen component and actin binding function (Fig. 2B-D).
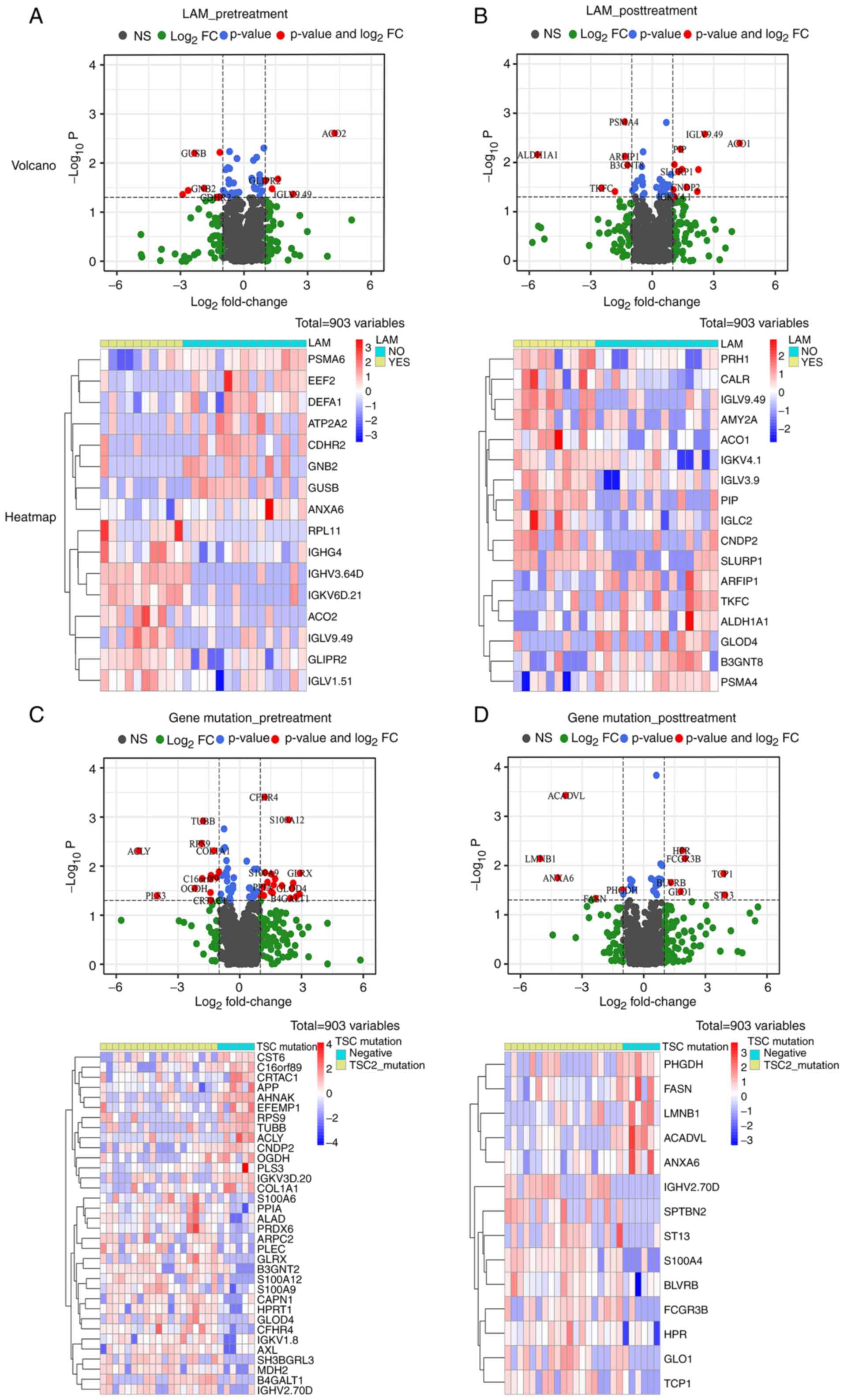
Relationship between plasma proteomics
with LAM and gene mutation status
Previous research has reported that serum proteins
such as VEGF-D may be prognostic markers not only for TSC-RAML
(8), but also for TSC-LAM
(26). Therefore, the impact of
LAM on plasma proteomics was explored in the present study. The
volcano plots and heatmaps in Fig. 3A
and B suggest that the existence of LAM does not appear to
influence the plasma proteomics because among the 903 proteins,
only 16 pretreatment and 17 post-treatment molecules were
statistically significant. It has been proposed that TSC2 gene
mutation and those producing premature termination codons may lead
to a severe phenotype (27,28).
For plasma proteomics, it appears that a TSC2 mutation prior to
treatment has a substantial influence on patients with 31 DEPs and
S100 family members, such as S100A6, -A9 and -A12, had a tendency
to be upregulated among patients with TSC2 mutation, which has been
proved to engage in the neutrophil and macrophage accumulation,
corresponding cytokine secretion and smooth muscle cell
proliferation (29-32). However, after receiving everolimus,
