Introduction
Pancreatic cancer is one of the most common
digestive system malignancies with a 5-year survival rate of 9% in
2008-2014 in the United States (1). At present, surgery is the most
efficient therapy for pancreatic cancer; however, only ~20% of
patients are diagnosed with surgically resectable disease (2). Furthermore, metastasis occurs in
patients undergoing extensive resection (3,4).
Metastatic response and progression of pancreatic cancer contribute
to poor clinical outcomes (5). The
pathogenesis of pancreatic cancer is complicated and involves
various processes and signaling pathways (6,7). It
is a priority to understand the mechanisms underlying the
development and progression of pancreatic cancer.
Anti-silencing function 1 (ASF1) comprises two
subtypes, ASF1A and ASF1B, which share a 70% sequence homology
(8,9). ASF1B is a histone H3/H4 chaperone
involved in DNA repair, replication and transcriptional regulation
(10). ASF1B controls chromatin
function and has been demonstrated to participate in tumorigenesis
(11). Previous studies have
determined that ASF1B is closely associated with the development
and progression of numerous cancer types, such as prostate, breast
and cervical cancer (12,13). In addition, ASF1B is frequently
highly expressed in pancreatic cancer specimens and is closely
associated with poor prognosis (14). Inhibition of ASF1B in pancreatic
cancer cells enhances cisplatin sensitivity, inhibits
micrometastasis and results in tumor cell death (14,15).
In addition, c-Myc serves as an essential cancer-related
transcription factor involved in diverse critical biological
processes of cancer development (16-18).
c-Myc has been reported to serve a critical role in modulating
pancreatic cancer progression (19,20);
however, the association between c-Myc and ASF1B in the modulation
of pancreatic cancer progression remains unclear.
Epigenetic regulation serves as a program by which
gene expression is modulated by events other than genomic
alterations (21). Due to the
extensive effect of epigenetic regulation on gene expression and
genomic stability, it serves a fundamental role in a number of
physiological and pathological processes, and influences
carcinogenesis from initiation to progression (22), thus serving as a potential driver
of tumorigenesis (21,23). As a common epigenetic regulator,
histone acetylation serves a crucial role in the modulation of
cancer development (24).
Furthermore, it has been reported that ASF1 promotes anti-viral
immune activity by associating with CREB-binding protein (CBP) to
mediate H3K56 acetylation (H3K56ac) at the promoter of interferon β
(25). Multi-site substrate
recognition in ASF1-related histone H3K56ac through regulator of
Ty1 transposition 109 has also been identified (26). CBP is an essential transcriptional
co-activator for a number of DNA-binding transcription factors and
acts as a histone acetyltransferase (27). The CBP-mediated acetylation of
H3K56 is involved in the modulation of cell proliferation and
cancer (28). However, the
association of ASF1B with CBP and H3K56ac in the development of
pancreatic cancer remains elusive.
The present study aimed to explore the role and the
underlying mechanism of ASF1B in the modulation of pancreatic
cancer. The present study revealed that ASF1B promoted pancreatic
cancer progression by activating c-Myc through CBP-mediated
H3K56ac.
Materials and methods
Clinical pancreatic cancer samples
A total of 31 clinical pancreatic cancer samples
were obtained from the Cancer Hospital Affiliated to Zhengzhou
University (Zhengzhou, China) between May 2019 and December 2020.
Clinicopathological information of patients is shown in Table I. The inclusion criteria were:
Patients were diagnosed by clinical and pathological examination by
two clinicians; no systemic or local therapy was administered
before the surgery; and written informed consent was obtained from
each patient before using the collected samples. The exclusion
criteria were: Patients with cirrhosis and coagulation dysfunction;
patients with serious cardiovascular and visceral diseases;
patients with a history of second primary malignant tumors; and
patients with mental illness or confused consciousness. The present
study included 19 male and 12 female patients with an age range of
49-75 years (median age, 63.45±7.54 years). The pancreatic cancer
tissues and corresponding para-neoplastic tissues obtained from the
patients were immediately frozen in liquid nitrogen, followed by
storage at −80°C prior to further analysis. The present study was
performed in accordance with the World Medical Association
guidelines and approved by the Ethics Committee of Cancer Hospital
Affiliated to Zhengzhou University (Ethics No. 2019-05-006;
Zhengzhou, China).
 | Table IClinicopathological information of
patients with pancreatic cancer. |
Table I
Clinicopathological information of
patients with pancreatic cancer.
| Variables | Cases, n (%) |
|---|
| Sex | |
| Male | 19 (61.29) |
| Female | 12 (38.71) |
| Age, years | |
| ≤60 | 10 (32.26) |
| >60 | 21 (67.74) |
| Histopathology | |
| Ductal
adenocarcinoma | 22 (70.97) |
| Other | 9 (29.03) |
| Location | |
| Head of
pancreas | 23 (74.19) |
| Other | 8 (25.81) |
| Grade | |
| G1 | 2 (6.45) |
| G2 | 11 (35.48) |
| G3 | 18 (58.06) |
| Stage | |
| I | 4 (12.90) |
| II | 9 (29.03) |
| III | 17 (54.84) |
| IV | 1 (3.23) |
Bioinformatics analysis
The data for ASF1B mRNA expression in pancreatic
cancer and normal specimens were from Gene Expression Profiling
Interactive Analysis (GEPIA) (29)
(http://gepia.cancer-pku.cn/). The Cancer
Genome Atlas (TCGA; ID, TCGA-PAAD; https://portal.gdc.cancer.gov/) data were used for
GEPIA. According to ASF1B expression, the top and bottom 25%
patients with pancreatic cancer were divided into high and low
expression groups. The overall survival of 90 patients with
pancreatic cancer was estimated using GEPIA with a cutoff value of
25%. Kaplan-Meier analysis was used for analyzing the difference of
overall survival between the high and low expression groups using
the log-rank test. Gene Set Enrichment Analysis (GSEA; version
4.2.1; h.all.v7.1.symbols.gmt; https://www.gsea-msigdb.org/gsea/index.jsp) was
performed using R (version 3.4.1; https://cran.r-project.org/index.html) and the R
clusterProfiler package (Bioconductor version 3.10; http://www.bioconductor.org/packages/3.10/bioc/html/clusterProfiler.html)
(30) to explore signaling
pathways.
