Introduction
Multidrug resistance can lead to the failure of
cancer chemotherapy and is a major cause of mortality in cancer
patients (1). Cancer cells acquire
resistance to drugs through various mechanisms, such as drug
effluxes and xenobiotic-mediated detoxification (2). Cisplatin is one of the most
frequently used drugs in a number of cancers; however, colon cancer
shows strong resistance to cisplatin (3). The autophagy-mediated cell survival
pathway has emerged as a prime mechanism for multidrug resistance
(4), but our understanding of
autophagy-mediated multidrug resistance is still lacking.
Glutathione S-transferases (GSTs) are multigene
family phase-II detoxifying enzymes activated during the resistance
to chemotherapeutic agents in several cancers, including colon,
esophageal and breast cancer (5-7). The
omega (Ω) class of GSTs (GSTOs) are unique among the GST family,
having a cysteine residue in their active site instead of tyrosine
or serine (8). A previous
publication suggested that GSTO1 serves a key role in
autophagy-apoptosis crosstalk. GSTO1 inhibits the JNK-mediated
apoptosis signaling pathway in macrophages (9). A previous publication documented the
upregulation of GSTO1 in chemotherapy-exposed breast cancer cells
(10). The knockdown of GSTO1 in
these cells reduced the formation of carboplatin-induced breast
cancer stem cells and decreased tumor initiation. The authors
suggested a novel interaction between GSTO1 and ryanodine receptor
1, which accelerates calcium release from the endoplasmic reticulum
(ER) and eventually activates the STAT3 pathway (10). Proteome-based analysis of
platinum-resistant human ovarian cancer cell lines revealed the
stable upregulation of GSTO1 expression compared with the parental
cells (11). Gene expression
profiling of proteasome inhibitor-resistant human myeloma cell
lines also revealed an upregulation of GSTO1 compared with the
parental cells (12).
Previous studies have suggested that autophagy and
drug resistance are causes of cancer metastasis (13-15).
Research on metastasis also suggested that the incidence of
autophagy increases more in metastasized tumors compared with the
levels of autophagy in the original tumor (13,15).
Autophagy also modulates tumor cell invasion and
epithelial-to-mesenchymal transition (13). Moreover, a previous publication
suggested that drug-resistant cancer cells show greater invasive
ability, leading to chemotherapy failure (14). The conclusion drawn from the
present study suggested that GSTO1 may control autophagy, which, in
turn, may control drug resistance in colon cancer cells and may be
involved in regulating metastasis.
The present study results suggested that GSTO1 may
be a crucial factor between cisplatin sensitization and resistance.
GSTO1 is a protein that prompts MDR cells into autophagy cell
survival. In addition, TNFαIP3/A20 was shown to interact very
strongly with GSTO1 in MDR cells. The GSTO1-TNFαIP3/A20 interaction
is one of the mechanisms of drug resistance, and the inhibition of
those mechanisms in MDR cells sensitizes them to cisplatin.
Materials and methods
Chemicals
5-Fluorouracil (5-FU), cisplatin, docetaxel,
vincristine, Hoechst 33342, bafilomycin A1 (BAF-A1),
dichloro-dihydrofluorescein diacetate (DCFH-DA), Fura-2/AM and
monodansylcadaverine (MDC) were purchased from MilliporeSigma (Merk
KGaA).
Cell culture, establishment of the MDR
HCT-116 cell line and treatment with BAF-A1
The HCT-116 human colorectal carcinoma cell line
(American Type Culture Collection) was maintained at 37°C and 5%
CO2 in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS, 25 mM HEPES and 1%
penicillin/streptomycin cocktail (Gibco; Thermo Fisher Scientific,
Inc.). Drug-resistant cells were generated using a stepwise
increase in treatment doses with docetaxel, vincristine, cisplatin
and 5-FU following a protocol from a previously published article
by our group (16). For the
initial dose, HCT-116 cells were treated with 0.2 nM docetaxel, 0.2
nM vincristine, 0.5 µM cisplatin and 0.5 µM 5-FU
until the cells became stable in these drug doses. Stable cells
were exposed three times to the drug combination over a three-day
period for 3-4 weeks, allowing growth recovery between cycles.
After completing three drug treatment cycles, the doses were
doubled, and the procedure was repeated until treatment with the
final drug concentrations was achieved over an 8-10-month period.
The MDR subline was maintained in complete RPMI-1640 medium
containing final drug concentrations of 10 nM docetaxel, 10 nM
vincristine and 10 µM cisplatin and 10 µM 5-FU.
To inhibit autophagy induction in MDR cells,
1×106 cells were treated with 10 nM of BAF-A1 for 24 h
at 37°C and 5% CO2 in complete media; 0.1% DMSO was used
as a vehicle. Cells were subsequently used for protein isolation
and western blot analysis.
Protein isolation and western blot
analysis
Whole-cell proteins were isolated using RIPA lysis
buffer (MilliporeSigma; Merck KGaA) according to the manufacturer's
protocol. The cytosolic and nuclear fractions were isolated using a
NE-PER Nuclear Protein Extraction Kit (Thermo Scientific; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The mitochondrial protein was obtained by isolating
the mitochondria from the cells using a Mitochondria Isolation kit
(MilliporeSigma; Merck KGaA); proteins were extracted from the
isolated mitochondria using a RIPA lysis buffer. Protein samples
were quantified using a BCA Protein Assay Kit (Cell Signaling
Technology, Inc.) according to the manufacturer's protocol.
Proteins (10 µg/lane for whole cell lysates;
20 µg/lane for nuclear lysates; and 30 µg/lane for
mitochondrial lysates) were resolved by 8-15% SDS-PAGE, based on
molecular weight of proteins, and transferred to a PVDF membrane
(Roche Diagnostics). The membranes were then incubated overnight
with primary antibodies at 4°C. The membranes were then incubated
with the respective secondary antibodies for 1 h at room
temperature and visualized by ECL Western Blotting detection
reagent (cat. no. RPN2109; Amersham; Cytiva) according to the
recommended procedure. β-actin, Lamin B1 and heat-shock protein 60
(HSP60) were used as markers for the whole cell, nucleus and
mitochondria, respectively. Tables SI
and SII list the primary and secondary antibodies used for
western blot analysis, respectively. Densitometric analysis of the
protein bands was performed using ImageJ (version 1.53f)
open-source program (National Institutes of Health). β-actin, Lamin
B1 and HSP60 were used as loading controls to normalize protein
expression in whole-cell lysates, nuclear lysates and mitochondrial
lysates, respectively.
Lactate dehydrogenase (LDH) assay
WT and MDR HCT-116 cells were seeded in a 48-well
plate at 1×105 cells/well and treated with 50 µM
cisplatin and vehicle for 12 h at 37°C and 5% CO2 in
complete medium. The cisplatin and vehicle-treated cell culture
supernatants were collected and LDH release from the cells was
quantified using an LDH cytotoxicity assay kit (cat. no. TOX7;
MilliporeSigma; Merck KGaA) according to the manufacturer's
protocol. The primary absorbance was measured at 490 nm and
background absorbance was measured at 690 nm using a microplate
reader (BioTek Instruments, Inc.). Actual absorbance was calculated
by subtracting background absorbance value from the primary
absorbance value.
Hoechst staining for chromatin
condensation
WT and MDR HCT-116 cells were seeded in a 4 well
chambered slide at 1×105 cells/well and treated with 50
µM cisplatin and vehicle for 12 h at 37°C and 5%
CO2 in complete medium. Briefly, the cisplatin- and
vehicle-treated cells were subsequently fixed with 4% formaldehyde
at room temperature, washed with PBS and incubated with Hoechst
33342 (1 µg/ml) at 37°C for 10 min. After washing with PBS,
the DNA chromatin morphological features were analyzed using a
Nikon Eclipse fluorescence microscope (Nikon Corporation).
Calcium measurement
WT and MDR HCT-116 cells were seeded in a 4-well
chambered slide at 1×104 cells/well. Calcium levels were
measured by incubating the cisplatin and vehicle-treated cells with
5 µM Fura-2/AM in Hank's balanced salt solution (HBSS)
buffer for 60 min at 37°C. The samples were then washed three times
with HBSS at 37°C and fluorescence micrographs were taken using a
Nikon Eclipse TS200 epifluorescence microscope (Nikon Corporation).
The corrected fluorescence intensity of the cells was measured
using ImageJ software version 1.53f (National Institutes of Health)
by subtracting the value of mean background fluorescence from total
mean fluorescence.
Determination of the total reactive
oxygen species (ROS) and mitochondrial superoxide level
The elevation of intracellular ROS induced by a
cisplatin treatment was detected by DCFH-DA. Briefly, WT and MDR
HCT-116 cells were seeded at 1×105 cells/well in 12-well
plates and treated with 50 µM cisplatin or vehicle for 12 h
at 37°C. The cells were washed twice with PBS and incubated with 10
µM of DCFH-DA for 15 min at 37°C. Fluorescent micrographs
were obtained using a Nikon Eclipse TS200 fluorescence microscope
(Nikon Corporation). The corrected fluorescence intensity of the
cells was measured using ImageJ software version 1.53f (National
Institutes of Health) by subtracting the value of mean background
fluorescence from total mean fluorescence.
Accumulation of mitochondrial superoxide generation
was determined using MitoSOX Red™ Mitochondrial Superoxide
Indicator (Invitrogen; Thermo Fisher Scientific, Inc.). Briefly,
the WT and MDR HCT-116 cisplatin- or vehicle treated cells were
incubated with 5 µM of a MitoSOX indicator for 10 min at
37°C, then washed twice with PBS; images were captured using a
Nikon Eclipse TS200 fluorescence microscope. Mito-ID green dye
(Enzo Life Sciences, Inc.) was used to stain the mitochondria,
regardless of their energetic state. The corrected fluorescence
intensity of the cells was measured using ImageJ software version
1.53f (National Institutes of Health) by subtracting the value of
mean background fluorescence from total mean fluorescence.