the number of DEPs was markedly reduced (Fig. 3C and D), indicating a reversible
pattern after treatment.
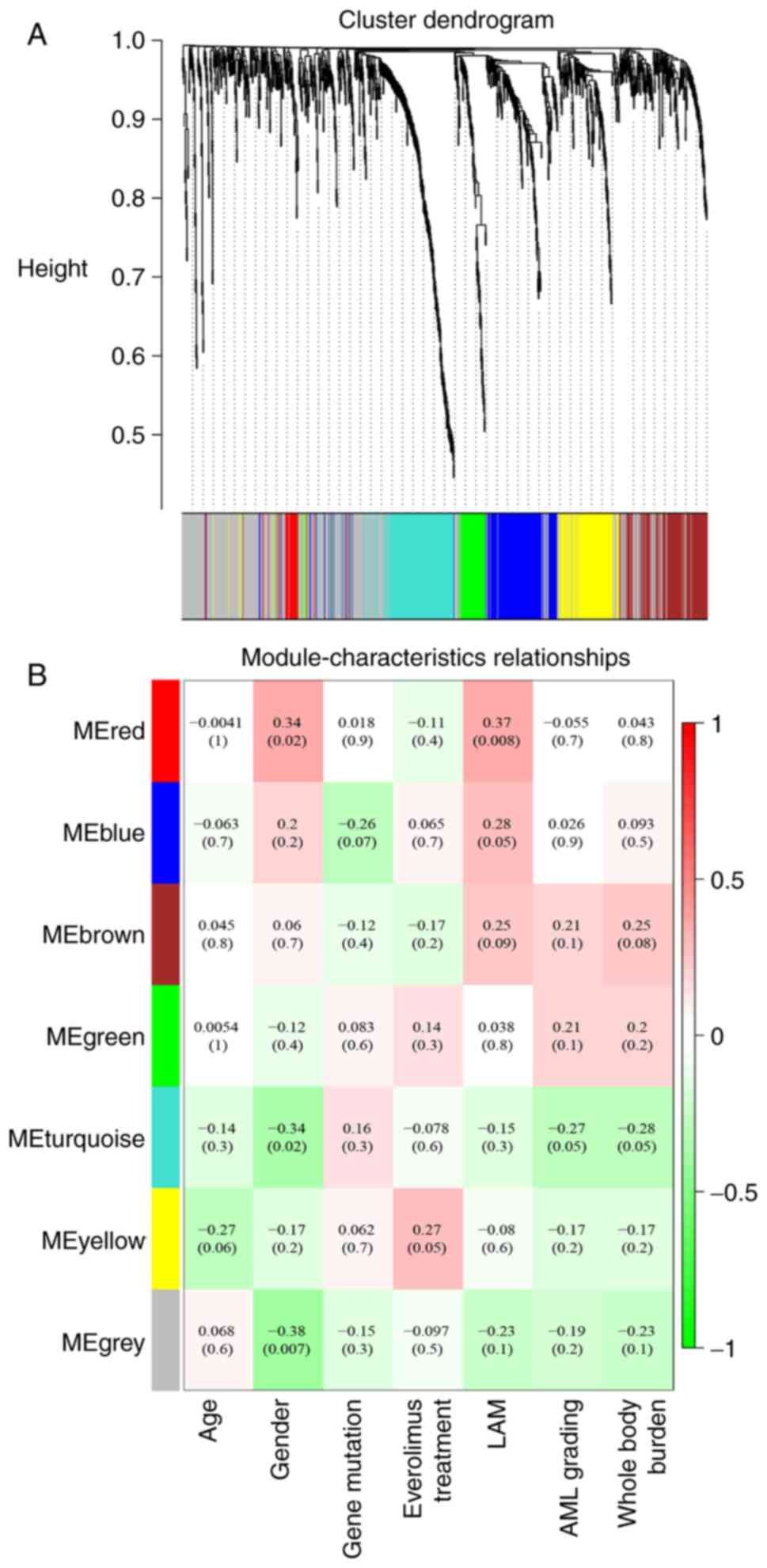
WGCNA analysis highlights relationship
between plasma proteomics and disease burden
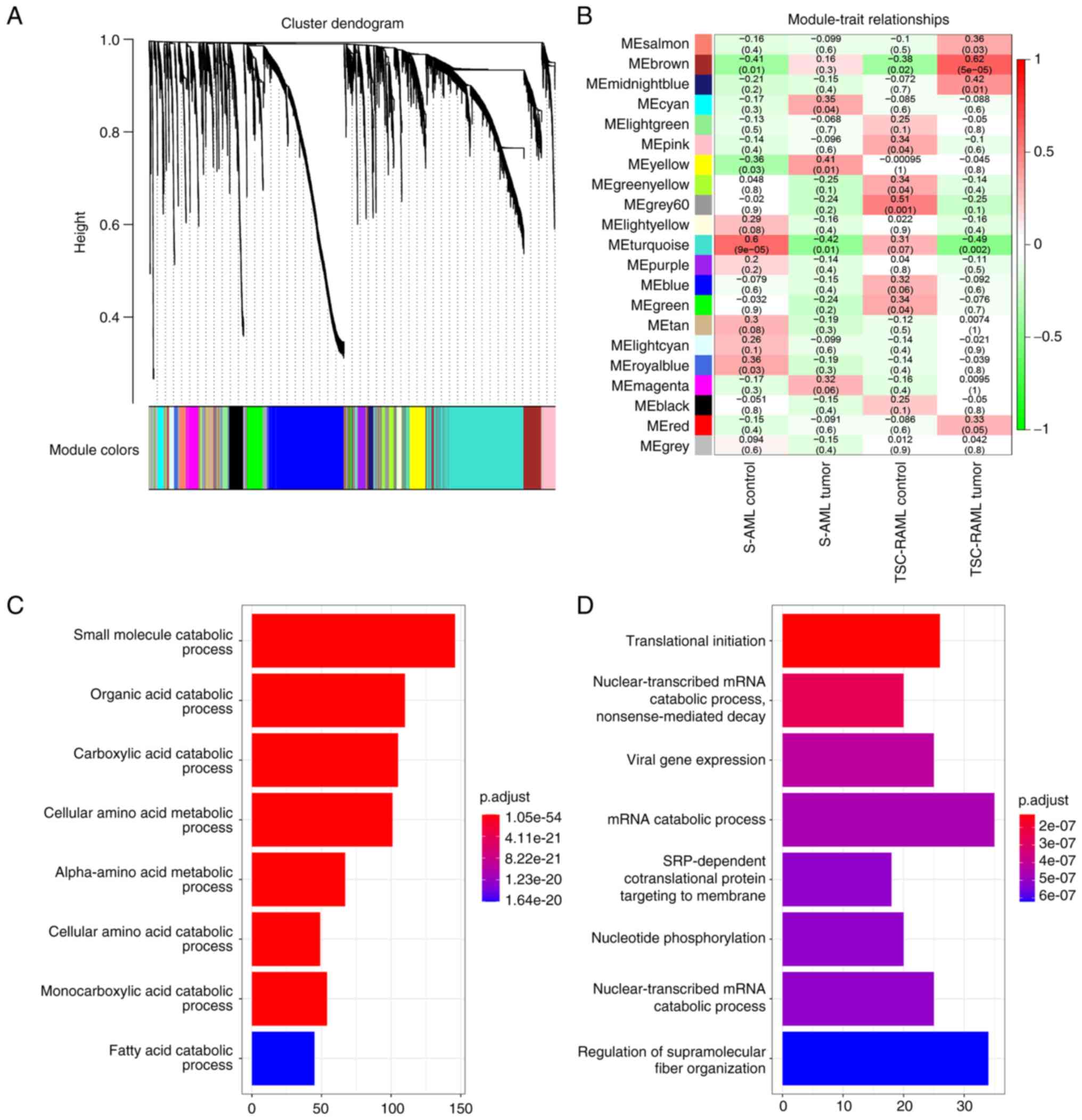
To assess the tumor burden-associated plasma
markers, all the proteins were divided into seven whole proteome
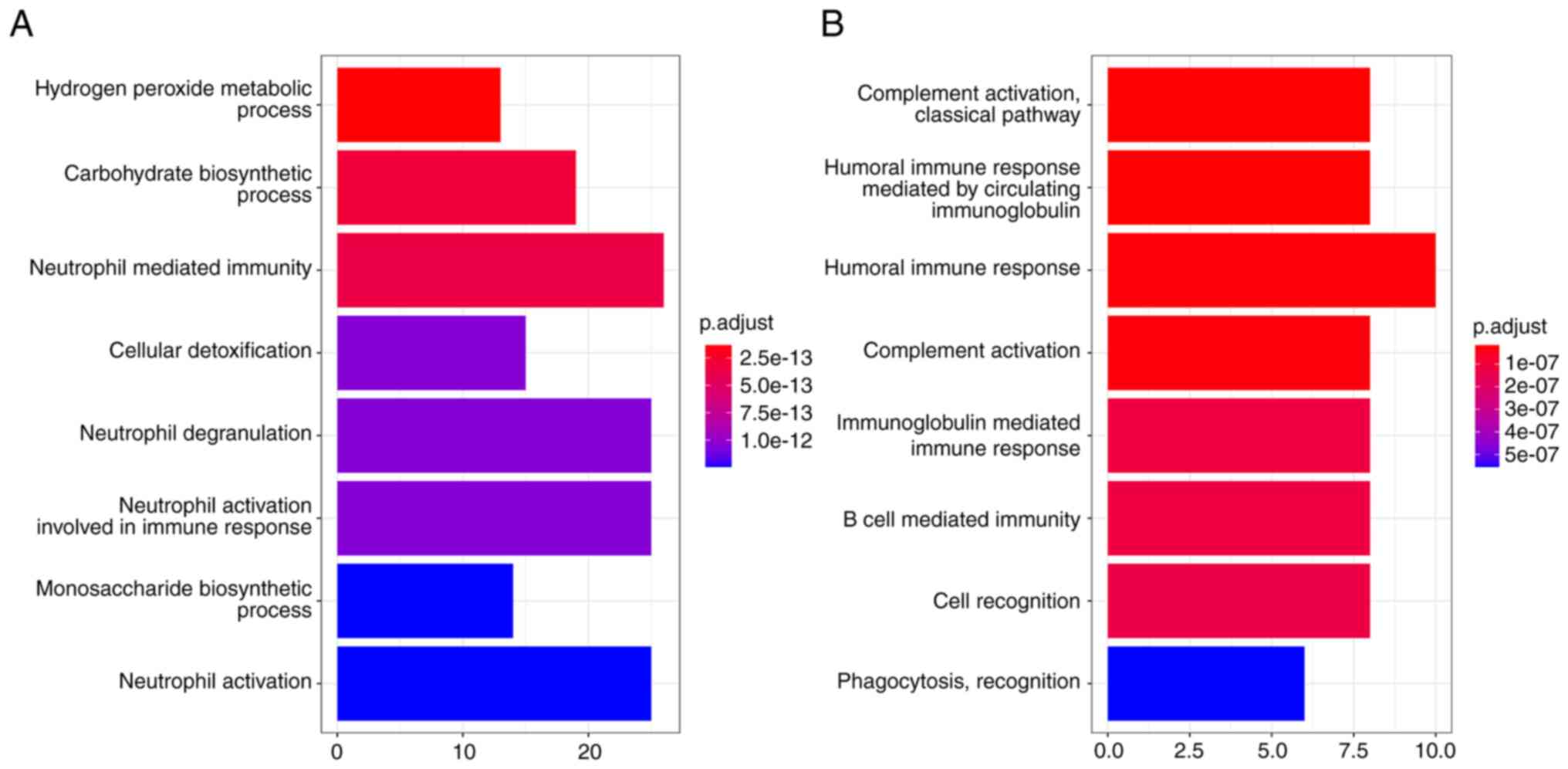
co-expression clusters using the WGCNA package (Fig. 4). The eigengene module (ME)
MEturquoise was negatively associated with the AML grading and
whole-body disease burden (WBDB), while MEred was positively
associated with LAM. Functional enrichment of MEturquoise suggested
that AML and the WBDB characteristic module were mostly enriched in
hydrogen peroxide metabolic process, carbohydrate biosynthetic
process and neutrophil-mediated immunity (Fig. 5A). However, the LAM-related module
MEred was enriched in the pathway of complement activation, humoral
immune response and B cell response (Fig. 5B). The analysis also indicated that
age and gene mutation status were not directly associated with any
module (Fig. 4B), suggesting the
limited effect of age and gene mutation status on plasma
proteomics.