Cell culture
PANC-1 (cat. no. CL-0184), CFPAC-1 (cat. no.
CL-0059), PaCa-2 (cat. no. CL-0627) and HPAF-II (cat. no. CL-0611)
pancreatic adenocarcinoma (PAAD) cell lines were purchased from
Procell Life Science & Technology Co., Ltd., and the SW1990
(cat. no. CRL-2172) cell line was purchased from American Type
Culture Collection. PANC-1 and SW1990 cells exhibiting epithelial
morphology were used throughout the study. PANC-1 cells were
cultured in DMEM (cat. no. 11965092; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (cat. no. SH30406.02;
HyClone; Cytiva) and 1% penicillin-streptomycin solution (Beyotime
Institute of Biotechnology). SW1990 cells were cultured in
Leibovitz's L-15 medium (cat. no. 30-2008; American Type Culture
Collection) supplemented with 10% FBS and 1%
penicillin-streptomycin solution. CFPAC-1 cells were cultured in
Iscove's Modified Dulbecco Medium (Procell Life Science &
Technology Co., Ltd.) supplemented with 10% FBS and 1%
penicillin-streptomycin solution. PaCa-2 cells were cultured in
DMEM supplemented with 10% FBS, 2.5% horse serum (Procell Life
Science & Technology Co., Ltd.) and 1% penicillin-streptomycin
solution. HPAF-II cells were cultured in Minimum Essential Medium
(Procell Life Science & Technology Co., Ltd.) supplemented with
10% FBS and 1% penicillin-streptomycin solution. All cell lines
were maintained at 37°C in a 5% CO2 air atmosphere and
100% air, respectively.
The CBP inhibitor curcumin and c-Myc inhibitor
10058-F4 were purchased from MedChemExpress. Curcumin and 10058-F4
were used to treat cells at a concentration of 100 µM for 24
h at 37°C.
Plasmids, small interfering RNA
(siRNA/si) and transfection
To construct Flag-tagged ASF1B, total RNAs was
extracted from PANC-1 cells using TRIzol (Invitrogen; Thermo Fisher
Scientific, Inc.). The full coding region of the ASF1B gene was
obtained by PCR with the specific primers of forward, 5′-CCC AAG
CTT ATG GCC AAG GTG TCG GTG CT-3′ (HindIII) and reverse,
5′-CGG GAT CCT TAG ATG CAG TCC ATG GAG T-3′ (BamHI) using
PrimeSTAR HS DNA Polymerase (Takara Biotechnology Co., Ltd.). The
thermocycling conditions were: 98°C for 10 sec, followed by 30
cycles of 55°C for 5 sec and 72°C for 1 min. The PCR products were
ligated into the p3XFLAG-CMV™-14 expression vector (MilliporeSigma)
using the In-Fusion HD Cloning Kit (Takara Biotechnology Co.,
Ltd.). Empty vector was used as the negative control. siRNAs
specific for ASF1B and CBP, and the non-targeting negative controls
were obtained from TsingKe Biological Technology. The Homo
sapiens siRNA sequences were as follows: si-ASF1B-1, 5′-GTT CAT
CCG AGT GGG CTA CTA-3′; si-ASF1B-2, 5′-GGT GAC CCG CTT CCA TAT
CAA-3′; and si-CBP, 5′-GCA AGT CAT GAA TGG ATC TCT-3′.
Non-targeting siRNAs were used as negative controls (siNC): 5′-GCT
CGT AAG CGT AGG TCT TCA-3′ (for si-ASF1B-1 and si-ASF1B-2); 5′-GCT
TAA GAT GAC GCA GTT CTA-3′ (for si-CBP). Cells were transfected
with vectors (2 µg for '+' group and 5 µg for '++'
group) or siRNAs (100 nM) using Lipofectamine 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) for 48 h at 37°C according to the
manufacturer's instructions. The time interval between transfection
and subsequent experimentation was <1 week.
Cell Counting Kit-8 (CCK-8) assay
Following transfection, PANC-1 and SW1990 cells
(1×105 cells/well) were plated in 96-well plates and
cultured for 0-72 h. Cell proliferation was measured using 10
µl/well CCK-8 (Dojindo Molecular Technologies, Inc.) for 2 h
at 37°C. The optical density value at 450 nm was measured using an
ELISA reader (Bio-Tek EL 800; BioTek Instruments, Inc.).
Colony formation
Following transfection with siRNAs, empty vector or
flag-tagged ASF1B, PANC-1 and SW1990 cells (1,000 cells/well) were
plated in 6-well plates and treated with 100 µM curcumin or
100 µM 10058-F4 for 24 h at 37°C. Non-treated cells were
used as the control group. The culture medium was replaced every 3
days. Colonies were washed twice with PBS, fixed with 4%
paraformaldehyde for 30 min at room temperature and stained with
0.5% crystal violet (Beyotime Institute of Biotechnology) for 1 h
at room temperature, and images were captured. Colonies with at
least 50 cells were counted using ImageJ software (version 5.0;
National Institutes of Health).
Cell apoptosis detection
Following transfection, PANC-1 and SW1990 cells
(2×105 cells/well) were plated in 6-well dishes. After
48 h, apoptotic cells were stained using the Annexin V-FITC Early
Apoptosis Detection Kit (Cell Signaling Technology, Inc.) according
to the manufacturer's instructions. Apoptotic cells were analyzed
using flow cytometry (Accuri-C6 plus; BD Biosciences). The number
of apoptotic cells was analyzed using FlowJo software (v7.6.5; Tree
Star, Inc.).
Transwell assays
For cell migration assays, 1×105
transfected PANC-1 and SW1990 cells were placed in the upper
chamber of a Corning® FluoroBlok™ 24-well Transwell
system (Costar; Corning, Inc.). The cells in the upper chamber were
maintained in DMEM or Leibovitz's L-15 base medium without serum.