Electrophoretic mobility shifts assay
(EMSA)
EMSA analyses were performed using an
Electrophoretic Mobility Shift Assay kit (cat. no. E33075;
Molecular Probes; Thermo Fisher Scientific, Inc.) according to the
manufacturer's recommendation. Nuclear extracts (1 µg),
aforementioned, were used to examine their binding capacity to the
DNA-binding motif of activator protein 1 (AP1). Table SIII lists the AP1 consensus
sequence used for EMSA, which was purchased from Santa Cruz
Biotechnology, Inc. (AP-1 Gel Shift Oligonucleotides; cat. no.
sc-2501). Briefly, 100 pmol AP1 consensus oligo was incubated with
nuclear extract in 1X binding buffer (750 mM KCl; 0.5 mM
dithiothreitol; 0.5 mM EDTA; 50 mM Tris, pH 7.4) at room
temperature for 20 min before loading onto a 6% non-denaturing
polyacrylamide gel. The EMSA bands were visualize by staining with
1X SYPRO® Ruby EMSA stain according to manufacturer
protocol.
Determination of autophagy using Cyto-ID
and MDC immunocytochemistry
A total of 1×105 WT and MDR cells
[including Mock (empty vector), GSTO1 activation CRISPR and
GSTO1 KD CRISPR transfected HCT-116 cells] were cultured on
coated glass coverslip and treated with 50 µM cisplatin or
vehicle for 12 h at 37°C and 5% CO2 in complete medium.
Cisplatin- or vehicle-treated cells were fixed with 4%
paraformaldehyde in PBS for 10 min at room temperature. The fixed
cells were stained using the components in the CYTO-ID®
Autophagy Detection Reagent (cat. no. ENZ-51031; Enzo Life
Sciences, Inc.) according to the manufacturer's protocol. Cyto-ID
is a proprietary reagent that labels the autophagy vacuoles
specifically. DAPI was used to stain the nucleus. Fluorescence
image acquisitions were performed using a Nikon Eclipse
fluorescence microscope (Nikon Corporation).
MDC (MilliporeSigma; Merck KGaA) was used to stain
the autophagy vacuoles in the GSTO1 KD plasmid transfected
MDR cells. GSTO1 KD plasmid contains a GFP as a selection
marker that restricts the use of Cyto-ID Green Detection Reagent in
this case. CYTO-ID® and MDC fluorescent intensity were
measured using ImageJ software version 1.53s (National Institutes
of Health).
Flow cytometry assays for autophagy and
apoptosis
Cell death and the development of acidic vacuoles
were quantified by flow cytometry (Beckman Coulter, Inc.). WT and
MDR HCT-116 cells [including Mock (empty vector), GSTO1 activation
CRISPR and GSTO1 KD CRISPR transfected HCT-116 cells] were seeded
at 1×105 cells/well in a 6-well plate and treated with
cisplatin or vehicle for 12 h at 37°C and 5% CO2 in
complete media.
The cells were harvested and washed twice with PBS.
An in situ Cell Death Detection Kit (cat. no. 11684795910;
Roche Applied Sciences) and CYTO-ID® Autophagy Detection
Kit (Enzo Life Sciences, Inc.) were used to quantify apoptosis
induction and the development of acidic vesicular organelles,
respectively. Green (510-530 nm) fluorescence emission from
~5×104 cells illuminated with blue (488 nm) excitation
light was measured using a Beckman Coulter Gallios flow cytometer
and analyzed by Kaluza Analysis Software version 2.0 (both from
Beckman Coulter, Inc.).
Immunoprecipitation and protein mass
fingerprinting
The aforementioned WT and MDR HCT-116 cell lysates
(1 mg in 500 µl of RIPA buffer) were immunoprecipitated with
2 µg of GSTO1 antibody, and the samples were rotated
overnight at 4°C. A total of 40 µl Protein A/G Sepharose
beads (Pierce™; Thermo Fisher Scientific, Inc.) were added to each
sample and rotated for 1 h at 4°C. The beads were washed three
times with RIPA lysis buffer, and 30 µl supernatant
fractions were collected by centrifugation at 2,000 × g for 10 min
and subsequently subjected to 10% SDS-PAGE followed by stained with
Coomassie blue. The proteins were identified by peptide mass
fingerprinting. Briefly, protein spots were excised, digested with
trypsin (Promega Corporation), and mixed with
α-cyano-4-hydroxycinnamic acid in 50% acetonitrile/0.1% TFA, and
subjected to MALDI-TOF analysis (Microflex LRF 20; Bruker
Daltonics; Bruker Corporation). Spectra were collected from 300
shots/spectrum over the m/z range 600-3000 and calibrated by
two-point internal calibration using Trypsin auto-digestion peaks
(m/z 842.5099, 2211.1046). The peak list was generated using Flex
Analysis 3.0 (Bruker Daltonics; Bruker Corporation). The threshold
used for peak-picking was as follows: 500 for a minimum resolution
of monoisotopic mass, 5 for the signal to background noise. The
MASCOT online database (Matrix Science, Ltd.) was used for protein
identification. The following parameters were used for the database
search: Trypsin as the cleaving enzyme; a maximum of one missed
cleavage; iodoacetamide (Cys) as a complete modification; oxidation
(Met) as a partial modification; monoisotopic masses; and a mass
tolerance of ±0.1 Da.
Generation of GSTO1-activated,
GSTO1-knockdown (KD) and TNFαIP3/A20-KD cells using
CRISPR-Cas9
CRISPR-Cas9-mediated genome editing was performed
using the GSTO1 CRISPR-Cas9 KD human plasmid (cat. no.
sc-404107), TNFαIP3/A20 CRISPR-Cas9 KD human plasmid (cat.
no. sc-400447-KO-2) and GSTO1 CRISPR activation human
plasmid (cat. no. sc-404107-ACT) purchased from Santa Cruz
Biotechnology, Inc. Human GSTO1 KD CRISPR plasmid mixture is
a pool of 3 different guide (g)RNA plasmids, each encoding the Cas9
nuclease and a target-specific 20 nucleotide (nt) gRNA in the
GSTO1 gene: gRNA 1 (5′-GCG TCT AGT CCT GAA GGC CA-3′), which
targets exon 2 and N-terminal domain of the protein; gRNA 2 (5′-ACA
ACT CTA AGA TCA TCT TC-3′), which targets exon 3 and N-terminal
domain of the protein; and gRNA 3 (5′-TCTAATAAAGCTTCCTACCA-3′),
which targets exon 6 and C-terminal domain of the protein
GSTO1. CRISPR/Cas9 KD plasmid disrupted gene expression by
causing a double-strand break in a 5′ constitutive exon within the
GSTO1 gene. A single gRNA (5′-AGG TCA GTG TCA CGG GAG GG-3′)
sequence was used for human GSTO1 CRISPR activation plasmid.
The human GSTO1 CRISPR activation plasmid is a synergistic
activation-mediator (SAM) transcription activation system that
specifically upregulates the GSTO1 gene. The GSTO1
CRISPR activation plasmid solution contains an equimolar ratio of
the following three plasmids: CRISPR/dCas9-VP64-Blast plasmid,
which encodes the deactivated Cas9 (dCas9) nuclease fused to the
transactivation domain VP64 and a blasticidin resistance gene;
MS2-P65-HSF1-Hygro plasmid, which encodes the MS2-p65-HSF1 fusion
protein and a hygromycin resistance gene; sgRNA (MS2)-Puro plasmid,
which encodes a target-specific 20 nt gRNA and a puromycin
resistance gene. The SAM complex activates transcription of
GSTO1 and upregulates GSTO1 gene expression.
Human TNFαIP3/A20 KD CRISPR plasmid is also a
pool of 3 different gRNA plasmids: gRNA 1 (5′-CAC GCA ACT TTA AAT
TCC GC-3′), which targets exon 3 and TRAF-binding domain of the
protein; gRNA 2 (5′-GTG AAC GTT GCC ACA ACG CC-3′), which targets
exon 7 and TNIP1-binding domain of the protein; and gRNA 3 (5′-CTT
GTG GCG CTG AAA ACG AA-3′), which targets exon 2 and TRAF-binding
domain of the protein. TNFαIP3/A20 CRISPR/Cas9 KD plasmid
disrupted gene expression by causing a double-strand break in a 5′
constitutive exon within the TNFαIP3/A20 gene. A total of
1×106 WT and MDR HCT-116 cells were seeded on a 6-well
plate in antibiotic free complete media. The cells were transfected
with 1 µg of CRISPR plasmid mixture using
UltraCruz® transfection reagent (Santa Cruz
Biotechnology, Inc.) for 24 h at 37°C. Transfection media was
replaced with complete growth media and cells were incubated at
37°C for an additional 48 h before selection using 1 µg/ml
puromycin (MilliporeSigma; Merck KGaA). Cells were selected for 2-4
weeks, dependent on growth recovery. Western blotting was used to
confirm the KD and activation efficiency. An empty vector was used
as the Mock group in both GSTO1-activation and -KD
experiments.
Statistical analysis
All data are presented as means ± SD using an
unpaired Student's t-test or one-way ANOVA (with Tukey's post hoc
test for multiple comparisons) using GraphPad Prism 9.0 software
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference. Each experiment was performed
at least three times unless indicated otherwise. Each western blot
was performed in duplicate.
Results
MDR HCT-116 cells exhibit higher GSTO1
expression and reduced cisplatin sensitization
ABCB1 expression is increased in cancer cells during
the development of anti-cancer drug resistance, which facilitates
the efflux of drugs from the cell (17). ABCB1 protein expression was
increased in MDR HCT-116 cells compared with the WT cells as
determined by western blotting (Fig.
1A); GSTO1 expression was also increased in the MDR cells
compared with WT cells. MDR HCT-116 cells showed resistance to the
50 µM cisplatin treatment compared with WT cells, as
determined by an LDH assay (Fig.
1B) and apoptosis assay using flow-cytometry (Figs. 1C, S1E, S1F and S2). Cisplatin treatment
significantly increased LDH release in WT cells compared with the
vehicle-treated cells (Fig. 1B).
LDH release in cisplatin-treated MDR cells was also significantly
higher compared with vehicle-treated MDR cells, although the
effectiveness of cisplatin was much lower in MDR cells compared
with the WT cells (Fig. 1B).