Tissue proteomics of patients with
TSC-RAML and WGCNA analysis
One of the problems in TSC-RAML mechanistic research
is the scarcity of tissue samples, as it is not generally
recommended to perform surgical interventions for TSC-RAML
(33). To validate the above
analytic results, nine tumor tissues and NATs from patients with
TSC-RAML and S-AML were collected and proteomics analysis was
performed with UPLC-MS. Compared with the plasma, protein diversity
in the tumor microenvironment was much higher with 6,178 proteins
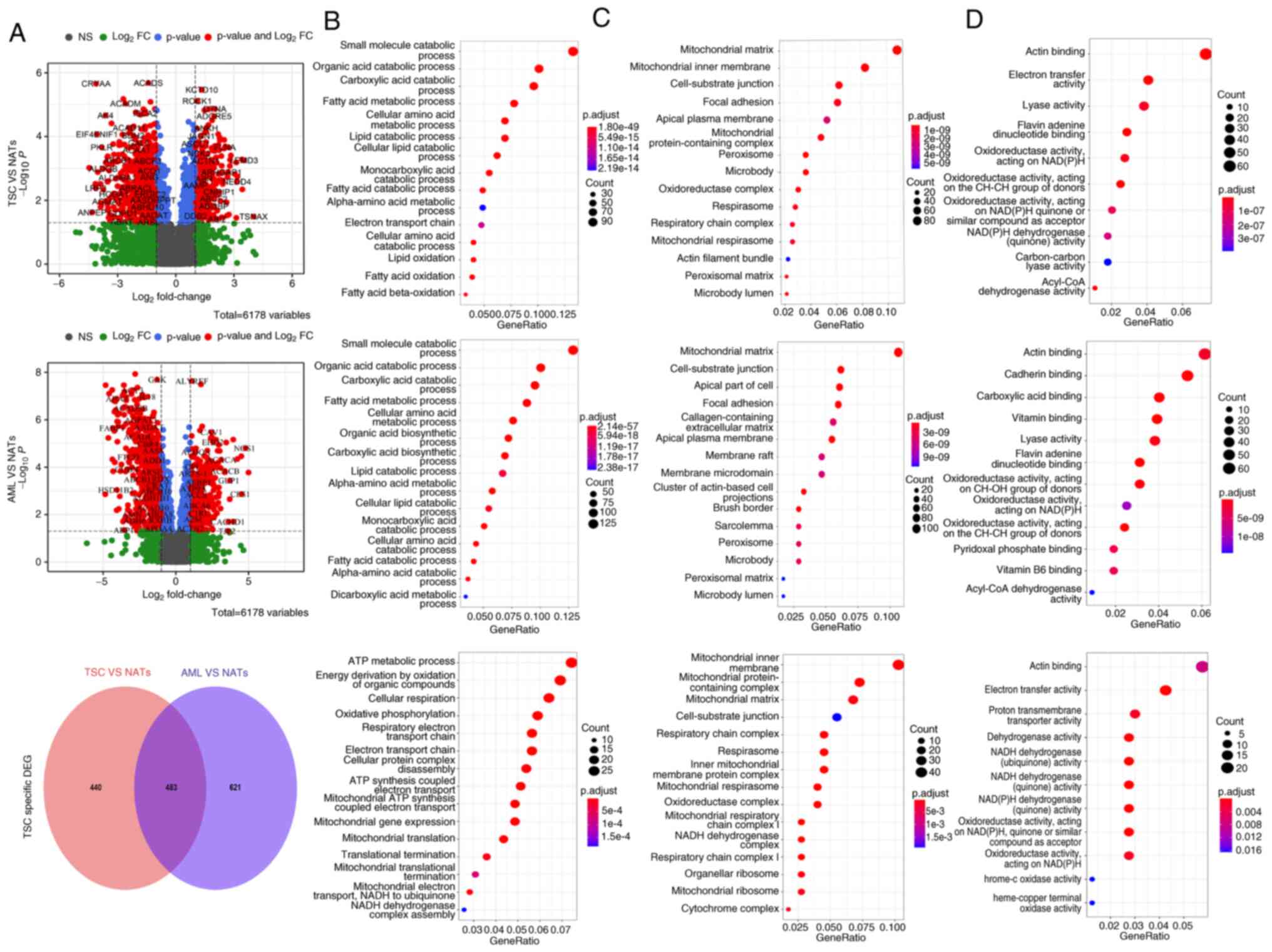
detected (Fig. 6).
From the heatmap in Fig. 6, a relative similarity between
TSC-RAML and S-AML tumors, as well as a similarity between TSC-AML
and S-AML NATs, was also observed. However, the difference between
tumors and NATs appeared distinct. Among the 6,178 total proteins,
there existed 307 significantly upregulated and 616 downregulated
proteins for TSC-RAML together with 417 upregulated and 687
downregulated proteins for S-AML (depicted in Fig. 7A, top and middle panels). GO
enrichment of DEPs further validated the above result, as both
TSC-RAML and S-AML had a similar enrichment result, namely altered
small molecule catabolic process, mitochondrial matrix component
and actin binding function. Both TSC-RAML and S-AML appeared to
have altered lipid metabolization, including lipid catabolic
process, cellular lipid catabolic metabolism and the fatty acid
catabolic process (Fig. 7B, top
and middle panels).
To explore TSC-RAML-specific protein patterns,
intersected proteins with S-AML were deleted and the enrichment
result indicated that ATP metabolic process, organic compound
oxidation, oxidative phosphorylation, mitochondrial-associated
compounds and actin binding were the most significantly
distinguished pathways involved (Fig.
7A-D, lower panel). Additional WGCNA analysis revealed that
MEbrown and MEturquoise were positively and negatively associated
with TSC-RAML, respectively (Fig. 8A
and B). Functional analysis of the two modules (Fig. 8C and D) highlighted the significant
terms of small molecule catabolic process, organic acid catabolic
process (MEbrown; all GO terms) and translational initiation and
nuclear-transcribed mRNA catabolic process (MEturquoise; all GO
terms), which have been reported previously by Lam et al
(33).
Tissue RNA sequencing data reveal
characteristic transcriptome of TSC-RAML and its unique tumor
micro-environment composition
Altered proteomics may reflect upstream changes in
RNA transcriptomes. Therefore, another cohort of TSC-RAML and NATs
was also tested (raw data are provided in Table SI). From the heatmap,
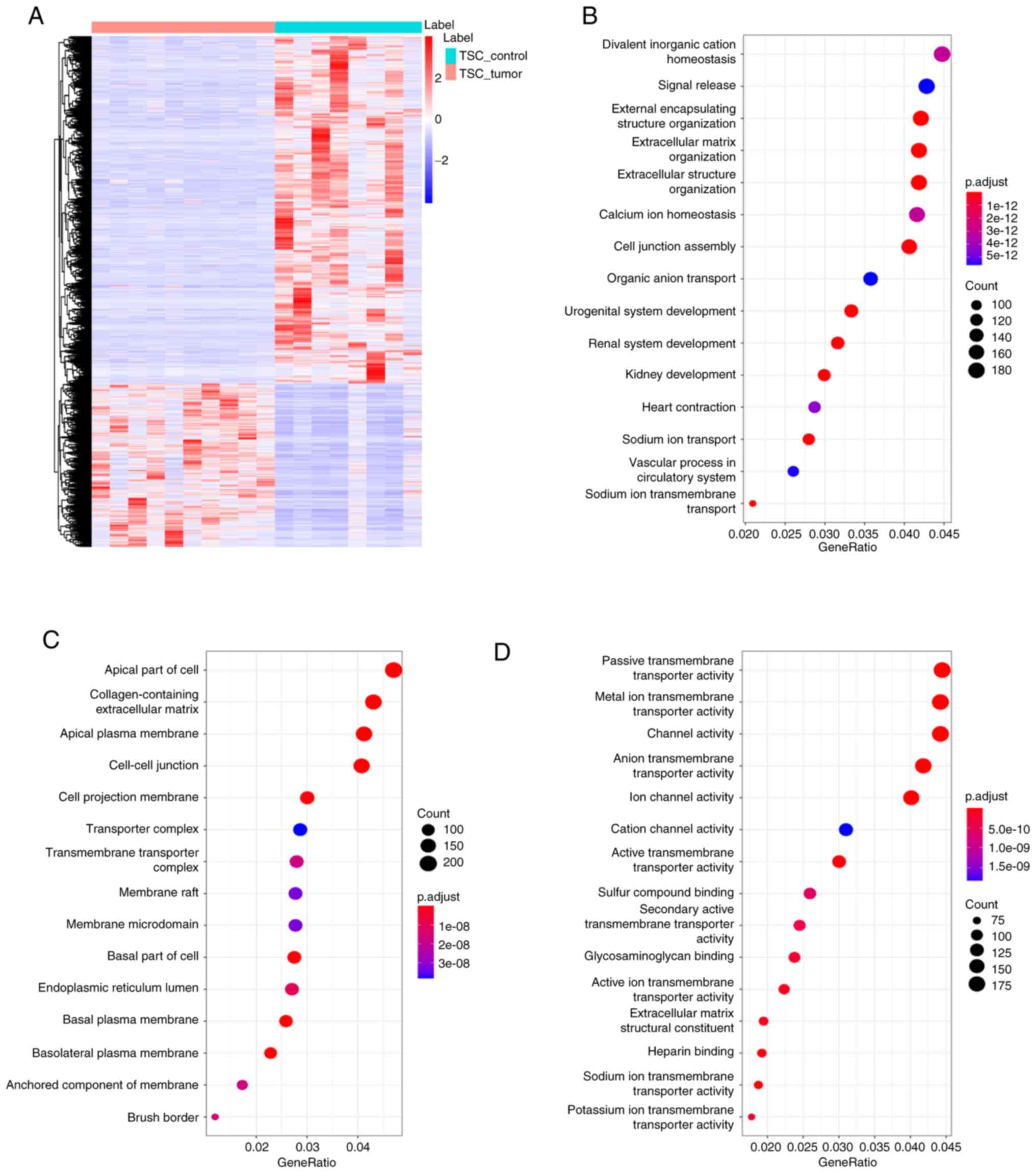
distinguished RNA profiling was clearly observed (Fig. 9A). GO enrichment indicated that the
altered RNAs were mostly enriched in the pathways of divalent
inorganic cation homeostasis, apical part of cell,
collagen-containing extracellular matrix and passive transmembrane
transporter activity (Fig.
9B-D).