The lower chamber was filled with DMEM or Leibovitz's L-15 medium
supplemented with 10% FBS. After 12 h of incubation at 37°C, the
cells that had migrated into the lower chamber were fixed with 4%
paraformaldehyde for 30 min at room temperature and stained with
0.5% crystal violet (Beyotime Institute of Biotechnology) for 30
min at room temperature. For cell invasion assays, the Transwell
system was pre-coated with 100 µl Matrigel (Invitrogen;
Thermo Fisher Scientific, Inc.) for 4 h at 37°C. The other steps
were the same as for the migration assay. Images were captured
under a light microscope (Eclipse 80i; Nikon Corporation). The
migrated and invaded cells in five randomly selected fields of view
in the lower chamber were counted to calculate cell migration and
invasion. Both the migration and invasion assays were performed in
triplicate.
Scratch test
Following transfection, PANC-1 and SW1990 cells
(3×105 cells/well) were plated overnight in 24-well
dishes to reach full confluence as a monolayer. A 20-µl
pipette tip was used to create a scratch across the well. Floating
cells were removed by washing three times with PBS. The cultures
were maintained at 37°C for 6 or 12 h in serum-free culture medium,
and then images were captured under a light microscope (Eclipse
80i; Nikon Corporation). The wound distance was measured using
ImageJ software (version 5.0; National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNAs was extracted from the human specimens
and PANC-1 and SW1990 cell lines using TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.). First-strand cDNA was synthesized using
the PrimeScript™ RT Master Mix (cat. no. RR036A; Takara
Biotechnology Co., Ltd.). The reverse transcription conditions were
37°C for 15 min followed by 85°C for 5 sec. qPCR was performed
using the TB Green Fast qPCR Mix (cat. no. RR430A; Takara
Biotechnology Co., Ltd.). Relative expression of the target genes
was calculated according to the 2−ΔΔCq method by
normalizing to the β-actin internal control (31). The Homo sapiens primer
sequences were as follows: ASF1B forward, 5′-GAT CAG CTT CGA GTG
CAG TG-3′ and reverse, 5′-TGG TAG GTG CAG GTG ATG AG-3′; c-Myc
forward, 5′-GGC TCC TGG CAA AAG GTC A-3′ and reverse, 5′-CTG CGT
AGT TGT GCT GAT GT-3′; and β-actin forward, 5′-GAT TCC TAT GTG GGC
GAC GA-3′ and reverse, 5′-CACAGGACTCCATGCCCAG-3′. The thermocycling
conditions were: 95°C for 30 sec, followed by 40 cycles of 95°C for
5 sec and 60°C for 10 sec.
Western blot analysis
Total proteins were extracted from the collected
tissue samples and cell lines using RIPA buffer (Cell Signaling
Technology, Inc.). Protein concentrations were measured using the
BCA Protein Quantification Kit (Abbkine Scientific Co., Ltd.).
Equal amounts (30 µg/lane) of protein extracts were
subjected to 10-12% SDS-PAGE and transferred to PVDF membranes
(MilliporeSigma). The membranes were blocked in 5% skimmed milk for
1 h at 37°C. Membranes were incubated with primary antibodies
against ASF1B (dilution, 1:1,000; cat. no. ab235358; Abcam), Bax
(dilution, 1:1,000; cat. no. ab32503; Abcam), Bcl-2 (dilution,
1:1,000; cat. no. ab182858; Abcam), c-Myc (dilution, 1:1,000; cat.
no. ab10910; Abcam), β-actin (dilution, 1:1,000; cat. no. ab8226;
Abcam), H3 (dilution, 1:1,000; cat. no. ab1791; Abcam) and H3K56ac
(dilution, 1:1,000; cat. no. PA5-121083; Thermo Fisher Scientific,
Inc.) overnight at 4°C. The membranes were then incubated with goat
anti-rabbit (cat. no. ab7090), anti-mouse (cat. no. ab97040) and
anti-rat (cat. no. ab7097) IgG H&L (HRP) secondary antibodies
(dilution, 1:1,000; Abcam) for 1 h at room temperature. The bands
were developed using BeyoECL Plus (Beyotime Institute of
Biotechnology) and semi-quantified using ImageJ software (version
5.0; National Institutes of Health). To determine the protein
half-life curves, the cells were treated with 25 µg/ml
cycloheximide (CHX; MilliporeSigma) for 0-4 h at 37°C.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a SimpleChIP Enzymatic
Chromatin IP Kit (Cell Signaling Technology, Inc.) according to the
manufacturer's instructions. Following transfection with si-ASF1B-2
or siNC, cells were incubated with 1% formaldehyde for 15 min at
room temperature, washed twice with PBS, centrifuged at 500 × g for
10 min at 4°C, and collected in SDS lysis buffer (Beyotime
Institute of Biotechnology). Chromatin prepared from the cells in a
15-cm dish was used to determine total DNA input and was incubated
overnight with 10 µg anti-H3K56ac (cat. no. PA5-121083;
Thermo Fisher Scientific, Inc.) or anti-IgG (cat. no. ab171870;
Abcam) at 4°C. In each immunoprecipitation, 10 µg of the
protein-chromatin complex was used. Following incubation with 70
µl protein G agarose beads (Upstate Biotechnology, Inc.) for
2 h at 4°C. The immune complexes were precipitated at 3,400 × g for
1 min at 4°C and washed with 1 ml low salt washing solution. After
three washes of low salt, 1 ml high salt solution was added to the
beads and incubated at 4°C for 5 min. The protein G agarose beads
were precipitated by centrifugation at 3,400 × g for 1 min at 4°C
and then the supernatant was used in the next step. The PCR
procedure was performed as aforementioned with the following primer
sequences for amplifying the c-Myc promoter: Forward, 5′-GTT TCA
ACT GTT CTC GTC GTT-3′ and reverse, 5′-TTC TTG TCC ATG CCA TAA
CCC-3′.