Moreover, cisplatin treatment increased apoptosis significantly in
WT cells compared with the vehicle-treated cells, but the effect
was lower in MDR cells (Figs. 1C,
S1E, S1F and S2).
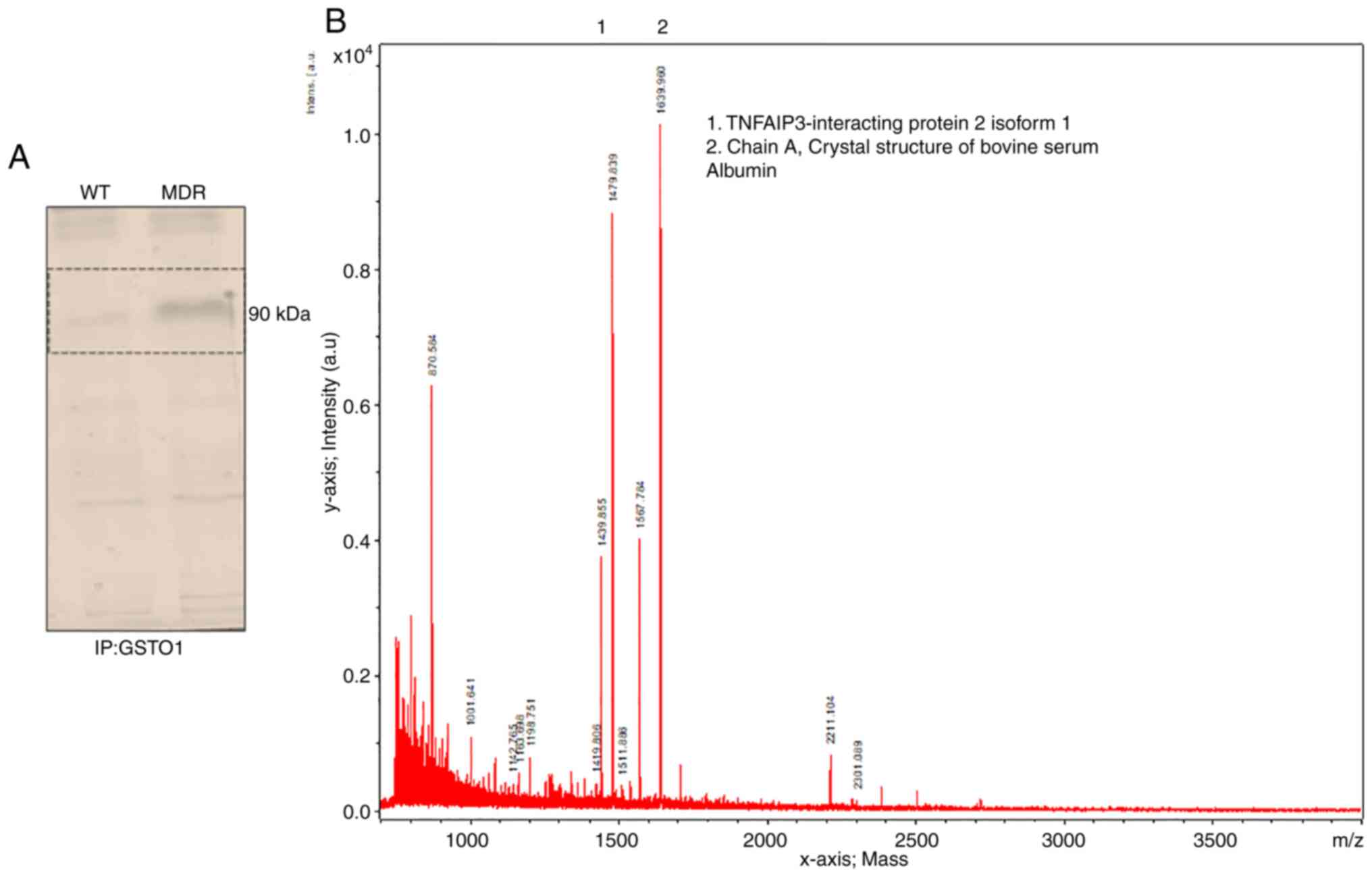
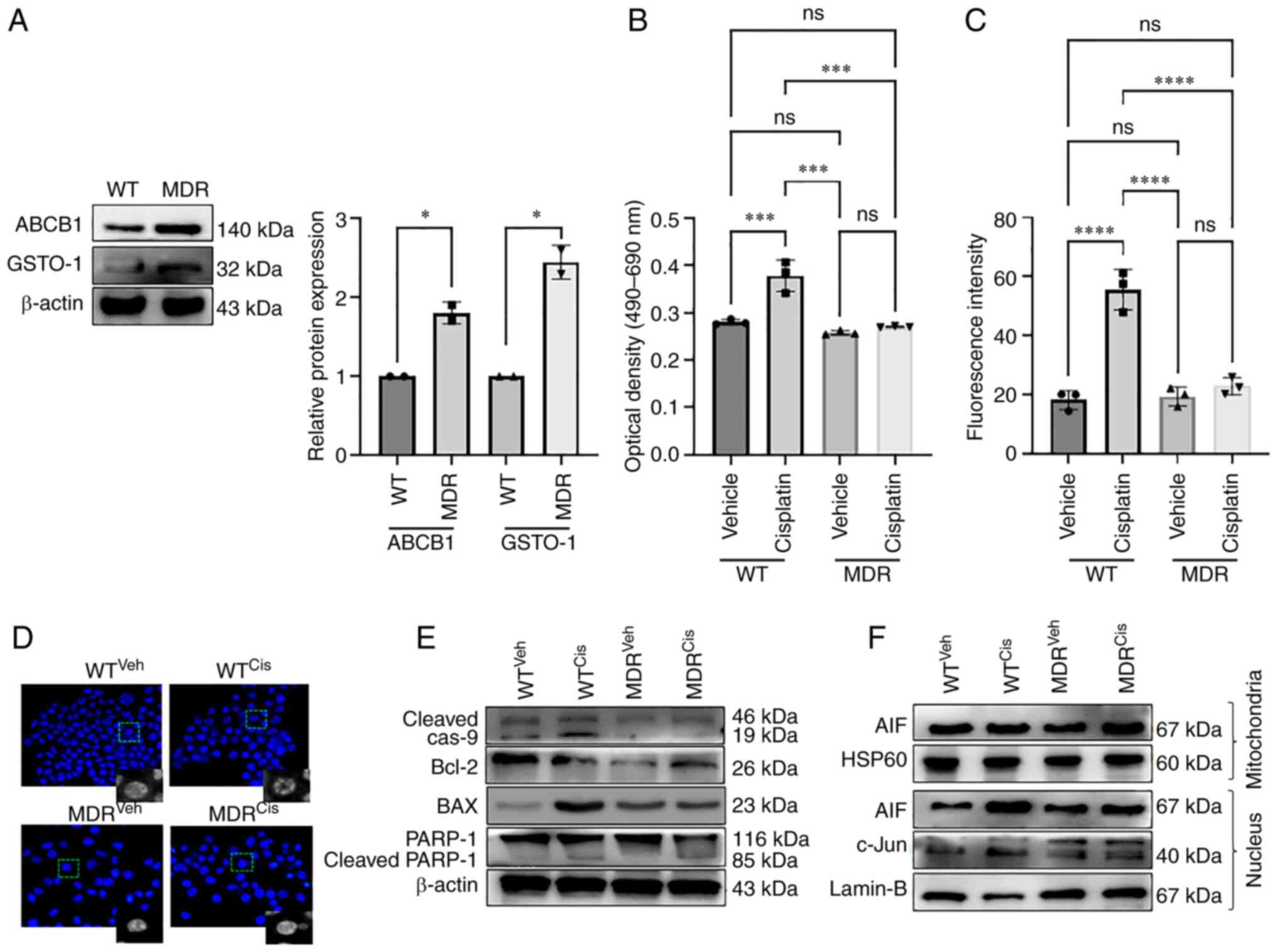 | Figure 1MDR HCT-116 cells are resistant to
cisplatin-induced apoptosis. (A) The protein expression levels of
the ABCB1 and GSTO1 proteins in WT and MDR HCT-116 cells were
analyzed by western blotting and quantified using ImageJ software.
Unpaired Student's t-test was used for statistical analysis. WT and
MDR cancer cells were treated with 50 µM cisplatin for 12 h,
and cell apoptosis was analyzed by (B) LDH assay and (C) ApoStrand™
apoptosis assay; one-way ANOVA was used for statistical analysis.
(D) Nuclear damage was analyzed by Hoechst 33342 staining (×200
magnification). (E) Western blotting of apoptosis markers. (F)
Mitochondrial and nuclear proteins were analyzed by western
blotting to determine the expression of AIF and c-Jun proteins.
*P<0.05; ***P<0.001;
****P<0.0001. AIF, apoptosis-inducing factor; cas,
caspase; cis, cisplatin; GSTO1, glutathione-S-transferase Ω-1; HSP,
heat-shock protein; LDH, lactate dehydrogenase; MDR, multidrug
resistant; MDR1, multidrug-resistance protein 1; ns, not
significant; PARP, poly(ADP-ribose) polymerase 1; veh, vehicle; WT,
wild-type. |
Cisplatin-treated WT cells showed morphological
deformities, mostly membrane blebbing compared with the
vehicle-treated WT, MDR and cisplatin-treated MDR cells (Fig. S1A). Total ROS generation was
increased in both cisplatin-treated WT and MDR cells compared with
their respective vehicle-treated cells; however, no significant
difference was noted in the cisplatin-treated MDR cells compared
with the cisplatin-treated WT cells (Fig. S1B). The mechanism of
cisplatin-mediated cytotoxicity is dependent on DNA-adduct
formation; that is, DNA damage-mediated cell death (18). Fig.
1D shows severe chromatin decondensation in the
cisplatin-treated WT cells, which was not visible in the MDR or
cisplatin-treated MDR cells. DNA damage by cisplatin treatment
leads to mitochondria-mediated apoptosis (18-20).