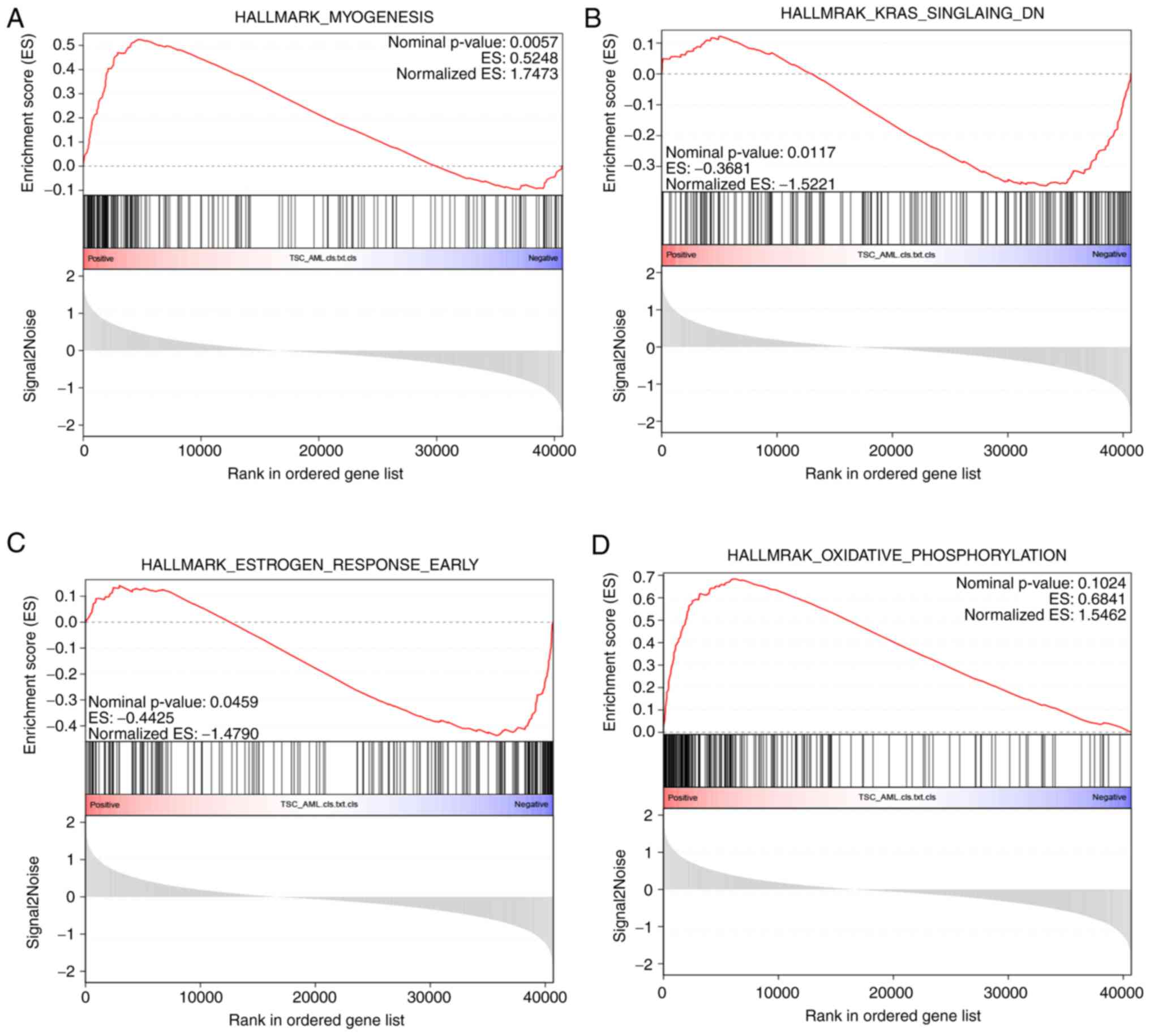
Compared with GO analysis, GSEA may better reveal
the statistically significant, concordant differences between two
biological states by ranking the molecules (34,35).
From the results, it was clear that myogenesis process (Fig. 10A) was significantly upregulated,
while K-ras and early-phase estrogen response were downregulated
(Fig. 10B and C). Another
important process in energy production, oxidation phosphorylation,
even though not statistically significant, exhibited an
upregulation tendency in TSC-RAML (Fig. 10D).
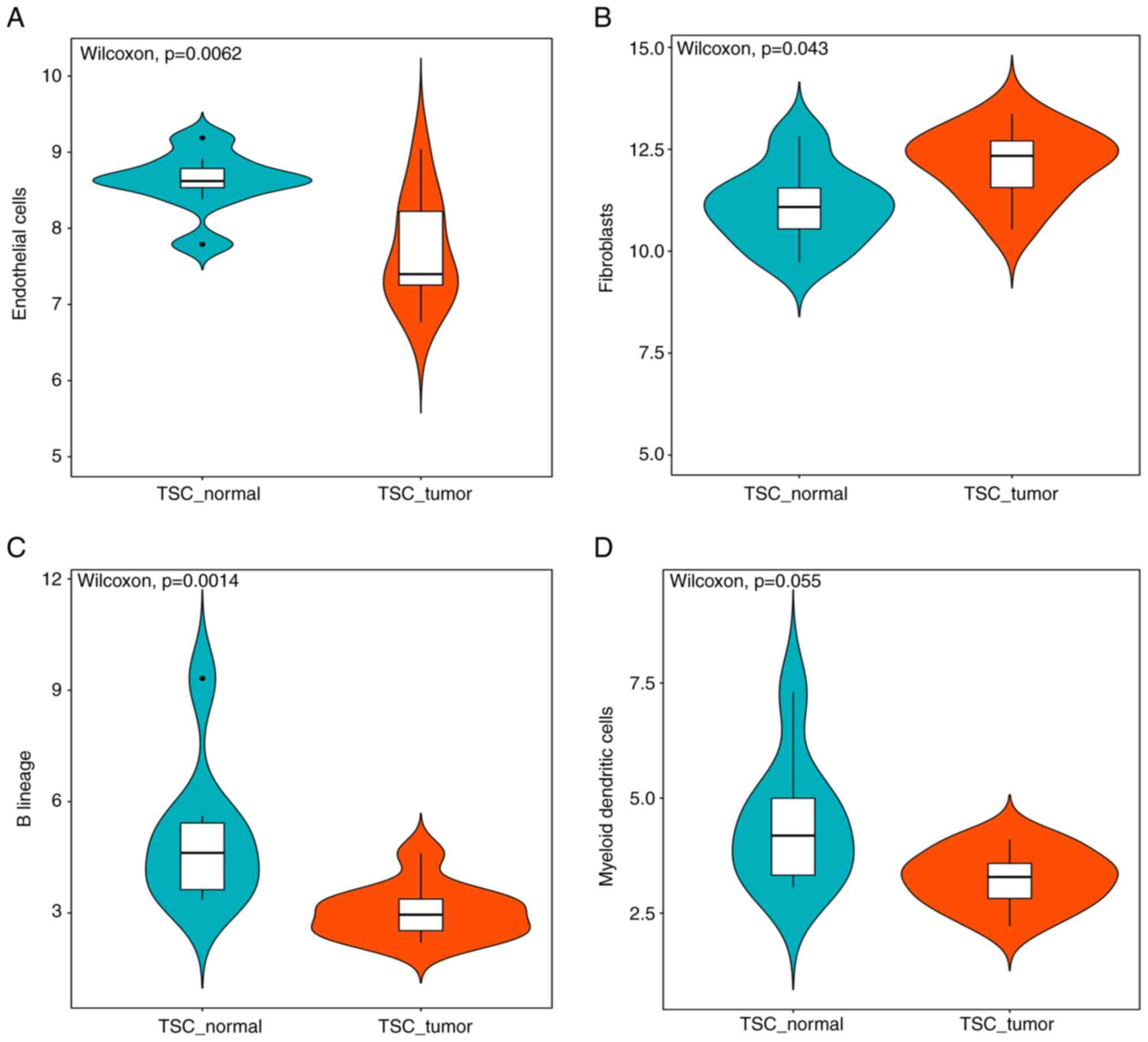
The R package 'MCFcounter' allows for the
calculation of the absolute abundance of eight immune and two
stromal cell populations within the tumor microenvironment from
transcriptomic data (22). It was
observed that the fibroblast subpopulation within the sample of
patients with TSC-RAML was significantly higher than that in NATs.
Furthermore, the endothelial cells and B lineage exhibited a
dramatic reduction, indicating the local immune insufficiency in
TSC-RAML (Fig. 11A-C). Due to the
critical role of antigen-presenting cells in the adaptive immune
response, dendritic cells had a decreasing tendency, although not
reaching statistical significance (P=0.055; Fig. 11D). Other cell compositional
changes without significance are presented in Fig. S2.