Pancreatic cancer xenograft
A total of 10 male BALB/c nude mice (8 weeks old;
22±2 g; Jinan Pengyue Laboratory Animal Breeding Co., Ltd.) were
divided into two groups (n=5 per group): Short hairpin RNA (sh)NC
and shASF1B. All animal experiments in the present study were
approved by the Ethics Committee of Cancer Hospital Affiliated to
Zhengzhou University (Ethics No. 2019-05-006; Zhengzhou, China) and
performed in accordance with the standards of Animal Care and Use
and the Animal Welfare Act. The tumor volume limit (≤2 cm in any
dimension) was the humane endpoint criterion for mouse euthanasia.
Mice were raised in specific pathogen-free conditions with ad
libitum access to food and water. Mice were kept at 22±2°C in a
humid environment (60±10%) with a 12/12 h light-dark cycle. After
acclimatization for 1 week, 5×106 PANC-1 cells in 150
µl PBS were injected subcutaneously into the right side of
the anterior flank of nude mice to establish in vivo tumor
models. PANC-1 cells were pre-infected with lentiviral vectors
(MOI, 20; Shanghai GeneChem Co., Ltd.) containing shNC (5′-ACC TCG
TTG TAC GAG CGT CGG AAC TAT CAA GAG TAG TTC CGA CGC TCG TAC AAC
TT-3′) or shASF1B (5′-ACC TCG ATC AGC TTC GAG TGC AGT GAT CAA GAG
TCA CTG CAC TCG AAG CTG ATC TT-3′). These sequences were cloned
into the GV115 vector (Shanghai GeneChem Co., Ltd.) and the
lentivirus was packaged in 293T cells. The third generation of 293T
cells (The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences) were inoculated into the 10-cm cell culture
dish (Corning, Inc.). After 24 h, when the cell density reached
70-80%, the plasmids including 20 µg GV vector, 15 µg
pHelper 1.0 and 10 µg pHelper 2.0 (the ratio was: 4:3:2)
were transfected using Lipofactamine 3000 (Thermo Fisher
Scientific, Inc.). The cells were cultured in the 5% CO2
incubator at 37°C for 48 h, then the cell supernatant was collected
for subsequent experiments. After centrifugation at 25,000 × g for
2 h at 4°C and filtration through 0.22-µm filters,
lentiviral particles (MOI, 20) were transduced into PANC-1 cells
for 24 h, the infected medium was replaced with fresh medium. After
72 h of continuous culture with puromycin (1 µg/ml) for
stable cell line selection, cells were cultured with puromycin (0.5
µg/ml) for amplification. Subcutaneous inoculation was
started after amplification. None of the mice died during the
experimental procedures. From 10 days after the injection, the
sizes of the subcutaneous tumors were measured on day 10, 12, 15,
17, 20, 22, 25, 27 and 30 using calipers. The tumor volumes were
calculated and the results for day 10, 15, 20, 25 and 30 are
presented. The length and width of the tumors were calculated using
calipers. Tumor volume=(length x width2)/2. At 30 days
after injection, the mice were sacrificed by cervical dislocation
with prior anesthesia by intraperitoneal injection of 3% sodium
pentobarbital (30 mg/kg). Death was verified to be respiratory
cardiac arrest for at least 10 min and the observation of pupil
dilation. Tumor tissues were washed with ice-cold PBS, weighed and
subjected to immunohistochemical staining.
Histological examination
Human para-neoplastic and pancreatic cancer tissues
were subjected to immunohistochemical staining. Tumor tissues were
collected from the xenograft mouse model. Tissues were sliced
(thickness, 0.1 mm) and then fixed with 4% paraformaldehyde at 4°C
for 2 h, permeabilized in 0.5% Triton X-100 at room temperature for
10 min and coated with paraffin. Tissue sections were heated at
62°C for 20 min, dewaxed in xylene for 20 min, rehydrated in
decreasing concentrations of ethyl alcohol (100, 100, 90, 80 and
70%) for 2 min each followed by boiling in 0.01 M citric acid (pH
6.0) for 10 min for antigen retrieval. The sections were then
incubated in 3% H2O2 for 10 min at room
temperature to block endogenous peroxidase/phosphatase activity.
The sections were blocked with 5% normal goat serum (Thermo Fisher
Scientific, Inc.) at room temperature for 15 min, followed by
incubation at 4°C overnight with primary antibodies against rabbit
anti-ASF1B (dilution, 1:20; cat. no. ab235358; Abcam) and rabbit
anti-Ki67 (dilution, 1:50; cat. no. ab16667; Abcam). A goat
anti-rabbit IgG H&L (Alexa Fluor 488) secondary anti-body
(dilution, 1:100; cat. no. ab150077; Abcam) was used for incubation
at 37°C for 1 h. The color was developed by adding 0.5 ml
3,3'-diaminobenzidine solution for 20 min at room temperature.
Images were captured under a light microscope (Eclipse 80i; Nikon
Corporation). Ki67-positive cells were counted using ImageJ
software (version 5.0; National Institutes of Health).
Apoptotic cells in the tumor tissues were detected
using a TUNEL assay kit (Beyotime Institute of Biotechnology). The
sections were fixed with 4% paraformaldehyde at 4°C for 1 h and
washed twice with PBS for 20 min at room temperature. The sections
were then incubated with 0.5% Triton X-100 and 3%
H2O2 at room temperature for 10 min followed
by incubation with 50 µl TUNEL reaction mixture (5 µl
TdT enzyme and 45 µl fluorescent labeling solution) for 1 h
at 37°C in the dark. After three washes with PBS, 1 µg/ml
DAPI (Beyotime Institute of Biotechnology) was used for nuclear
staining at room temperature for 10 min. The slides were mounted
with antifade mounting medium (Wuhan Servicebio Technology Co.,
Ltd.). Images were captured using a fluorescence microscope
(DMI6000B; Leica Microsystems GmbH). Images were captured using a
fluorescence microscope (DMI6000B; Leica Microsystems GmbH). Five
fields were selected for counting TUNEL-positive cells using ImageJ
software (version 5.0; National Institutes of Health).