The total ROS generation following cisplatin treatment led to an
increase in ROS in the mitochondria of WT and MDR cells compared
with their respective vehicle-treated controls; however, no
significant difference was noted between the two cisplatin-treated
groups (Fig. S1C and D).
Cisplatin treatment increased the cleavage of
caspase 9 and poly(ADP-ribose) polymerase 1, and increased Bax
expression in the WT cells compared with the vehicle-treated WT
cells and MDR cells (Figs. 1E and
S3A). Expression of cleaved
PARP-1 was also significantly higher in cisplatin-treated MDR cells
compared with the vehicle-treated MDR cells, although the level was
much lower than that of cisplatin-treated WT cells (Figs. 1E and S3A). Bcl2 expression levels was not
significantly different between vehicle-treated WT cells and
cisplatin-treated WT cells, nor between cisplatin-treated WT cells
and cisplatin-treated MDR cells (Figs.
1E and S3A). Moreover, Bcl2
expression was significantly reduced in vehicle and
cisplatin-treated MDR cells compared with the vehicle-treated cells
(Figs. 1E and S3A). A previous publication also
demonstrated a reduction of Bcl2 expression during autophagy
activation (21). The
translocation of the apoptosis-inducing factor (AIF) from the
mitochondria to the nucleus induces apoptosis in drug-treated
cancer cells (22). The
nuclear/mitochondrial expression ratio of AIF was significantly
increased in the cisplatin-treated WT cells compared with the
vehicle-treated WT cells and cisplatin-treated MDR cells (Figs. 1F, S3B and C). Cisplatin treatment also
reduced c-Jun nuclear expression (Figs. 1F and S3D) and binding of AP1 transcription
factor with its consensus DNA sequence in the cisplatin-treated MDR
cells compared with the cisplatin-treated WT cells (Fig. S4A and B), a well-documented mode
of action of cisplatin-mediated cell death (23). AP1 binding with its DNA sequence
was also reduced in vehicle-treated compared with the
cisplatin-treated WT cells. Moreover, cisplatin-treated WT cells
showed similar AP1 binding as seen in vehicle and cisplatin-treated
MDR cells (Figs. S4A and B).
MDR HCT-116 cells show an unfolded
protein response (UPR)-mediated autophagy
Previous studies suggest that the elevated levels of
mitochondrial ROS can promote both autophagy and apoptosis
(9,24,25).
Cisplatin-treatment activated calcium signaling in MDR cells
(Fig. 2A and B). Significantly
higher calcium deposition was noted in the cisplatin-treated MDR
cells compared with the vehicle-treated MDR and the
cisplatin-treated WT cells (Fig. 2A
and B). Cellular calcium level was also higher in
cisplatin-treated WT cells compared with the vehicle-treated WT
cells (Fig. 2A and B), although
that level was much lower than in cisplatin-treated MDR cells. A
previous study demonstrated that cellular calcium level determined
cellular fate towards apoptosis or autophagy (26). High calcium can activate ROS in
mitochondria (27), similar to
what was observed in cisplatin-treated WT cells (Fig. 2A and B). Calcium-induced UPR is a
well-studied mechanism that ultimately leads to autophagy (24,28).
Expression levels of the UPR protein markers glucose-regulated
protein 78 (GRP78), CHOP and PKR-like endoplasmic reticulum kinase
(PERK) were increased in the cisplatin-induced MDR cells compared
with the cisplatin-induced WT cells (Figs. 2C and S5A). Interestingly, no significant
difference was noted when inositol-requiring kinase 1α (IRE1α)
expression was compared between cisplatin-treated MDR and WT cells
(Figs. 2C and S5A). The present study also noticed a
significantly higher expression of IRE1α and CHOP in
cisplatin-treated WT cells compared with the vehicle-treated WT
cells, suggesting a mild ER stress mediated-apoptosis (29,30).
No significant difference in GRP78, IRE1α, CHOP and PERK protein
expression levels were noted in vehicle-compared with
cisplatin-treated MDR cells (Figs.
2C and S5A).
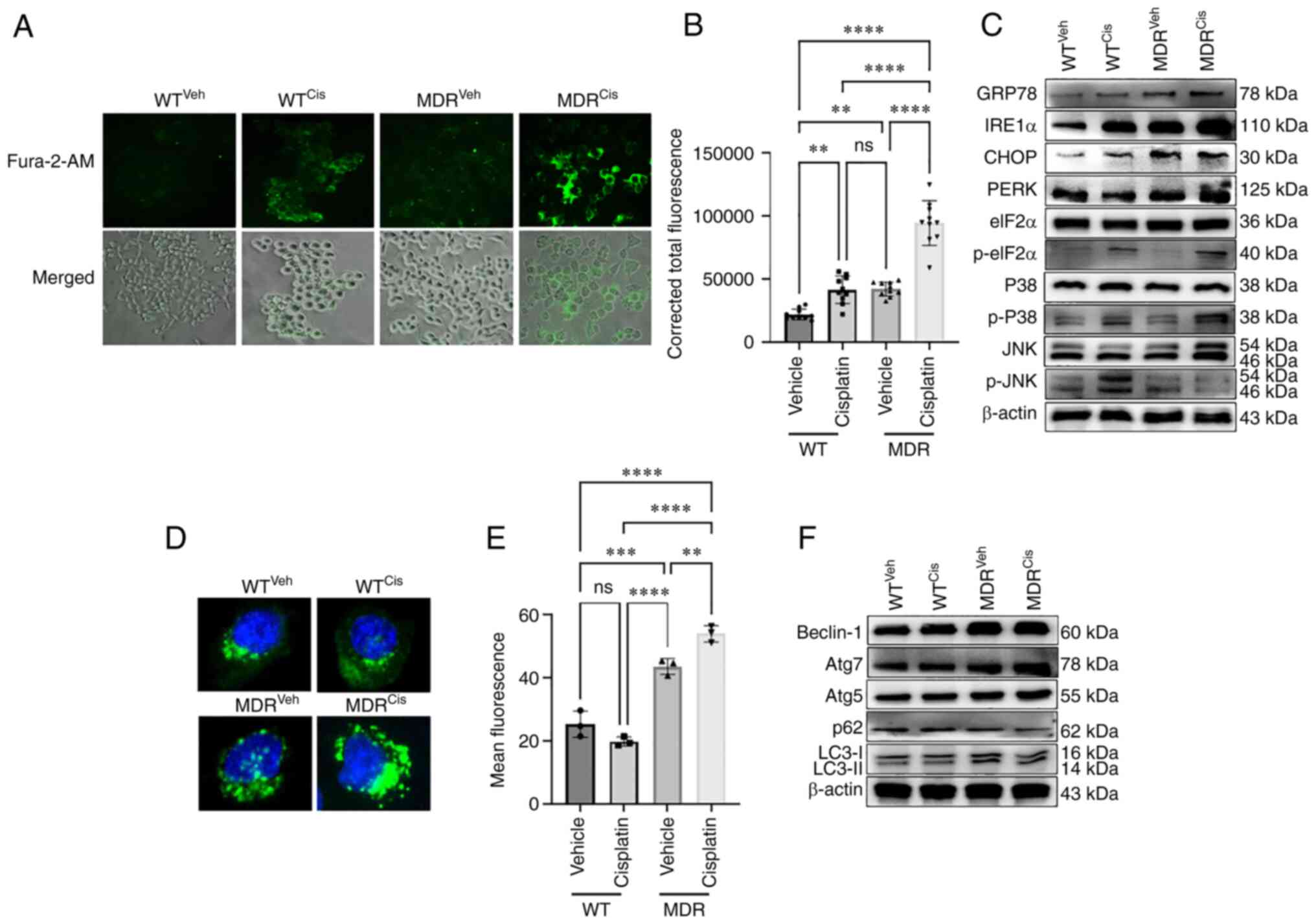 | Figure 2MDR HCT-116 cells exhibit an increase
in calcium signaling-dependent UPR and autophagy. WT and MDR
HCT-116 cells were treated with 50 µM cisplatin or vehicle
for 12 h and subsequently (A) stained with Fura-2/AM to determine
the cellular calcium release (×200 magnification) and (B)
quantified using ImageJ (n=400 cells); one-way ANOVA was used for
statistical analysis. (C) Western blotting analysis of ER stress-
and UPR-related markers in WT and MDR cells with or without
cisplatin exposure. (D) Fluorescence images of autophagic vesicles
stained with Cyto-ID® Autophagy reagent (green) in WT
and MDR cells (×1,000 magnification); nuclei were stained with
Hoechst 33342 (blue). (E) Quantification of fluorescence intensity
presented of cells presented in part (D); one-way ANOVA was used
for statistical analysis. (F) Representative immunoblots of
autophagy marker protein expressions in whole-cell protein lysates.
**P<0.01; ***P<0.001;
****P<0.0001. ATG, autophagy-related gene; cis,
cisplatin; eIF2α, eukaryotic initiation factor 2α; ER, endoplasmic
reticulum; GRP78, glucose-regulated protein 78; IRE1α,
inositol-requiring kinase 1α; MDR, multidrug resistant; ns, not
significant; p-, phosphorylated; PERK, PKR-like endoplasmic
reticulum kinase; UPR, unfolded protein response; veh, vehicle; WT,
wild-type. |
The phosphorylation of p38 and eukaryotic initiation
factor 2α (eIF2α) were reported as positive markers for ER
stress-mediated UPR mechanism, even though both are also expressed
during apoptosis (31,32). p38 and eIF2α proteins can be
phosphorylated during activation of apoptosis or autophagy
(33,34). The phosphorylation of eIF2α was
higher in the cisplatin-treated WT and MDR cells compared with the
vehicle-treated WT and MDR cells (Figs. 2C, S5A and B). Interestingly,
phosphorylation of p38 showed no significant difference between
vehicle and cisplatin-treated WT cells nor between
cisplatin-treated WT and MDR cells. However, p-p38/p38 ratio was
significantly higher in cisplatin-treated MDR cells compared to the
cisplatin-treated WT cells (Figs.
2C, S5A and B). JNK
phosphorylation was lower in the cisplatin-treated MDR cells
compared with the cisplatin-treated WT cells (Figs. 2C, S5A and B).