Single-cell RNA sequencing identifies
TSC-RAML-like tumor cells and pseudo-time trajectories analysis
reveals the developmental routine of tumor cells
The above analysis highlighted abnormal stromal and
immune cell composition within the tumor microenvironment, although
direct evidence is lacking. Based on the previously published
article (13), a second-time
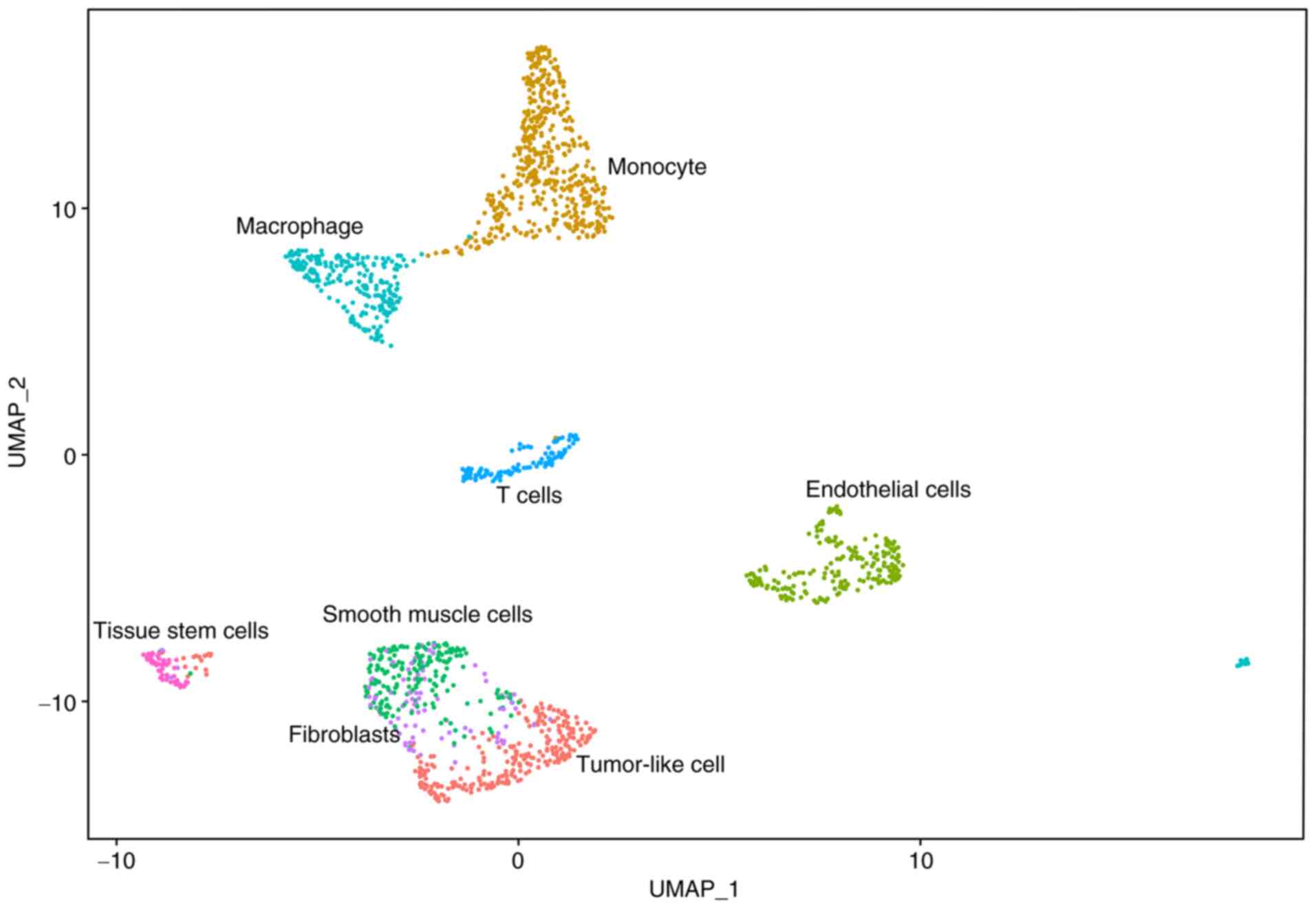
analysis was performed. After quality control, 1,675 high-quality
cells with 17,076 RNAs were selected and then classified into 10
subgroups. After sequentially automatic and manual annotation, the
10 subgroups were further divided into eight cell types (Fig. 12). A heatmap of characteristic
markers for each cell type is presented in Fig. S3.
The source of TSC-RAML cells has not been identified
until now (33), although TSC-LAM
has been suggested to have a uterine source (13). According to clinical markers and
previous research (13), cells
with high expression of PMEL, CTSK, FIGF, ESR1, HOXA11 and SLC35F1
were defined as tumor-like cells and the UMAP result is presented
in Fig. S4. In terms of cell
composition, accumulative monocytes and macrophages were observed
together with a reduced number of T cells within the tumor
microenvironment (percentage for monocytes, macrophages and T cells
is 29, 15 and 7%, respectively). The functional enrichment of
monocytes and macrophages suggested overactive neutrophil
activation, increased secretory granules and increased actin
binding process (depicted in Fig.
S5). Another striking point is the similar RNA expression
pattern of smooth muscle cells, fibroblasts and tumor-like cells
(depicted in Fig. 12). The
functional enrichment regarding the top 200 RNAs of tumor-like
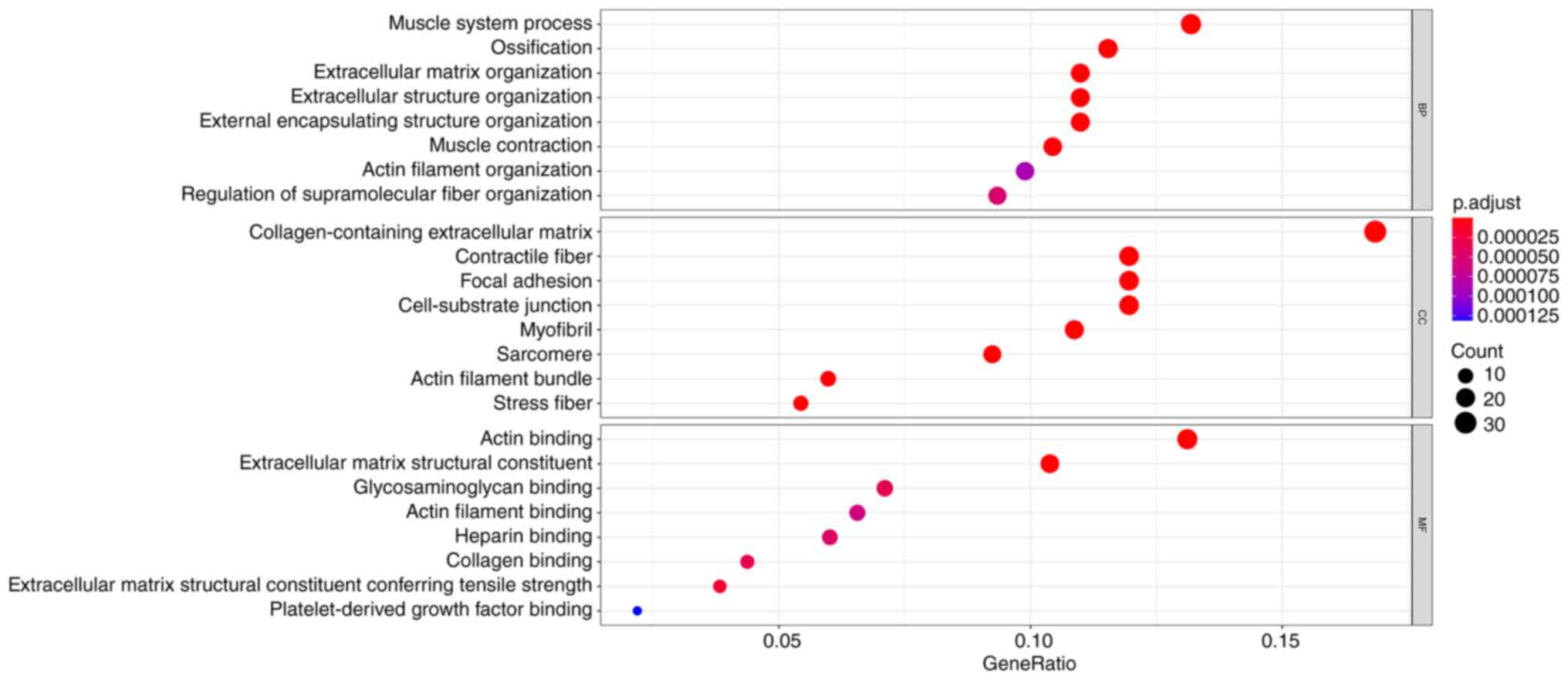
cells indicated that the differentially expressed genes (DEGs) are
mainly involved in the muscle system process, ossification,
collagen-containing extracellular matrix and actin binding
(Fig. 13). Fibroblasts with
TSC−/− have been widely applied in basic research to
explore TSC mechanisms (36,37).
From the GO analysis of fibroblasts, it was indicated that most of
the DEGs were enriched in the process of ATP metabolic process,
oxidative phosphorylation, mitochondrial composition and cadherin
binding (Fig. S6).