A HE staining kit (Beyotime Institute of
Biotechnology) was used to detect the histology of the harvested
tumor tissues. Tissues were fixed with 4% paraformaldehyde for 24 h
at room temperature, coated with paraffin and sectioned into
4-µm section. The sections were stained with hematoxylin for
10 min at room temperature followed by incubation with eosin for 30
sec at room temperature. Images were captured under a light
microscope (Eclipse 80i; Nikon Corporation).
Statistical analysis
Data are presented as the mean ± SD. In vitro
data were obtained from three independent experiments. Statistical
analyses were performed using GraphPad Prism 7 software (GraphPad
Software, Inc.). The paired and unpaired t-test was used for
two-group comparisons. One-way ANOVA followed by Tukey's post hoc
test and two-way ANOVA followed by Bonferroni's post hoc test were
used for multiple-group comparisons. Brown-Forsythe and Bartlett's
tests were used to assess the normality and variance homogeneity.
The correlation between ASF1B and c-Myc expression was analyzed
using the linear regression test. P<0.05 was considered to
indicate a statistically significant difference.
Results
ASF1B is highly expressed in pancreatic
cancer and is closely associated with poor prognosis
To evaluate the potential association between ASF1B
and pancreatic cancer, ASF1B expression was analyzed in PAAD
samples from TCGA. ASF1B expression was significantly increased in
primary tumor samples compared with in normal samples (Fig. 1A). Kaplan-Meier analysis based on
the PAAD samples from TCGA revealed that a high level of ASF1B was
associated with poor overall survival of patients with pancreatic
cancer (Fig. 1B). In the present
study, 31 patients with pancreatic cancer were enrolled, and their
tumor tissues were collected during surgery. RT-qPCR indicated that
ASF1B mRNA expression was significantly higher in pancreatic cancer
tissues than in adjacent normal controls (Fig. 1C). Immunohistochemical staining
revealed that ASF1B was highly expressed in pancreatic cancer
tissues compared with the adjacent normal controls (Fig. 1D). Consistently, ASF1B protein
expression was higher in 12 randomly selected pancreatic cancer
tissues than in paired adjacent normal controls (Fig. 1E). ASF1B expression was higher in
PANC-1 and SW1990 cells than the other three cell lines (Fig. 1F), and thus, these two cell lines
were used in subsequent experiments.
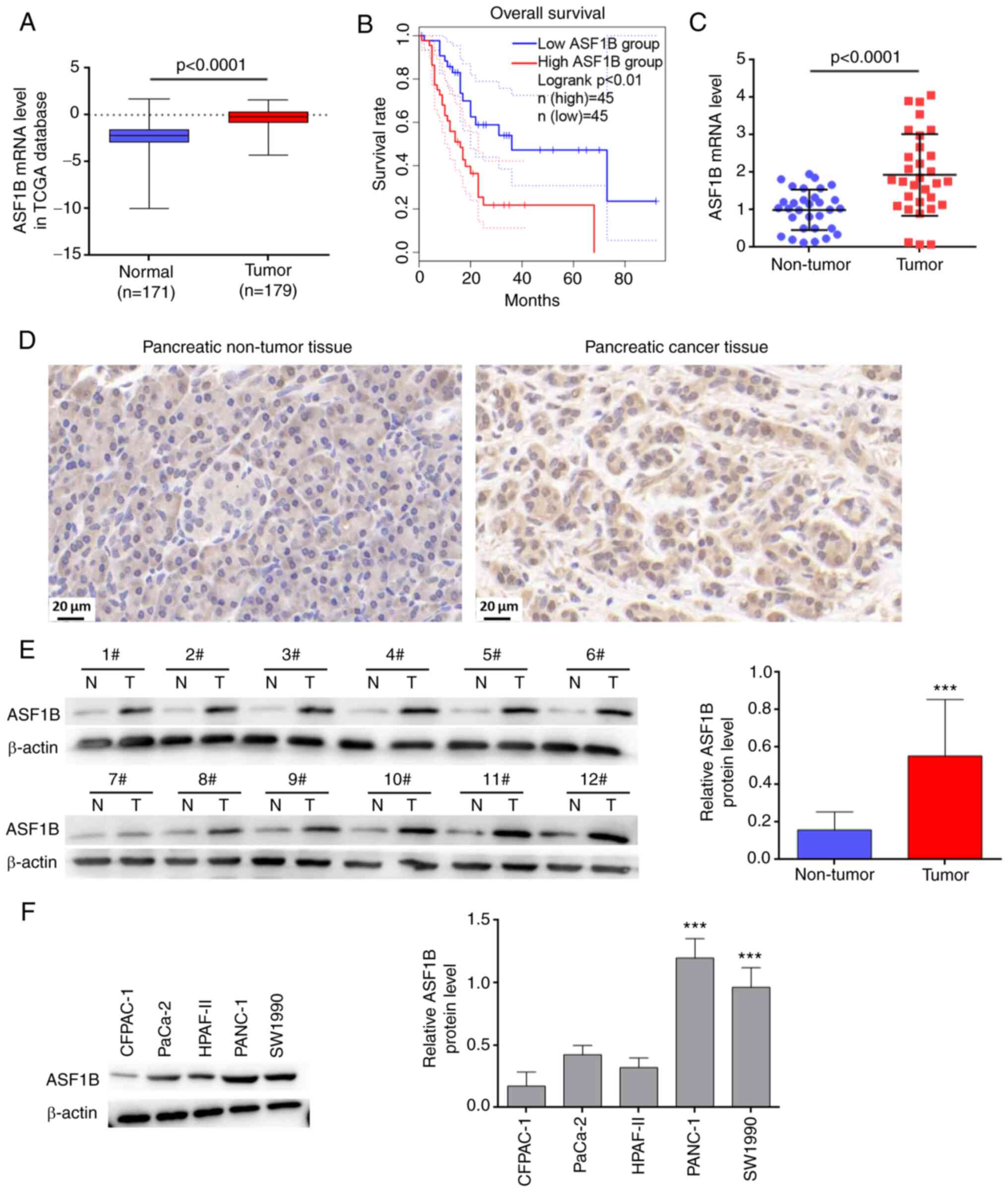 | Figure 1ASF1B is highly expressed in
pancreatic cancer and its high expression is closely associated
with poor prognosis. (A) ASF1B expression in PAAD samples (n=179)
and normal controls (n=171) was analyzed using data from TCGA. (B)
Kaplan-Meier analysis based on the PAAD samples from TCGA revealed
an association between ASF1B expression and overall survival of
patients with pancreatic cancer (n=45 per group). (C) mRNA levels
of ASF1B in 31 paired pancreatic cancer and adjacent tissues. (D)
Immunohistochemical staining of ASF1B in pancreatic cancer tissues
and the paired adjacent normal controls. Scale bar, 20 µm.