Furthermore, immunofluorescence and flow cytometry
confirmed an increase in autophagic flux in MDR cells compared with
WT cells, regardless of the treatments. A significant increase in
autophagy activation was observed in vehicle- and cisplatin-treated
MDR cells compared with the vehicle- and cisplatin-treated WT cells
(Figs. 2D and E); autophagic flux
was higher upon cisplatin treatment compared with vehicle treatment
in MDR cells. Similarly, flow cytometry also demonstrated higher
autophagic flux in MDR cells compared with the WT cells
irrespective of treatment. Notably, cisplatin treatment further
increased autophagic flux in MDR cells compared with in
vehicle-treated MDR cells (Figs. S6A,
S6B and S7). The protein expression levels of autophagy markers
Beclin-1, autophagy-related gene (ATG)-7, ATG5 and LC3II increased,
whereas the expression of ubiquitin-binding protein p62 (p62; also
known as sequestosome-1) decreased in cisplatin-treated MDR cells
compared with the cisplatin-treated WT cells (Figs. 2F, S6C and D). Treatment with autophagy
inhibitor BAF-A1 sensitized the MDR cells to cisplatin, inducing
the expression of apoptosis and depleting the levels of autophagy
markers (Fig. S8). BAF-A1
treatment significantly increased p62 expression in MDR cells
compared with the vehicle-only- and cisplatin-only-treated group
(Fig. S8A and B); however, BAF-A1
and cisplatin co-treatment further increased p62 level compared
with all other groups. LC3II/LC3I ratio was decreased in
BAF-A1-treated cells with or without cisplatin co-treatment
compared with the vehicle-only- and cisplatin-only-treated cells
(Figs. S8A and C). Moreover,
BAF-A1 and cisplatin co-treatment increased expression levels of
the apoptosis markers cleaved caspase 9, cleaved caspase 3 and
cytochrome c compared with the vehicle, only BAF-A1and only
cisplatin-treated cells (Figs. S8A
and D-F).
CRISPR-Cas9 activation of GSTO1 in WT
HCT-116 cells activates autophagy
To mimic the drug-resistance condition in WT cells,
GSTO1 was activated and checked to determine if it could
induce autophagy. Figs. 3A and
S9E demonstrated that CRISPR-Cas9
genome editing successfully induced GSTO1 expression in vehicle-
and cisplatin-treated WT cells compared with the Mock groups.
GSTO1 activation induced the expression of autophagy marker
proteins Beclin-1 and LC3II in cisplatin-treated,
GSTO1-activated WT cells compare with the vehicle- or
cisplatin-treated Mock cells (Figs.
3A and S10A). Interestingly,
Beclin-1 and LC3II expression levels were significantly higher in
cisplatin-treated, GSTO1-activated cells compared with the
vehicle-treated, GSTO1-activated cells, which could be
attributed to a connection between GST-mediated drug-efflux and
autophagy activation (35).
Apoptosis marker cleaved caspase 9 was significantly reduced
marginally in the GSTO1-activated, cisplatin-treated WT
cells compared with the cisplatin-treated Mock WT cells (Figs. 3A and S10A).
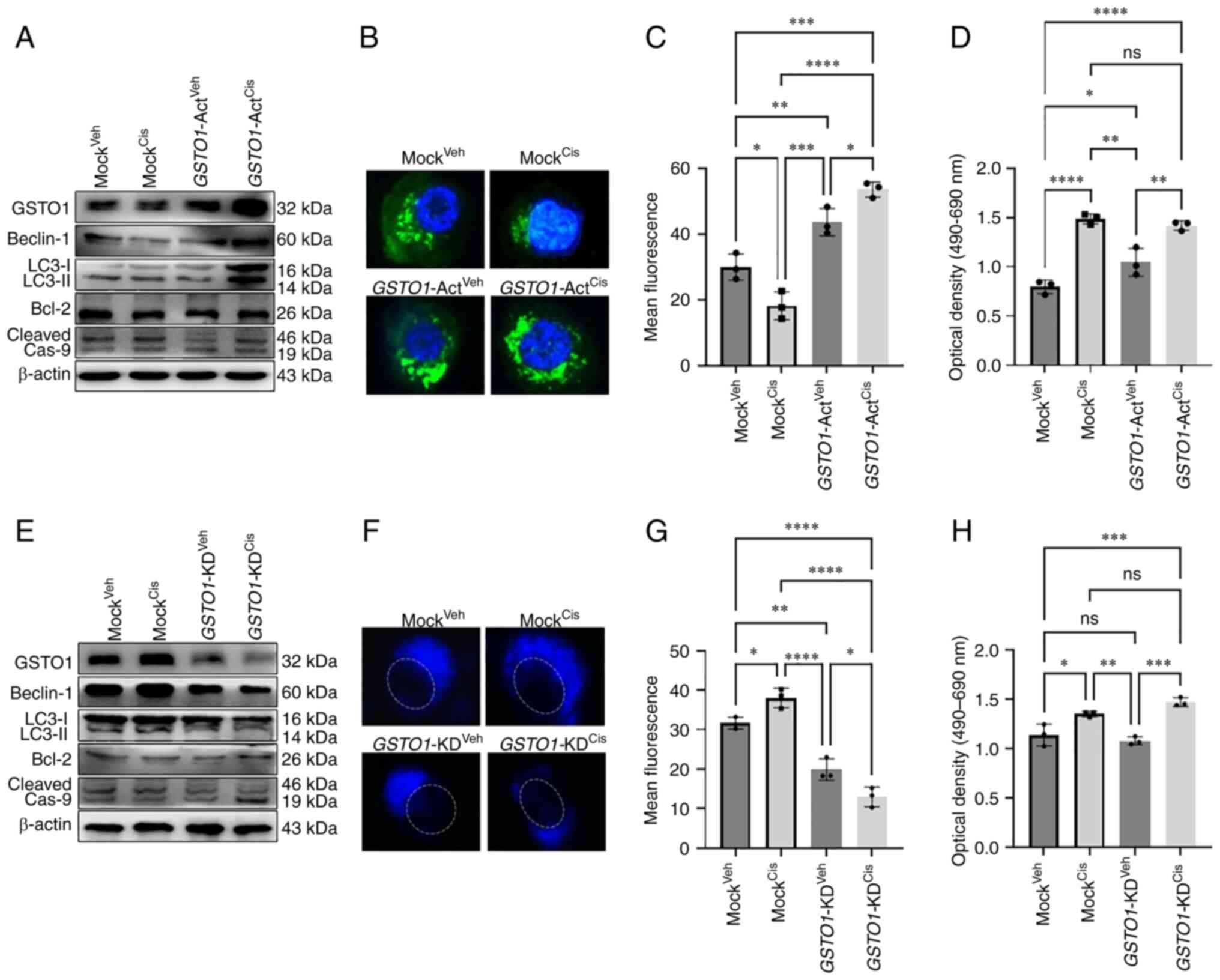 | Figure 3GSTO1 activation in WT HCT-116
cells induces autophagy following cisplatin treatment, whereas
GSTO1 inhibition in MDR cells induces apoptosis.
GSTO1 was overexpressed using GSTO1-Act CRISPR
plasmid transfected in WT HCT-116 cells. (A) Western blotting was
performed to determine the protein expression levels of GSTO1
expression as well as autophagy and apoptosis marker proteins. (B)
Fluorescence images of autophagic vesicles stained with
Cyto-ID® Autophagy reagent (green) in Mock- and
GSTO1-Act plasmid-transfected WT cells (×1,000
magnification); nuclei are stained with DAPI (blue). (C)
Quantification of fluorescence intensity of the data presented in
part (B); one-way ANOVA was used for statistical analysis. (D) Cell
death was analyzed using an LDH ELISA assay; one-way ANOVA was used
for statistical analysis. GSTO1 was inhibited using the
GSTO1-KD CRISPR/Cas9 plasmid in MDR cells. (E) Western
blotting was used to assess protein expression levels of GSTO1 as
well as autophagy and apoptosis marker proteins. (F) Representative
fluorescence images of autophagic vesicles stained with
monodansylcadaverine (blue); the dashed white line indicates the
outline of the nucleus (×1,000 magnification). (G) Quantification
of fluorescence intensity of the data presented part (F); one-way
ANOVA was used for statistical analysis. (H) Cell death was
analyzed using an LDH ELISA assay; one-way ANOVA was used for
statistical analysis. *P<0.05;
**P<0.01; ***P<0.001;
****P<0.0001. Act, activation; cas, caspase; cis,
cisplatin; GSTO1, glutathione-S-transferase Ω-1; KD, knockdown;
LDH, lactate dehydrogenase; MDR, multidrug resistant; ns, not
significant; veh, vehicle; WT, wild-type. |
Unexpectedly, Beclin-1 expression was reduced
considerably in cisplatin-treated Mock CRISPR-transfected cells
(Figs. 3A and S10A) compared with WT cisplatin-treated
cells (Figs. 2F and S6C). Notably, a slight increase in the
number of floating cells in the Mock cisplatin-treated
CRISPR-transfected group compared with the WT cisplatin-treated
group (data not shown). Presumably, the puromycin selection
procedure might increase cisplatin sensitivity in Mock CRISPR cells
compared with WT cells, leading to necroptosis and decreased
Beclin-1 expression. However, a recent study also revealed that
puromycin selection might occasionally impede protein expression
(36).
Immunofluorescence and flow cytometric analysis
showed that GSTO1 activation and cisplatin treatment
resulted in higher autophagy flux compared with that observed in
the cisplatin-treated Mock cells. Cisplatin treatment produced more
autophagic vesicles in GSTO1-activated cells compared with
the cisplatin-treated Mock cells (Figs. 3B, 3C, S9C, S9D
and S11). The LDH assay revealed a slight reduction of
apoptosis in the cisplatin-treated, GSTO1-activated cells
compared with the cisplatin-treated Mock cells, but the difference
was not significant (Fig. 3D).
Cisplatin treatment induced significant amount of apoptotic cell
death in Mock group compared with the vehicle-treated Mock and
GSTO1-ACT groups (Fig. 3D).