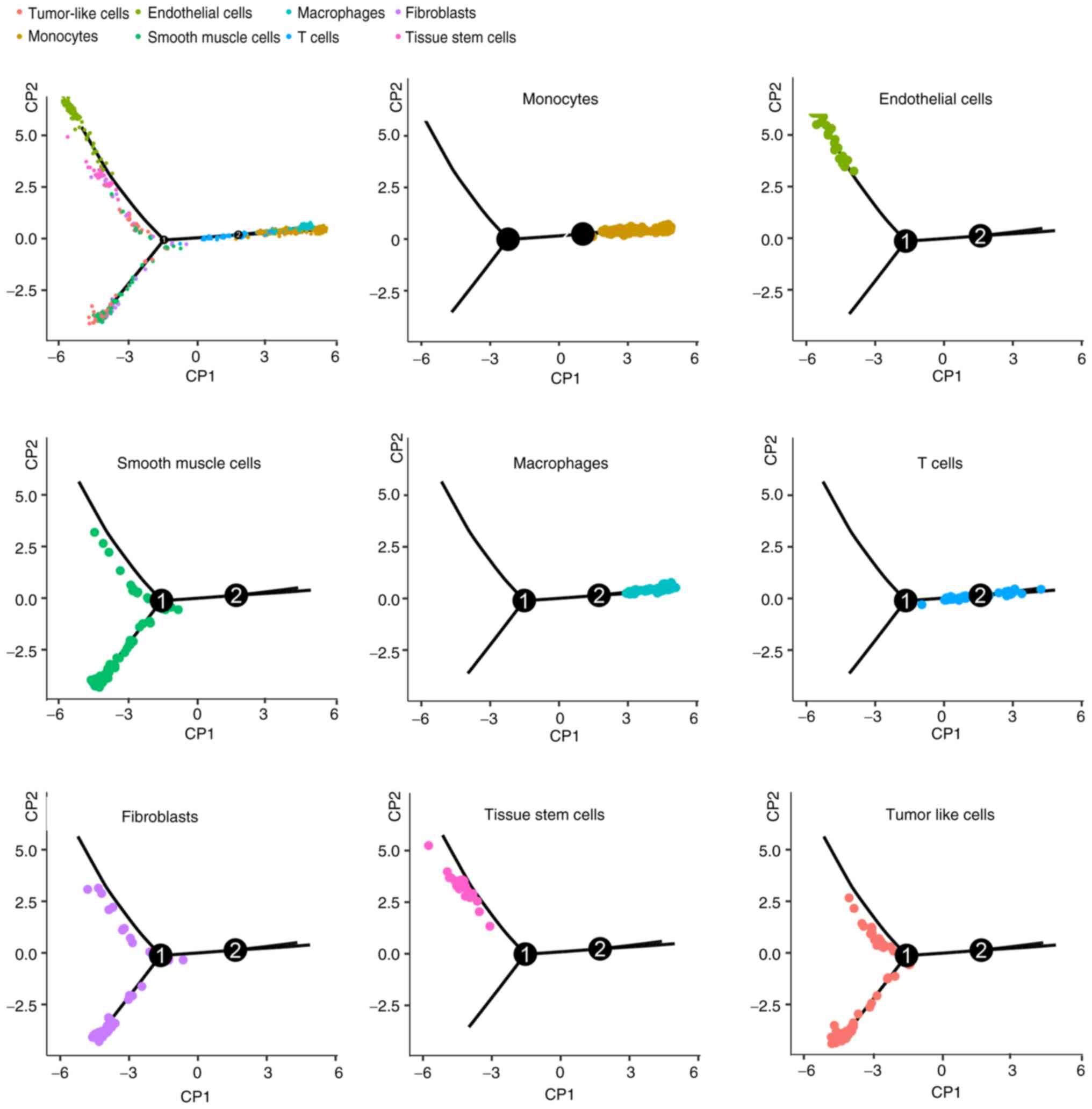
Another way to explore the source and development of
tumor cells is to simulate the differentiation lineage. Using the R
package 'monocle' (19),
developmental pseudo-time analyses of different cell types were
drawn (Fig. 14), from which
similar development between fibroblasts, smooth muscle and
tumor-like cells was observed. This result is in accordance with
the composition of TSC-RAML, namely vessels, smooth muscle and
adipose (33).
Discussion
By retrospectively analyzing TSC-RAML, S-AML and
renal cyst multi-omics data, the present study gained insight into
the mechanisms of TSC-RAML, which may be summarized as follows: i)
Plasma proteins such as MMP9 and CCL5 may be possible markers for
TSC-RAML; ii) plasma proteomics enriched in neutrophil-mediated
immunity, hydrogen peroxide and carbohydrate metabolic processes
were associated with renal tumor grading and whole disease burden;
and iii) tissue transcriptome revealed increased
monocyte-macrophages within the tumor microenvironment, which may
be crucial for TSC-RAML tumorigenesis and development.
In addition to TSC-RAML plasma proteomics analysis,
the tissue transcriptome was also examined and indicated elevated
MMP9. MMPs, containing a group of zinc-dependent endopeptidases,
are responsible for remodeling the extracellular matrix collagen
and elastin under pathological and physiological conditions
(38). Previous studies (24,39)
have also reported higher MMP9 mRNA and protein expression in TSC
brain tubers compared to controls and peritubular brain tissues. In
LAM, one of the most common pulmonary manifestations of TSC, MMP-9
has been detected at high levels in both serum and urine (40). This has been associated with loss
of pulmonary function in LAM (38,40).
Through Src kinase activation, MMP9 has been suggested to promote
the invasiveness of TSC2-null embryonic fibroblasts in vitro
(40,41). The inhibitor of MMP9 and MMP2,
doxycycline, has demonstrated an anti-migration effect on
TSC2-negative mouse embryonic fibroblasts (MEF) through
downregulating RhoA-GTPase activity and phosphorylation of focal
adhesion kinase (36). All
evidence suggests that MMPs have a role in the tumorigenesis of
TSC-related diseases. A recent study has also suggested that an
axis containing CD147-MMP-VEGF increases tumor angiogenesis
(40). This appears to be in
accordance with the plasma elevated MMPs and VEGF-D related to TSC.
In the present study, it was first observed that MMP9 was elevated
not only in the blood of patients with TSC-RAML but also in the
tissue transcriptome. Although further research is required to
validate the present results, MMP9 may be suggested as a possible
marker and drug target for TSC-RAML.
In addition to MMP9, CCL5, which is a member of the
CC subfamily of chemokines, also appeared to have good
distinguishability. CCL5 was able to be expressed on most
inflammatory cell types, although monocytes and T cells are the
most common CCL5-expressing cells (25). Accumulating evidence suggests that
CCL5 participates in numerous processes, including inflammation,
cancers, viral infection and immune responses (25). Recent research has also indicated
that plasminogen activator inhibitor-1 is able to regulate CCL5 and
promote cell migration and angiogenesis and inhibit cell apoptosis
through MMPs (25), suggesting
there is a synergistic function between CCL5 and MMPs in
tumorigenesis. William et al (42) reported that treatment of wild-type
murine marrow-derived macrophages with mTORC1 inhibitors promoted
4E-BP1/2 activation and reduced CCL5 secretion, indicating that
overactive mTOR and downstream 4E-BP1/2 may increase the production
of CCL5. Accumulating evidence suggested that, as a chemoattractant
factor, CCL5 was able to recruit tumor-associated macrophages
(TAMs) to the tumor beds, thus facilitating tumor metastasis
(43-45). In addition, TAMs have been proven
to increase the secretion of collagen and thus potentiate the cell
migration and increase tissue stiffness (43). Based on the results of the present
study, plasma CCL5 was elevated and a large quantity of monocytes
and macrophages was deposited within the tumor microenvironment
according to single-cell RNA sequencing. Therefore, it may be
proposed that high levels of CCL5 are necessary for modulating and
recruiting monocyte-macrophages towards the tumor site and thus
promoting tumor growth, although further experiments are required
to confirm this finding.
Although a large number of studies have focused on
genotype-phenotype correlation, no definite conclusion regarding
TSC-RAML has been made, unless the TSC2 mutation is much more
severe than that of TSC1 (27,28,46,47).
A relatively large systematic review including 261 patients with
TSC suggested that TSC1 missense mutation and the mutation of TSC2
encoding TAD1 were associated with a high risk of TSC-RAML
(28). The present study attempted
to find the proteins associated with phenotype. The results
suggested that the proteins involved in metabolization,
neutrophil-mediated immunity and extracellular matrix organization
were significantly associated with renal tumor grading and
whole-body disease burden. The present results were consistent with
established plasma markers, namely elevated MMPs (reflecting the
extracellular matrix organization) and CCL5, which participates in
immune responses. In the aspect of metabolization, Bottolo et
al (48) indicated that
metabolites involving fatty acid and sphingolipid metabolism were
associated with the severity of lung disease, whole-body disease
burden and disease activity. Feng et al (49) observed that secreted
lysophospholipase D autotaxin (ATX) was upregulated in human renal
angiomyolipoma-derived TSC2-deficient cells and inhibiting ATX was
able to suppress TSC2 loss-associated oncogenicity in vitro
and in vivo and induce apoptosis in TSC2-deficient cells.
The tissue proteomic analysis of the present study indicated that
both TSC-RAML and S-AML harbored a dysregulated lipid metabolism,
further validating the notion that targeted lipid metabolism may be
another breakthrough in treating TSC-related disease. From the RNA
sequencing data, altered energy production was also observed,
further validating the above analysis. It is generally thought that
mTOR has a critical role in the regulation of metabolism and energy
production (33). Although there
was no difference in the components of TSC-RAML and S-AML,
proteomics analysis revealed distinctive alterations of plasma and
tissue proteins. The RNA transcriptome analysis indicated that,
compared with NATs, most of the differential RNAs were
downregulated and the functional enrichment also revealed altered
matrix organization and collagen-containing extracellular
matrix.
Increasing evidence suggests that the cell
composition within the tumor microenvironment has a critical role
in tumorigenesis. From the RNA transcriptome data, the composition
of 8 various cell types was determined in the present study.
Fibroblasts were indicated to be higher in TSC-RAML compared with
NATs. A previous in vitro study suggested that TSC2-null
fibroblast-like cells grown from human TSC skin hamartomas were
able to induce normal human keratinocytes to form hair follicles
and stimulate changes in hamartomata, suggesting that the
fibroblast-like cells may be the source of the TSC tumor (50). In experiments using
TSC−/− cell lines, MEFs with TSC2 knockout are still
most widely used, indicating the similarity between fibroblasts and
tumor cells in terms of biological behaviour (36,51).