(E) Protein levels of ASF1B in 12 paired pancreatic cancer and
adjacent tissues. ***P<0.001 vs. non-tumor. (F)
Protein levels of ASF1B in CFPAC-1, PaCa-2, HPAF-II, PANC-1 and
SW1990 cells (n=3). ***P<0.001 vs. CFPAC-1, PaCa-2
and HPAF-II groups. Data in (A) were analyzed using the unpaired
t-test. Data in (F) were analyzed using one-way ANOVA followed by
Tukey's post hoc test. Other data were analyzed using the paired
t-test. ASF1B, anti-silencing function 1B; PAAD, pancreatic
adenocarcinoma; TCGA, The Cancer Genome Atlas; N, non-tumor; T,
tumor. |
Silencing of ASF1B inhibits pancreatic
cancer cell proliferation, migration and invasion, and induces
apoptosis
Considering that ASF1B is highly expressed in
pancreatic cancer, the present study aimed to investigate the
effects of ASF1B silencing on pancreatic cancer cells. To this end,
two different siRNA sequences specific to ASF1B were transfected
into PANC-1 and SW1990 cells. ASF1B protein expression was
significantly decreased by ASF1B siRNAs compared with siNC
(Fig. 2A). Transfection of PANC-1
and SW1990 cells with ASF1B siRNAs significantly inhibited
proliferation (Fig. 2B) and colony
formation (Fig. 2C) compared with
those of the cells transfected with siNC. Transfection of cells
with ASF1B siRNAs significantly increased the apoptosis of PANC-1
and SW1990 cells compared with transfection with siNC (Fig. 2D). In addition, Bax was upregulated
and Bcl-2 was downregulated following transfection with ASF1B
siRNAs compared with the siNC group (Fig. 2E).
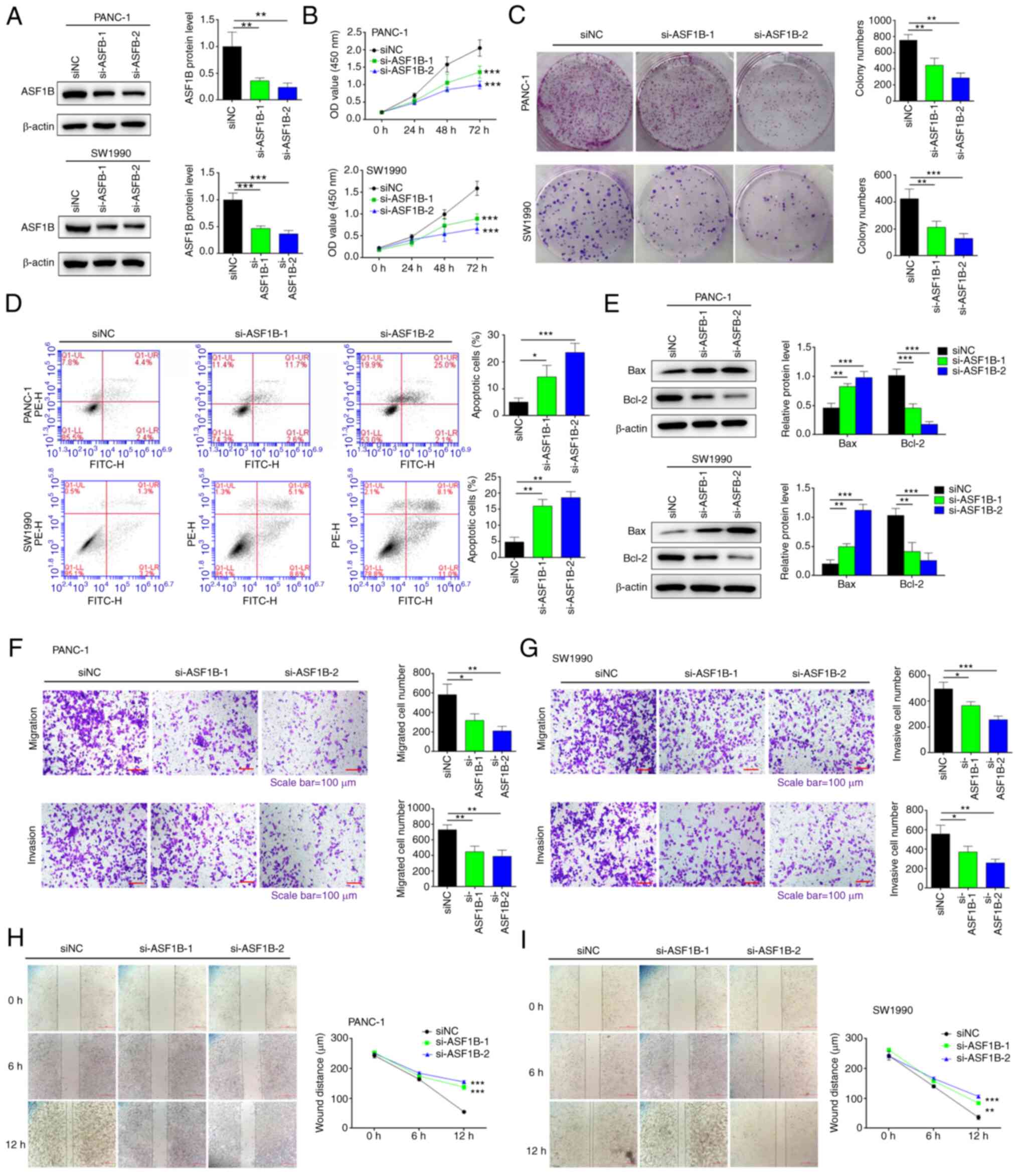 | Figure 2Silencing of ASF1B inhibits
pancreatic cancer cell proliferation, migration and invasion, and
induces apoptosis. (A) PANC-1 and SW1990 cells were transfected
with ASF1B siRNAs (si-ASF1B-1 and si-ASF1B-2) or siNC. Protein
levels of ASF1B were measured by western blotting. (B) Cell
proliferation, (C) colony formation, (D) apoptosis, and (E)
expression levels of Bax and Bcl-2 were detected by Cell Counting
Kit-8 assays, colony formation assays, flow cytometry and western
blotting, respectively. (F) PANC-1 and (G) SW1990 cells were
transfected with ASF1B siRNAs (si-1 and si-2) or siNC. Cell
migration and invasion were detected by transwell assays.