Protein expression levels of the UPR-related protein markers GRP78,
IRE1α and PERK were not changed in either cisplatinor
vehicle-treated GSTO1 activated cells compared with the cisplatin-
or vehicle-treated Mock cells (Fig.
S9A and B).
CRISPR-Cas9 inhibition of GSTO1 in MDR
HCT-116 cells inhibits autophagy and sensitizes them to cisplatin
treatment
Figs. 3E and
S9E confirmed the GSTO1
inhibition efficiency in MDR cells. Although GFP fluorescence
showed a satisfactory transfection efficiency of Control and
GSTO1-KD CRISPR-Cas9 plasmid (Fig. S9F), residual GSTO1 was detected in
western blotting analysis. Therefore, we used the term 'knockdown',
rather than knockout. The inhibition of GSTO1 in MDR cells
reduced the expression levels of the autophagy markers Beclin-1 and
LC3II compared with their expressions in the Mock MDR cells
(Figs. 3E and S10B). Protein expression of the
apoptosis marker cleaved caspase 9 was increased in
GSTO1-inhibited cisplatin-treated MDR cells compared with
the cisplatin-treated Mock MDR cells (Figs. 3E and S10B). Consistent with the western
blotting results, autophagy was also reduced in the GSTO1-KD
MDR cells compared with the Mock MDR group (Fig. 3F and G). The LDH assay further
confirmed the sensitization of GSTO1-inhibited MDR cells to
cisplatin (Fig. 3H).
Cisplatin-treatment significantly induced LDH release in
GSTO1-KD cells compared with the cisplatin-treated Mock
cells, a vehicle-treated Mock or GSTO1-KD cells (Fig. 3H). GSTO-KD and cisplatin
treatment in MDR cells inhibited GRP78, IRE1α and PERK compared
with the Mock cisplatin-treated cells (Fig. S9G and H).
GSTO1-TNFαIP3/A20 interaction is a novel
mechanism towards drug resistance
The GSTO1 pull-down assay showed an interacting
protein band in MDR cells but not in WT cells (Fig. 4A). Subsequently, MALDI-TOF mass
spectrometry identified TNFαIP3/A20 as a putative novel binding
partner of GSTO1 in MDR cells (Fig.
4B). Immunoprecipitation of the GSTO1 protein and subsequent
immunoblotting for TNFαIP3/A20 in MDR cells confirmed an
interaction between GSTO1 and TNFαIP3/A20 (Fig. 5A). TNFαIP3/A20 expression was
increased in both vehicle- and cisplatin-treated MDR cells compared
with the vehicle- and cisplatin-treated WT cells (Fig. 5B and C). Moreover, interaction with
GSTO1 also increased in the MDR cells compared with the WT cells,
particularly following cisplatin treatment (Fig. 5D and E). Furthermore, TNFαIP3/A20
and GSTO1 interaction was reduced in GSTO1-KD MDR cells
compared with Mock-transfected cells (Fig. 5F). Western blotting analysis showed
that TNFαIP3/A20 protein expression may be dependent on the GSTO1
expression status, as significantly reduced TNFαIP3/A20 expression
was detected in the GSTO1-KD cells (Figs. 5G and S12). GSTO1 and
TNFαIP3/A20 single-gene or double-gene KD combined
with cisplatin co-treatment showed significantly reduced Beclin-1
expression compared with cisplatin-treated Mock cells. No
significant differences were identified between GSTO1 and
TNFαIP3/A20 single-gene KD and double-gene KD groups
(Figs. 5G and S12). LC3II/LC3I ratio was also reduced
significantly in GSTO1 and TNFαIP3/A20 single-gene or
double-gene KD cisplatin-treated cells compared with
cisplatin-treated Mock cells (Figs.
5G and S12). GSTO1 and
TNFαIP3/A20 double-gene KD cisplatin-treated cells showed
significantly lower LC3II/LC3I ratio compared with GSTO1 KD
cisplatin-treated cells. Expression levels of cleaved caspase 9
were increased in GSTO1 single-gene KD or GSTO1 and
TNFαIP3/A20 double-KD cisplatin-treated cells compared with
the cisplatin-treated Mock cells (Figs. 5G and S12); no significant difference was
identified between the cisplatin-treated TNFαIP3/A20-KD and
the cisplatin-treated Mock groups. GSTO1 and
TNFαIP3/A20 double-gene KD cisplatin-treated cells showed
significantly higher cleaved caspase 9 expression compared with the
GSTO1 and TNFαIP3/A20 single-gene KD cisplatin-treated cells
(Figs. 5G and S12). Notably, only GSTO1 and
TNFαIP3/A20 double-gene KD cisplatin-treated cells showed
significantly higher Bax expression compared with the other three
groups (Figs. 5G and S12). Overall, the results suggested that
the GSTO1-TNFαIP3/A20 axis may be important for drug resistance in
colon cancer cells, and the inhibition of this axis may sensitize
MDR cells to cisplatin.
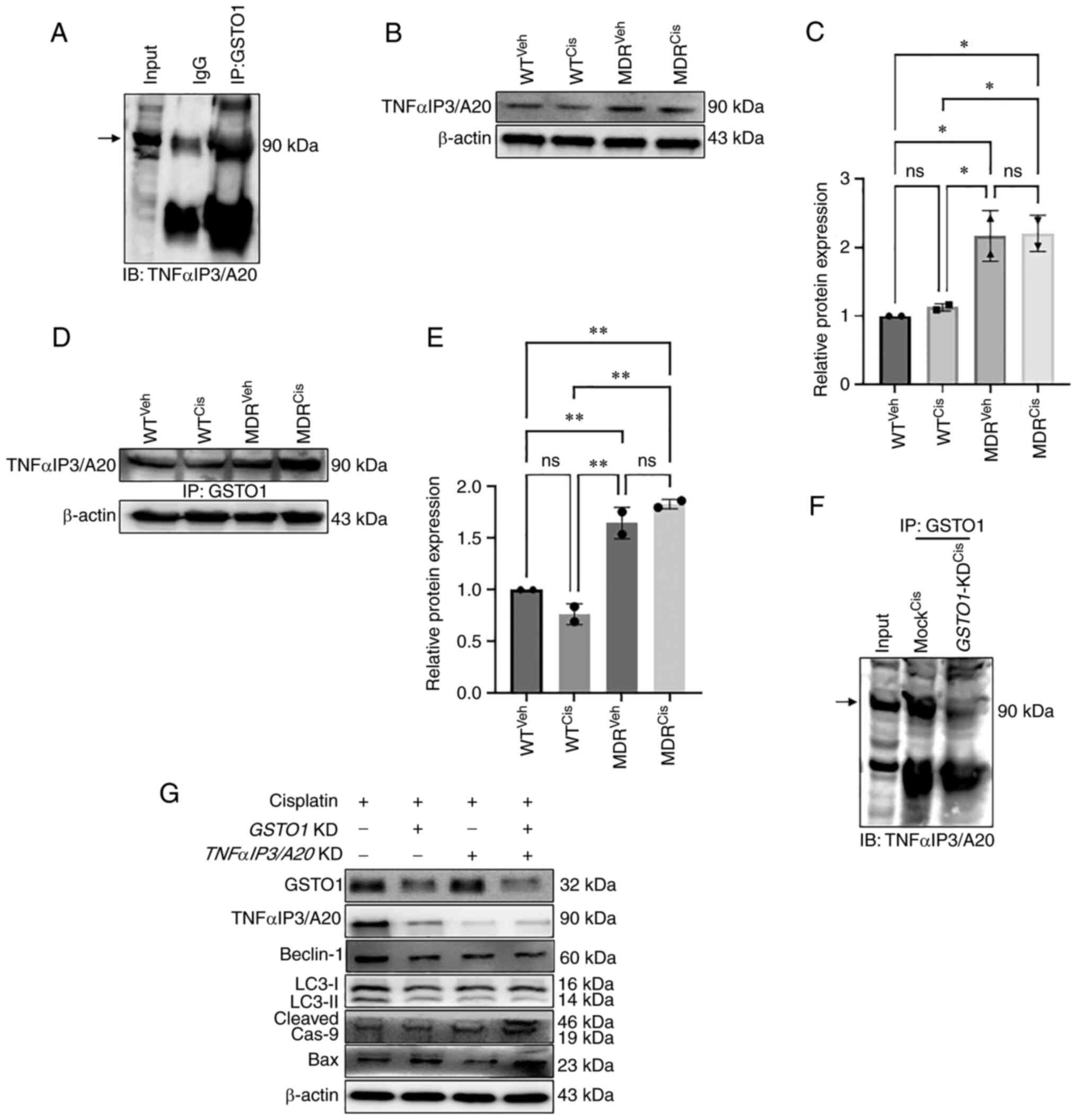 | Figure 5Inhibition of GSTO1-TNFαIP3/A20
interaction sensitizes MDR HCT-116 cells to cisplatin. (A) Co-IP
was used to determine the interaction of endogenous GSTO1 and
TNFαIP3/A20 in the whole cell lysate of MDR HCT116 cells. (B)
Western blotting and (C) densitometric analysis showing that
TNFαIP3/A20 expression was increased in MDR cells compared with the
WT cells irrespective of treatments; one-way ANOVA was used for
statistical analysis. (D) GSTO1 pull-down, western blotting for
TNFαIP3/A20 and (E) densitometric analysis showing that the
GSTO1-TNFαIP3/A20 interaction is increased in MDR cells compared
with WT cells irrespective of treatments; one-way ANOVA was used
for statistical analysis. (F) GSTO1 co-IP in cis-treated Mock and
GSTO1-KD MDR cells shows that the GSTO1-TNFαIP3/A20
interaction is reduced in GSTO1-KD cells. (G) Representative
western blotting images of GSTO1, TNFαIP3/A20 and apoptosis- and
autophagy-related protein markers in GSTO1 and
TNFαIP3/A20 single-KD and double-KD MDR cells showing that
the double KD had a more notable effect compared with either single
KD. *P<0.05; **P<0.01. GSTO1,
glutathione-S-transferase Ω-1; cas, caspase; cis, cisplatin; IB,
immunoblot; IP, immunoprecipitation; KD, knockdown; MDR, multidrug
resistant; ns, not significant; TNFαIP3/A20, TNF-α-induced protein
3/zinc-finger protein A20; veh, vehicle; WT, wild-type. |
Discussion
Cisplatin is commonly used as a chemotherapeutic
agent in a number of cancers, including colon cancer (37). Cisplatin induces oxidative damage
of DNA, mitochondrial apoptosis, and cell death (38). However, some cancers, including
colon, lung and ovarian cancer, can acquire resistance to cisplatin
chemotherapy, leading to treatment failure (39-41).