Another experiment discovered that a population of stromal
cells/fibroblasts from the kidney labelled by Prx1 was able to form
a cyst from the loop of Henle after ablation of the Tsc1 gene,
indicating the contributive role of fibroblasts in patients with
TSC (52). From the cell
annotation for patients with TSC-RAML, the tumor-like cells were
also identified, which had similarities with fibroblasts and smooth
muscle cells. The pseudo-time analysis revealed a similar
developing tendency of the three cell clusters, indicating their
same source. All of the evidence suggested that the tumor cells, as
well as fibroblasts and smooth muscle cells, may originate from the
same progenitor cells.
Contrary to the increased fibroblasts, other cell
types, including endothelial cells, B cells and DC cells, were
decreased in TSC-RAML, suggesting an immune inhibitory state within
the tumor microenvironment. In line with the RNA transcriptome, the
single-cell RNA sequencing revealed a high proportion of monocytes
and macrophages but low proportion of T cells. As reported, M2-like
tumor-associated macrophages have critical roles in facilitating
epithelial-mesenchymal transition, angiogenesis and
immunosuppression (53). A
previous in vitro experiment indicated that fibroblast-like
cells from angiofibroma and periungual fibromas were able to
secrete higher levels of monocyte chemoattractant protein-1 (MCP-1)
mRNA and protein than fibroblasts from the same patient's normal
skin (54). Furthermore,
conditioned medium from cultured TSC skin tumor cells was
chemotactic for human monocytic cells and neutralizing antibody
against MCP-1 inhibited the chemotactic activity (54). In line with this, Eker rat
embryonic fibroblasts with TSC2 deletion produced 28 times higher
MCP-1 protein compared with the wild-type (54). In a mouse model with TSC1 deletion,
it was demonstrated that macrophages were refractory to
IL-4-induced M2 polarization and evoked increased inflammatory
responses to pro-inflammatory stimuli, suggesting the critical role
of the mTOR pathway in regulating macrophage polarization (55). All of the above results validated
the role of monocyte-macrophages in tumorigenesis and suggested a
new therapeutic approach for TSC-related disease.
As exploratory research, the present study has the
following limitations. First, the sample size in the present cohort
was relatively small and therefore, it remains to be further
determined whether the present results are able to be widely
applied to patients with TSC-RAML. Due to the rarity of TSC-RAML,
studies with large sample sizes are difficult to perform. In
addition, the relatively small sample size may have introduced
selection bias and affected the validity of the present results.
Furthermore, the fact that the present study is a single-center
study without external validation may be another limitation.
Therefore, our group has started enrolling patients with TSC-RAML
from different centers to validate the present findings in external
cohorts. In the end, although the present results revealed the
unique tumor microenvironment of TSC-RAML, further in vitro
and in vivo experiments are required to elucidate the
internal mechanism.
To conclude, the present results suggested that
plasma proteins such as CCL5 and MMP9 may be useful diagnostic
markers and disease burden markers. The unique inflammatory tumor
microenvironment, particularly the monocyte-macro-phages, also
potentially has a crucial role in the development of TSC-RAML.
Targeting the plasma diagnostic markers and inflammatory cells
within the tumor microenvironment may be another breakthrough in
TSC-RAML treatment.
Supplementary Data
Availability of data and materials
The RNA sequencing matrix may be obtained from
Table SI and the raw data are
being deposited in the Genome Sequence Archive (https://ngdc.cncb.ac.cn/bioproject/) under the
accession number 'PRJCA011152'.
Authors' contributions
YuZ and LJ conceived the project and organized all
the experiments. ZW and XL conducted the experiments and wrote the
original manuscript. WW, YaZ, XW, ZL and YL collected the
biological samples and clinical information and interpreted the
data. JW, SS, JX, HS, YY and WS analyzed the data. All the authors
have read and approved the manuscript. ZW and YuZ checked and
confirmed the authenticity of the raw data.
Ethics approval and consent to
participate
The study was performed in accordance with the
Declaration of Helsinki and was approved by the Institutional
Review Board of Peking Union Medical College Hospital and the
Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences (Beijing, China; approval no. KS2020127). Written informed
consent was obtained from each patient for the use of their
biological samples for scientific research and publication of all
the related data.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
Not applicable.
Abbreviations:
|
AML
|
angiomyolipoma
|
|
DEGs
|
differentially expressed genes
|
|
DEPs
|
differentially expressed proteins
|
|
GO
|
Gene Ontology
|
|
GSEA
|
gene set enrichment analysis
|
|
LAM
|
lymphangiomyomatosis
|
|
MCP-1
|
monocyte chemoattractant
protein-1
|
|
NATs
|
non-tumor normal tissues
|
|
PMEL
|
premelanosome protein
|
|
S-AML
|
sporadic angiomyolipoma
|
|
SEGA
|
subependymal giant cell
astrocytoma
|
|
TSC
|
tuberous sclerosis complex
|
|
TSC-RAML
|
TSC-related AML
|
|
UPLC-MS
|
ultra-performance liquid
chromatography-mass spectrometry
|
|
WBDB
|
whole-body disease burden
|
|
WGCNA
|
weighted gene correlation network
analysis
|
References
|
1
|
Curatolo P, Bombardieri R and Jozwiak S:
Tuberous sclerosis. Lancet. 372:657–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henske EP, Jóźwiak S, Kingswood JC,
Sampson JR and Thiele EA: Tuberous sclerosis complex. Nat Rev Dis
Primers. 2:160352016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rebaine Y, Nasser M, Girerd B, Leroux C
and Cottin V: Tuberous sclerosis complex for the pulmonologist. Eur
Respir Rev. 30:2003482021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Crino PB, Nathanson KL and Henske EP: The
tuberous sclerosis complex. N Eng J Med. 355:1345–1356. 2006.
View Article : Google Scholar
|
|
5
|
Amin S, Lux A, Calder N, Laugharne M,
Osborne J and O'callaghan F: Causes of mortality in individuals
with tuberous sclerosis complex. Dev Med Child Neurol. 59:612–617.
2017. View Article : Google Scholar
|
|
6
|
Shepherd CW, Gomez MR, Lie JT and Crowson
CS: Causes of death in patients with tuberous sclerosis. Mayo Clin
Proc. 66:792–796. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saffari A, Brösse I, Wiemer-Kruel A,
Wilken B, Kreuzaler P, Hahn A, Bernhard MK, van Tilburg CM,
Hoffmann GF, Gorenflo M, et al: Safety and efficacy of mTOR
inhibitor treatment in patients with tuberous sclerosis complex
under 2 years of age-a multicenter retrospective study. Orphanet J
Rare Dis. 14:962019. View Article : Google Scholar
|
|
8
|
Bissler JJ, Kingswood JC, Radzikowska E,
Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N,
Brakemeier S, de Vries PJ, et al: Everolimus for angiomyolipoma
associated with tuberous sclerosis complex or sporadic
lymphangi-oleiomyomatosis (EXIST-2): A multicentre, randomised,
double-blind, placebo-controlled trial. Lancet. 381:817–824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hatano T and Egawa S: Renal angiomyolipoma
with tuberous sclerosis complex: How it differs from sporadic
angiomyolipoma in both management and care. Asian J Surg.
43:967–972. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Northrup H and Krueger DA; International
Tuberous Sclerosis Complex Consensus Group: Tuberous sclerosis
complex diagnostic criteria update: Recommendations of the 2012
iinternational tuberous sclerosis complex consensus conference.
Pediatr Neurol. 49:243–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eijkemans MJ, van der Wal W, Reijnders LJ,
Roes KC, van Waalwijk van Doorn-Khosrovani SB, Pelletier C,
Magestro M and Zonnenberg B: Long-term follow-up assessing renal
angiomyolipoma treatment patterns, morbidity, and mortality: An
observational study in tuberous sclerosis complex patients in the
Netherlands. Am J Kidney Dis. 66:638–645. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao F, Kataoka M, Liu N, Liang T, Huang
ZP, Gu F, Ding J, Liu J, Zhang F, Ma Q, et al: Therapeutic role of
miR-19a/19b in cardiac regeneration and protection from myocardial
infarction. Nat Commun. 10:18022019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo M, Yu JJ, Perl AK, Wikenheiser-Brokamp
KA, Riccetti M, Zhang EY, Sudha P, Adam M, Potter A, Kopras EJ, et
al: Single-cell transcriptomic analysis identifies a unique
pulmonary lymphangioleiomyomatosis cell. Am J Respir Crit Care Med.
202:1373–1387. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hao Y, Hao S, Andersen-Nissen E, Mauck WM
III, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al:
Integrated analysis of multimodal single-cell data. Cell.
184:3573–3587.e29. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stuart T, Butler A, Hoffman P, Hafemeister
C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P and
Satija R: Comprehensive integration of single-cell data. Cell.