Magnification, ×100; scale bar, 100 µm. The wound distance
of (H) PANC-1 and (I) SW1990 cell cultures was detected using a
scratch test. Magnification, ×40; scale bar, 200 µm. Data
are presented as the mean ± SD (n=3). *P<0.05,
**P<0.01, ***P<0.001 vs. siNC group.
Data in (B) were analyzed using two-way ANOVA followed by
Bonferroni's post hoc test. Other data were analyzed using one-way
ANOVA followed by Tukey's post hoc test. Brown-Forsythe and
Bartlett's tests were used to assess the normality and variance
homogeneity. ASF1B, anti-silencing function 1B; NC, negative
control; OD, optical density; siRNA/si, small interfering RNA. |
The migration and invasion of PANC-1 and SW1990
cells following transfection with ASF1B siRNAs or siNC were
detected using a transwell assay. As shown in Fig. 2F and G, the migration and invasion
of PANC-1 and SW1990 cells were significantly inhibited following
transfection with ASF1B siRNAs compared with the siNC group.
Consistently, ASF1B siRNA transfection significantly inhibited
wound healing at 12 h of incubation compared with the siNC group
(Fig. 2H and I).
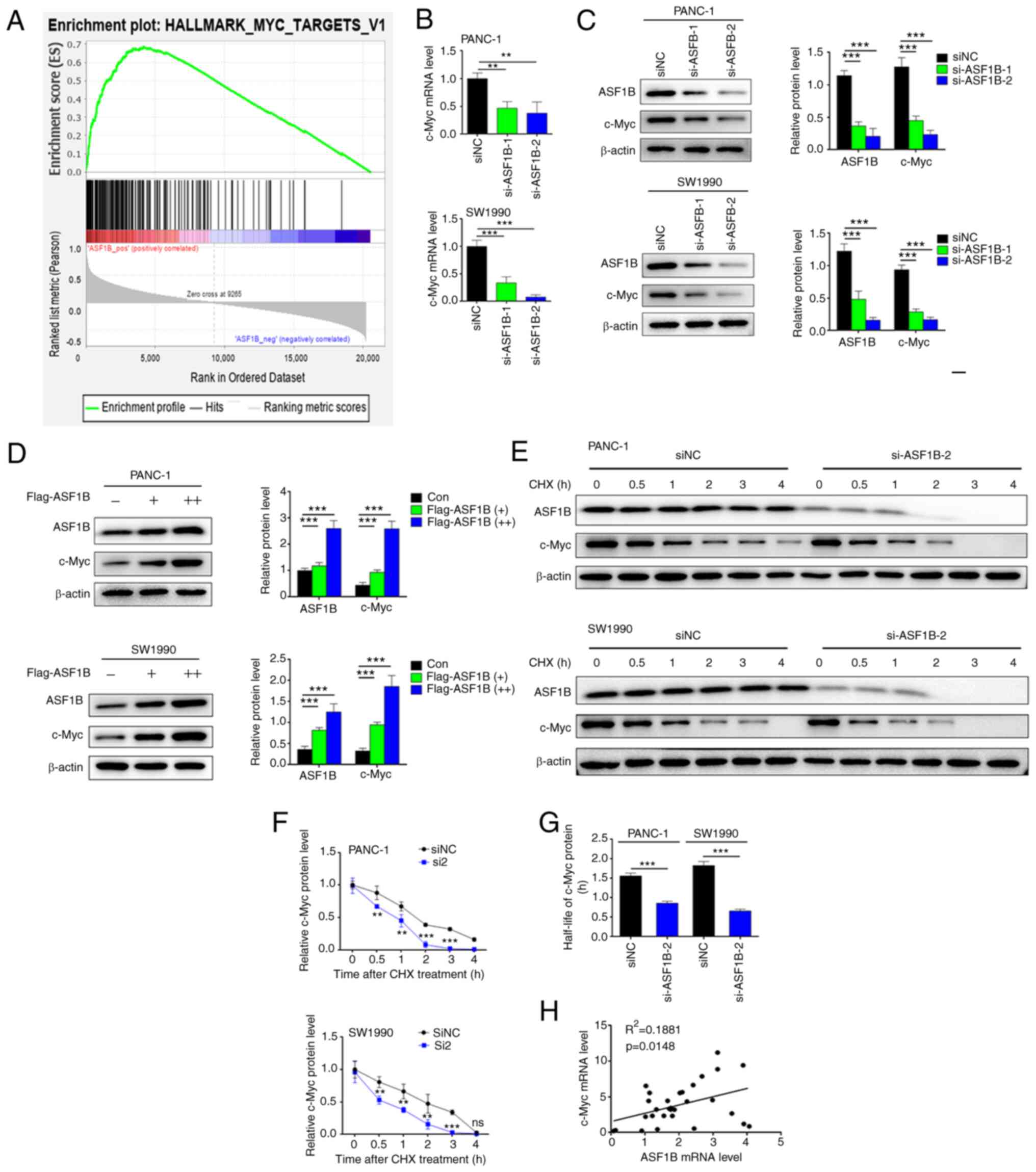
ASF1B induces c-Myc expression in
pancreatic cancer cells
Furthermore, the underlying mechanism by which ASF1B
exerts its effects on pancreatic cancer cells was explored. GSEA
predicted a positive association between ASF1B and the c-Myc
signaling pathway (Fig. 3A). The
association between ASF1B and the c-Myc signaling pathway was
confirmed in subsequent experiments. The mRNA and protein
expression levels of c-Myc were downregulated in cells transfected
with ASF1B siRNAs compared with siNC (Fig. 3B and C). However, c-Myc protein
levels were upregulated in cells transfected with Flag-tagged ASF1B
overexpression plasmid compared with the negative control (Fig. 3D). The stability of c-Myc was
detected after treatment with the protein synthesis inhibitor, CHX.