Cancer cell resistance to a particular drug is often accompanied by
resistance to other drugs, a condition known as multidrug
resistance, which significantly reduces cancer chemotherapy
efficacy and overall survival of patients (42). GST family enzymes are responsible
for acquired resistance either by drug efflux or drug inactivation;
GST family proteins protect cellular macromolecules from oxidants
(43,44). A previous study showed that GSTs
inactivate cisplatin by thiol conjugation (18). Although isoforms of GST enzyme have
been reported to serve a role in drug resistance (5-7),
little is known about the role of GSTO1 in drug resistance. Also,
there are no previous reports showing that GSTO1 is increased in
chemoresistant colon cancer tissues. In this study, the molecular
mechanism of GSTO1-mediated drug resistance was investigated. The
results demonstrated a higher expression of both ABCB1 and GSTO1 in
MDR cells compared with WT HCT-116 cells. Although cisplatin
effectively induced the total cellular ROS and mitochondrial ROS in
both WT and MDR cells, the ROS in WT cells led to
mitochondria-dependent apoptosis, whereas it activated calcium
signaling in MDR. Interestingly, transcription factor AP1 showed
significantly higher binding with its DNA sequence in
cisplatin-treated WT, MDR and vehicle-treated MDR cells compared
with the vehicle treated WT cells. AP1 is a transcription factor
that binds with consensus DNA sequences across the genome and AP1
binding with its consensus DNA increased upon apoptosis activation
(19). A previous publication also
documented that AP1 is essential for autophagosomes formation
(45).
We hypothesized that calcium signaling may aid MDR
cells bypass cisplatin by activating UPR-mediated autophagy
activation. Therefore, the role of calcium in chemoresistant colon
cancer needs to be studied in further detail, including GSTO1
knockout in an animal model.
The overexpression of GSTO1 in WT cells
induced autophagy-mediated cell survival following cisplatin
treatment. Therefore, targeting GSTO1 in drug-resistant cells could
be an attractive target for resensitizing MDR colon cancer cells to
cisplatin. A recent publication demonstrated that YTH
N6-methyladenosine RNA binding protein 1 resensitized
drug-resistant colon cancer cells to cisplatin (46). A recent report documented a novel
role of GSTP1 in adriamycin resistance in breast cancer cells by
activating autophagy (47). The
overexpression of GSTP1 in MCF-7 cells activated autophagy, whereas
inhibition of GSTP1 reduced autophagy in drug-resistant cells
(47). Furthermore, GSTO1
inhibition in MDR cells inhibits autophagy through UPR modulation
and sensitizes the cells to cisplatin. Hence, GSTO1 may be an
important target for reversing drug resistance in MDR cells.
The present study results indicated that, in MDR
HCT-116 cells, GSTO1 interacts with TNFαIP3/A20, and that
TNFαIP3/A20 was reduced in GSTO-KD MDR cells. GSTO1
expression may regulate TNFαIP3/A20 expression in MDR cells.
However, GSTO1 expression was not dependent on the TNFαIP3/A20
status in cells. GSTO1-TNFαIP3/A20 double-KD MDR cells were
more effective than either GSTO1 or TNFαIP3/A20
single KD in lowering the expression levels of apoptosis markers
upon cisplatin treatment. TNFαIP3/A20 is a ubiquitin-editing enzyme
that negatively regulates NF-κB and plays a major role in the
biology of cells (48,49). Notably, a previous study suggested
that TNFαIP3/A20 mediates the resistance to DNA-damage repair in
cancer (50). Furthermore, a
previous publication also suggested that TNFαIP3/A20 is a major
factor in tamoxifen resistance in breast cancer cells (51).
The contribution of the GST family proteins to
cancer and chemoresistance is well known, but the available
information on GSTO1 is unclear. The present study examined a novel
interaction between GSTO1 and TNFαIP3/A20. The results suggested
that targeting both is more effective than targeting either alone,
in relation to chemosensitivity. Including specific inhibitors for
both GSTO1 and TNFαIP3/A20 in chemotherapy may help prevent drug
resistance, but a detailed mechanistic study on mice or human
subjects is required. In future studies, GSTO1 and
TNFαIP3/A20 double-knockout mice will be generated to
examine acquired drug resistance in an in vivo colon cancer
model. Furthermore, a detailed study on the role of the
inflammatory pathway related to TNFαIP3/A20 in chemoresistant
cancer cells is also required because TNFαIP3/A20 is an
inflammation pathway-related protein (49).
In conclusion, the GSTO1-TNFαIP3/A20 interaction
may be vital during acquired drug resistance. A previous study
reported TNFαIP3/A20 promotes survival of CD4 T cells by promoting
autophagy (52); TNFαIP3/A20 was
also overexpressed in HCT-116 MDR cells in the present study. Both
GSTO1 and TNFαIP3/A20 were reported to be upregulated during drug
resistance (10-12,50,51).
The present study identified a novel interaction between these two
proteins in acquired MDR cells. Therefore, targeting the
GSTO1-TNFαIP3/A20 axis may help overcome MDR. 4. S13 represents a schematic diagram of the
mechanism of GSTO1-mediated drug resistance in colon cancer cells.
WT HCT-116 cells showed apoptotic cell death upon cisplatin
treatment, whereas MDR cells survived upon cisplatin treatment by
activating autophagy. GSTO1 was upregulated and interacted with
TNFαIP3/A20 in MDR cells. Activation of GSTO1 in WT cells activated
autophagy and inhibition of GSTO1 in MDR cells sensitized them to
cisplatin treatment and leading to apoptosis.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
SP designed the study, performed experiments,
interpreted results and wrote the manuscript. MB contributed to
performing western blotting and data analysis. SCK designed the
study, interpreted the results and reviewed and edited the
manuscript. All authors have read and approved the final
manuscript. SP, MB and SCK confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was partially supported by the Daegu University
Research Grant, 2020.
Abbreviations:
|
ATG
|
autophagy-related gene
|
|
AP1
|
activator protein 1
|
|
eIF2α
|
eukaryotic initiation factor 2α
|
|
ER
|
endoplasmic reticulum
|
|
GRP78
|
glucose-regulated protein 78
|
|
GSTO1
|
glutathione-S-transferase Ω-1
|
|
IRE1α
|
inositol-requiring kinase 1α
|
|
MDR
|
multi-drug resistant
|
|
p62
|
ubiquitin-binding protein p62
|
|
PERK
|
PKR-like endoplasmic reticulum
kinase
|
|
TNFαIP3/A20
|
TNF-α-induced protein 3/zinc-finger
protein A20
|
|
UPR
|
unfolded protein response
|
References
|
1
|
Wang X, Zhang H and Chen X: Drug
resistance and combating drug resistance in cancer. Cancer Drug
Resist. 2:141–160. 2019.PubMed/NCBI
|
|
2
|
Bukowski K, Kciuk M and Kontek R:
Mechanisms of multidrug resistance in cancer chemotherapy1. Int J
Mol Sci. 21:32332020. View Article : Google Scholar
|
|
3
|
Köberle B and Schoch S: Platinum complexes
in colorectal cancer and other solid tumors. Cancers (Basel).
13:20732021. View Article : Google Scholar
|
|
4
|
Mele L, Del Vecchio V, Liccardo D, Prisco
C, Schwerdtfeger M, Robinson N, Desiderio V, Tirino V, Papaccio G
and La Noce M: The role of autophagy in resistance to targeted
therapies. Cancer Treat Rev. 88:1020432020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dang DT, Chen F, Kohli M, Rago C, Cummins
JM and Dang LH: Glutathione S-transferase pi1 promotes
tumorigenicity in HCT116 human colon cancer cells. Cancer Res.
65:9485–9494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogino S, Konishi H, Ichikawa D, Matsubara
D, Shoda K, Arita T, Kosuga T, Komatsu S, Shiozaki A, Okamoto K, et
al: Glutathione S-transferase Pi 1 is a valuable predictor for
cancer drug resistance in esophageal squamous cell carcinoma.
Cancer Sci. 110:795–804. 2019. View Article : Google Scholar
|
|
7
|
Su F, Hu X, Jia W, Gong C, Song E and
Hamar P: Glutathion S transferase pi indicates chemotherapy
resistance in breast cancer. J Surg Res. 113:102–108. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Board PG, Coggan M, Chelvanayagam G,
Easteal S, Jermiin LS, Schulte GK, Danley DE, Hoth LR, Griffor MC,
Kamath AV, et al: Identification, characterization, and crystal
structure of the omega class glutathione transferases. J Biol Chem.
275:24798–24806. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Paul S, Jakhar R, Bha rdwaj M and Kang SC:
Glutathione-S-transferase omega 1 (GSTO11) acts as mediator of
signaling pathways involved in aflatoxin B1-induced
apoptosis-autophagy crosstalk in macrophages. Free Radic Biol Med.
89:1218–1230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu H, Chen I, Shimoda LA, Park Y, Zhang C,
Tran L, Zhang H and Semenza GL: Erratum: Chemotherapy-induced
Ca2+ release stimulates breast cancer stem cell
enrichment. Cell Rep. 18:1946–1957. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan XD, Pan LY, Yuan Y, Lang JH and Mao N:
Identification of platinum-resistance associated proteins through
proteomic analysis of human ovarian cancer cells and their
platinum-resistant sublines. J Proteome Res. 6:772–780. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Boussac H, Kassambara A, Machura A,
Chemlal D, Gourzones C, Requirand G, Robert N, Vincent L, Herbaux
C, Bruyer A and Moreaux J: Genomic characterization of in vitro
acquired-resistance to proteasome inhibitors. Blood. 138(Suppl 1):
S26512021. View Article : Google Scholar
|
|
13
|
Mowers EE, Sharifi MN and Macleod KF:
Autophagy in cancer metastasis. Oncogene. 36:1619–1630. 2017.