177:1888–1902.e21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aran D, Looney AP, Liu L, Wu E, Fong V,
Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al:
Reference-based analysis of lung single-cell sequencing reveals a
transitional profibrotic macrophage. Nat Immunol. 20:163–172. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z,
Feng T, Zhou L, Tang W, Zhan L, et al: clusterProfiler 4.0: A
universal enrichment tool for interpreting omics data. Innovation
(Camb). 2:1001412021.
|
|
19
|
Trapnell C, Cacchiarelli D, Grimsby J,
Pokharel P, Li S, Morse M, Lennon NJ and Livak KJ: The dynamics and
regulators of cell fate decisions are revealed by pseudotemporal
ordering of single cells. Nat Biotechnol. 32:381–386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Langfelder P and Horvath S: Fast R
functions for robust correlations and hierarchical clustering. J
Statis Softw. 46:i112012.
|
|
22
|
Becht E, Giraldo NA, Lacroix L, Buttard B,
Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman
C, Fridman WH and de Reyniès A: Estimating the population abundance
of tissue-infiltrating immune and stromal cell populations using
gene expression. Genome Biol. 17:2182016. View Article : Google Scholar
|
|
23
|
Bongaarts A, de Jong JM, Broekaart DW, van
Scheppingen J, Anink JJ, Mijnsbergen C, Jansen FE, Spliet WG, den
Dunnen WFA, Gruber VE, et al: Dysregulation of the MMP/TIMP
proteolytic system in subependymal giant cell astrocytomas in
patients with tuberous sclerosis complex: Modulation of MMP by
MicroRNA-320d in vitro. J Neuropathol Exp Neurol. 79:777–790. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Broekaart DW, van Scheppingen J, Anink JJ,
Wierts L, van Het Hof B, Jansen FE, Spliet WG, van Rijen PC,
Kamphuis WW, de Vries HE, et al: Increased matrix
metalloproteinases expression in tuberous sclerosis complex:
Modulation by microRNA 146a and 147b in vitro. Neuropathol Appl
Neurobiol. 46:142–159. 2020. View Article : Google Scholar :
|
|
25
|
Zeng Z, Lan T, Wei Y and Wei X: CCL5/CCR5
axis in human diseases and related treatments. Genes Dis. 9:12–27.
2022. View Article : Google Scholar
|
|
26
|
McCormack FX, Inoue Y, Moss J, Singer LG,
Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM,
et al: Efficacy and safety of sirolimus in
lymphangioleiomyomatosis. N Eng J Med. 364:1595–1606. 2011.
View Article : Google Scholar
|
|
27
|
Muto Y, Sasaki H, Sumitomo M, Inagaki H,
Kato M, Kato T, Miyai S, Kurahashi H and Shiroki R:
Genotype-phenotype correlation of renal lesions in the tuberous
sclerosis complex. Hum Genome Var. 9:52022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang N, Wang X, Tang Z, Qiu X, Guo Z,
Huang D, Xiong H and Guo Q: The correlation between tuberous
sclerosis complex genotype and renal angiomyolipoma phenotype.
Front Genet. 11:5757502021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng JB, Sedgewick AJ, Finnegan AI,
Harirchian P, Lee J, Kwon S, Fassett MS, Golovato J, Gray M,
Ghadially R, et al: Transcriptional programming of normal and
inflamed human epidermis at single-cell resolution. Cell Rep.
25:871–883. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Holzinger D, Foell D and Kessel C: The
role of S100 proteins in the pathogenesis and monitoring of
autoinflammatory diseases. Mol Cell Pediatr. 5:72018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pruenster M, Vogl T, Roth J and Sperandio
M: S100A8/A9: From basic science to clinical application. Pharmacol
Ther. 167:120–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Spiekerkoetter E, Guignabert C, de Jesus
Perez V, Alastalo TP, Powers JM, Wang L, Lawrie A, Ambartsumian N,
Schmidt AM, Berryman M, et al: S100A4 and bone morphogenetic
protein-2 codependently induce vascular smooth muscle cell
migration via phospho-extracellular signal-regulated kinase and
chloride intracellular channel 4. Circ Res. 105:639–647. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lam HC, Siroky BJ and Henske EP: Renal
disease in tuberous sclerosis complex: Pathogenesis and therapy.
Nat Rev Nephrol. 14:704–716. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1α-responsive genes involved
in oxidative phosphorylation are coordinately downregulated in
human diabetes. Nat Genet. 34:267–273. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ng HY, Oliver BG, Burgess JK, Krymskaya
VP, Black JL and Moir LM: Doxycycline reduces the migration of
tuberous sclerosis complex-2 null cells-effects on RhoA-GTPase and
focal adhesion kinase. J Cell Mol Med. 19:2633–2646. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pollizzi K, Malinowska-Kolodziej I,
Doughty C, Betz C, Ma J, Goto J and Kwiatkowski DJ: A hypomorphic
allele of Tsc2 highlights the role of TSC1/TSC2 in signaling to AKT
and models mild human TSC2 alleles. Hum Mol Genet. 18:2378–2387.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Terraneo S, Lesma E, Ancona S, Imeri G,
Palumbo G, Torre O, Giuliani L, Centanni S, Peron A, Tresoldi S, et
al: Exploring the role of matrix metalloproteinases as biomarkers
in sporadic lymphangioleiomyomatosis and tuberous sclerosis
complex. A pilot study. Front Med (Lausanne). 8:6059092021.
View Article : Google Scholar
|
|
39
|
Li S, Yu S, Zhang C, Shu H, Liu S, An N,
Yang M, Yin Q and Yang H: Increased expression of matrix
metalloproteinase 9 in cortical lesions from patients with focal
cortical dysplasia type IIb and tuberous sclerosis complex. Brain
Res. 1453:46–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ancona S, Orpianesi E, Bernardelli C,
Chiaramonte E, Chiaramonte R, Terraneo S, Di Marco F and Lesma E:
Differential modulation of matrix metalloproteinases-2 and -7 in
LAM/TSC cells. Biomedicines. 9:17602021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tyryshkin A, Bhattacharya A and Eissa NT:
SRC kinase is a novel therapeutic target in
lymphangioleiomyomatosis. Cancer Res. 74:1996–2005. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
William M, Leroux LP, Chaparro V, Graber
TE, Alain T and Jaramillo M: Translational repression of Ccl5 and
Cxcl10 by 4E-BP1 and 4E-BP2 restrains the ability of mouse
macrophages to induce migration of activated T cells. Eur J
Immunol. 49:1200–1212. 2019.PubMed/NCBI
|
|
43
|
Walens A, DiMarco AV, Lupo R, Kroger BR,
Damrauer JS and Alvarez JV: CCL5 promotes breast cancer recurrence
through macrophage recruitment in residual tumors. eLife.
8:e436532019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Soria G and Ben-Baruch A: The inflammatory
chemokines CCL2 and CCL5 in breast cancer. Cancer Lett.
267:271–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rabe DC, Walker ND, Rustandy FD, Wallace
J, Lee J, Stott SL and Rosner MR: Tumor extracellular vesicles
regulate macrophage-driven metastasis through CCL5. Cancers
(Basel). 13:34592021. View Article : Google Scholar
|
|
46
|
Farach LS, Pearson DA, Woodhouse JP,
Schraw JM, Sahin M, Krueger DA, Wu JY, Bebin EM, Lupo PJ, Au KS, et
al: Tuberous sclerosis complex genotypes and developmental
phenotype. Pediatr Neurol. 96:58–63. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ogórek B, Hamieh L, Hulshof HM, Lasseter
K, Klonowska K, Kuijf H, Moavero R, Hertzberg C, Weschke B, Riney
K, et al: TSC2 pathogenic variants are predictive of severe
clinical manifestations in TSC infants: Results of the EPISTOP
study. Genet Med. 22:1489–1497. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bottolo L, Miller S and Johnson SR:
Sphingolipid, fatty acid and phospholipid metabolites are
associated with disease severity and mTOR inhibition in
lymphangioleiomyomatosis. Thorax. 75:679–688. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feng Y, Mischler WJ, Gurung AC, Kavanagh
TR, Androsov G, Sadow PM, Herbert ZT and Priolo C: Therapeutic
targeting of the secreted lysophospholipase D autotaxin suppresses
tuberous sclerosis complex-associated tumorigenesis. Cancer Res.
80:2751–2763. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li S, Thangapazham RL, Wang JA, Rajesh S,
Kao TC, Sperling L, Moss J and Darling TN: Human TSC2-null
fibroblast-like cells induce hair follicle neogenesis and hamartoma
morphogenesis. Nat Commun. 2:2352011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
García-Aguilar A, Guillén C, Nellist M,
Bartolomé A and Benito M: TSC2 N-terminal lysine acetylation status
affects to its stability modulating mTORC1 signaling and autophagy.
Biochim Biophys Acta. 1863:2658–2667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu Z, Wu H, Md S, Yu G, Habib SL, Li B and
Li J: Tsc1 ablation in Prx1 and Osterix lineages causes renal
cystogenesis in mouse. Sci Rep. 9:8372019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu K, Lin K, Li X, Yuan X, Xu P, Ni P and
Xu D: Redefining tumor-associated macrophage subpopulations and
functions in the tumor microenvironment. Front Immunol.
11:17312020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li S, Takeuchi F, Wang JA, Fuller C,
Pacheco-Rodriguez G, Moss J and Darling TN: MCP-1 overexpressed in
tuberous sclerosis lesions acts as a paracrine factor for tumor
development. J Exp Med. 202:617–624. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Byles V, Covarrubias AJ, Ben-Sahra I,
Lamming DW, Sabatini DM, Manning BD and Horng T: The TSC-mTOR
pathway regulates macrophage polarization. Nat Commun. 4:28342013.
View Article : Google Scholar : PubMed/NCBI
|