View Article : Google Scholar :
|
|
14
|
Cao W, Wei W, Zhan Z, Xie D, Xie Y and
Xiao Q: Regulation of drug resistance and metastasis of gastric
cancer cells via the microRNA647-ANK2 axis. Int J Mol Med.
41:1958–1966. 2018.PubMed/NCBI
|
|
15
|
Malek E, Jagannathan S and Driscoll JJ:
Correlation of long non-coding RNA expression with metastasis, drug
resistance and clinical outcome in cancer. Oncotarget. 5:8027–8038.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhardwaj M, Cho HJ, Paul S, Jakhar R, Khan
I, Lee SJ, Kim BY, Krishnan M, Khaket TP, Lee HG and Kang SC:
Vitexin induces apoptosis by suppressing autophagy in multi-drug
resistant colorectal cancer cells. Oncotarget. 9:3278–3291. 2017.
View Article : Google Scholar
|
|
17
|
Wang J, Seebacher N, Shi H, Kan Q and Duan
Z: Novel strategies to prevent the development of multidrug
resistance (MDR) in cancer. Oncotarget. 8:84559–84571. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Novel strategies to prevent the
development of multidrug resistance (MDR) in cancer. Oncogene.
31:1869–1883. 2012. View Article : Google Scholar
|
|
19
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Florea AM and Büsselberg D: Cisplatin as
an anti-tumor drug: Cellular mechanisms of activity, drug
resistance and induced side effects. Cancers (Basel). 3:1351–1371.
2011. View Article : Google Scholar
|
|
21
|
Chen Y, Zhang W, Guo X, Ren J and Gao A:
The crosstalk between autophagy and apoptosis was mediated by
phosphorylation of Bcl-2 and beclin1 in benzene-induced
hematotoxicity. Cell Death Dis. 10:7722019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Daugas E, Susin SA, Zamzami N, Ferri KF,
Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D,
Penninger J and Kroemer G: Mitochondrio-nuclear translocation of
AIF in apoptosis and necrosis. FASEB J. 14:729–739. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koo MS, Kwon YG, Park JH, Choi WJ, Billiar
TR and Kim YM: Signaling and function of caspase and c-Jun
N-terminal kinase in cisplatin-induced apoptosis. Mol Cells.
13:194–201. 2002.PubMed/NCBI
|
|
24
|
Lin Y, Jiang M, Chen W, Zhao T and Wei Y:
Cancer and ER stress: Mutual crosstalk between autophagy, oxidative
stress and inflammatory response. Biomed Pharmacother.
118:1092492019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bertero E and Maack C: Calcium signaling
and reactive oxygen species in mitochondria. Circ Res.
122:1460–1478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sukumaran P, Nascimento Da Conceicao V,
Sun Y, Ahamad N, Saraiva LR, Selvaraj S and Singh BB: Calcium
signaling regulates autophagy and apoptosis. Cells. 10:21252021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bertero E and Maack C: Calcium signaling
and reactive oxygen species in mitochondria. Circ Res.
122:1460–1478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carreras-Sureda A, Pihán P and Hetz C:
Calcium signaling at the endoplasmic reticulum: Fine-tuning stress
responses. Cell Calcium. 70:2018. View Article : Google Scholar
|
|
29
|
Huang R, Hui Z, Wei S, Li D, Li W, Daping
W and Alahdal M: IRE1 signaling regulates chondrocyte apoptosis and
death fate in the osteoarthritis. J Cell Physiol. 237:118–127.
2022. View Article : Google Scholar :
|
|
30
|
Hu H, Tian M, Ding C and Yu S: The C/EBP
homologous protein (CHOP) transcription factor functions in
endoplasmic reticulum stress-induced apoptosis and microbial
infection. Front Immunol. 9:30832019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang Q, Li F, Shi K, Wu P, An J, Yang Y
and Xu C: Involvement of p38 in signal switching from autophagy to
apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4
cells. Cell Death Dis. 5:e12702014. View Article : Google Scholar
|
|
32
|
Lumley EC, Osborn AR, Scott JE, Scholl AG,
Mercado V, McMahan YT, Coffman ZG and Brewster JL: Moderate
endoplasmic reticulum stress activates a PERK and p38-dependent
apoptosis. Cell Stress Chaperones. 22:43–54. 2017. View Article : Google Scholar :
|
|
33
|
Xu Y, Sun Q, Yuan F, Dong H, Zhang H, Geng
R, Qi Y, Xiong X, Chen Q and Liu B: RND2 attenuates apoptosis and
autophagy in glioblastoma cells by targeting the p38 MAPK
signalling pathway. J Exp Clin Cancer Res. 39:1742020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koromilas AE: M(en)TORship lessons on life
and death by the integrated stress response. Biochim Biophys Acta
Gen Subj. 1863:644–649. 2019. View Article : Google Scholar
|
|
35
|
Pljesa-Ercegovac M, Savic-Radojevic A,
Matic M, Coric V, Djukic T, Radic T and Simic T: Glutathione
transferases: Potential targets to overcome chemoresistance in
solid tumors. Int J Mol Sci. 19:37852018. View Article : Google Scholar
|
|
36
|
Guo C, Fordjour FK, Tsai SJ, Morrell JC
and Gould SJ: Choice of selectable marker affects recombinant
protein expression in cells and exosomes. J Biol Chem.
297:1008382021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang W, Yan Y, Chen M, Luo G, Hao J, Pan
J, Hu S, Guo P, Li W, Wang R, et al: Aspirin enhances the
sensitivity of colon cancer cells to cisplatin by abrogating the
binding of NF-κB to the COX-2 promoter. Aging (Albany NY).
12:611–627. 2020. View Article : Google Scholar
|
|
38
|
Kleih M, Böpple K, Dong M, Gaißler A,
Heine S, Olayioye MA, Aulitzky WE and Essmann F: Direct impact of
cisplatin on mitochondria induces ROS production that dictates cell
fate of ovarian cancer cells. Cell Death Dis. 10:8512019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang W, Wang Z, Cai G and Huang P:
Downregulation of circ_0071589 suppresses cisplatin resistance in
colorectal cancer by regulating the MiR-526b-3p/KLF12 axis. Cancer
Manag Res. 13:2717–2731. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun M, He L, Fan Z, Tang R and Du J:
Effective treatment of drug-resistant lung cancer via a nanogel
capable of reactivating cisplatin and enhancing early apoptosis.
Biomaterials. 257:1202522020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun X, Wang S, Gai J, Guan J, Li J, Li Y,
Zhao J, Zhao C, Fu L and Li Q: SIRT5 promotes cisplatin resistance
in ovarian cancer by suppressing DNA damage in a ROS-dependent
manner via regulation of the Nrf2/HO-1 pathway. Front Oncol.
9:7542019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang YK, Wang YJ, Lei ZN, Zhang GN, Zhang
XY, Wang DS, Al Rihani SB, Shukla S, Ambudkar SV, Kaddoumi A, et
al: Regorafenib antagonizes BCRP-mediated multidrug resistance in
colon cancer. Cancer Lett. 442:104–112. 2019. View Article : Google Scholar
|
|
43
|
Uozaki H, Horiuchi H, Ishida T, Iijima T,
Imamura T and Machinami R: Overexpression of resistance-related
proteins (metallothioneins, glutathione-S-transferase pi, heat
shock protein 27, and lung resistance-related protein) in
osteosarcoma: Relationship with poor prognosis. Cancer.
79:2336–2344. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Singh RR and Reindl KM: Glutathione
S-transferases in cancer. Antioxidants (Basel). 10:7012021.
View Article : Google Scholar
|
|
45
|
Guo Y, Chang C, Huang R, Liu B, Bao L and
Liu W: AP1 is essential for generation of autophagosomes from the
trans-Golgi network. J Cell Sci. 125:1706–1715. 2012.PubMed/NCBI
|
|
46
|
Chen P, Liu XQ, Lin X, Gao LY, Zhang S and
Huang X: Targeting YTHDF1 effectively re-sensitizes
cisplatin-resistant colon cancer cells by modulating GLS-mediated
glutamine metabolism. Mol Ther Oncolytics. 20:228–239. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dong X, Yang Y, Zhou Y, Bi X, Zhao N,
Zhang Z, Li L, Hang Q, Zhang R, Chen D, et al: Glutathione
S-transferases P1 protects breast cancer cell from
adriamycin-induced cell death through promoting autophagy. Cell
Death Differ. 26:2086–2099. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Abbasi A, Forsberg K and Bischof F: The
role of the ubiquitin-editing enzyme A20 in diseases of the central
nervous system and other pathological processes. Front Mol
Neurosci. 8:212015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Das T, Chen Z, Hendriks RW and Kool M:
A20/tumor necrosis factor α-induced protein 3 in immune cells
controls development of autoinflammation and autoimmunity: Lessons
from mouse models. Front Immunol. 9:1042018. View Article : Google Scholar
|
|
50
|
Yang C, Zang W, Tang Z, Ji Y, Xu R, Yang
Y, Luo A, Hu B, Zhang Z, Liu Z and Zheng X: A20/TNFAIP3 regulates
the DNA damage response and mediates tumor cell resistance to
DNA-damaging therapy. Cancer Res. 78:1069–1082. 2018. View Article : Google Scholar
|
|
51
|
Vendrell JA, Ghayad S, Ben-Larbi S,
Dumontet C, Mechti N and Cohen PA: A20/TNFAIP3, a new
estrogen-regulated gene that confers tamoxifen resistance in breast
cancer cells. Oncogene. 26:4656–4667. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Matsuzawa Y, Oshima S, Takahara M,
Maeyashiki C, Nemoto Y, Kobayashi M, Nibe Y, Nozaki K, Nagaishi T,
Okamoto R, et al: TNFAIP3 promotes survival of CD4 T cells by
restricting MTOR and promoting autophagy. Autophagy. 11:1052–1062.
2015. View Article : Google Scholar : PubMed/NCBI
|