As shown in Fig. 3E, ASF1B siRNA
regulated c-Myc stability in PANC-1 and SW1990 cells. The protein
levels and half-life of c-Myc protein were decreased significantly
in the ASF1B siRNA group compared with the siNC group (Fig. 3F and G). Furthermore, a linear
regression test revealed a positive correlation between ASF1B and
c-Myc mRNA expression in 31 pancreatic cancer tissues
(R2=0.1881; P=0.0148; Fig.
3H).
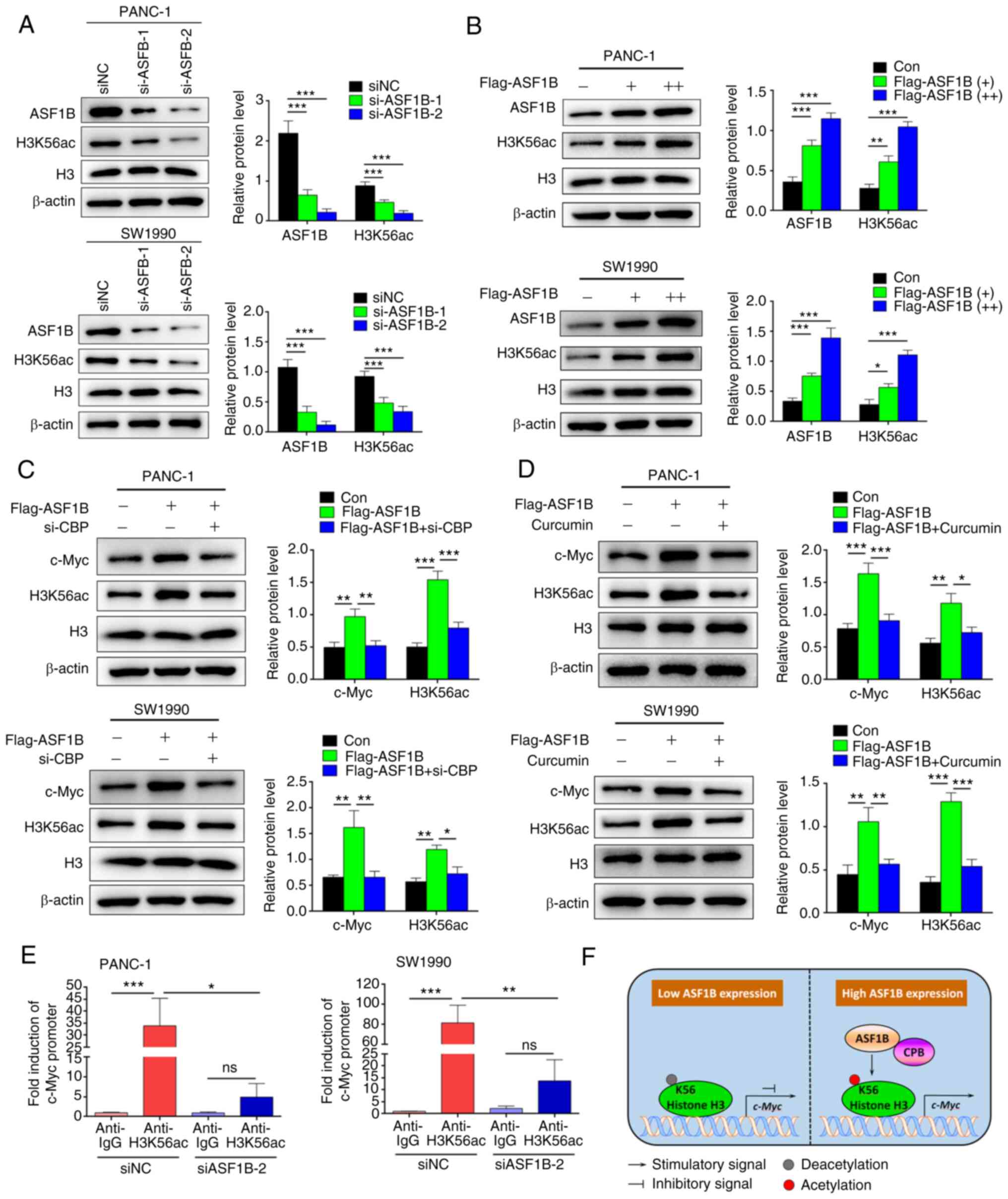
ASF1B induces c-Myc expression through
CBP-mediated H3K56ac
The present study next investigated whether ASF1B
upregulates c-Myc expression by inducing H3K56ac. In PANC-1 and
SW1990 cells, silencing of ASF1B by specific siRNAs significantly
inhibited the protein levels of acetylated H3K56 (Fig. 4A). Overexpression of ASF1B by
transfection with Flag-tagged plasmids increased the expression of
acetylated H3K56 (Fig. 4B),
indicating that ASF1B is a key regulator of H3K56ac. Silencing of
acetyltransferase CBP with specific siRNAs inhibited the expression
of acetylated H3K56 and c-Myc induced by ASF1B (Fig. 4C). The proof of successful
transfection of si-CBP is shown in Fig. S1. Consistently, the CBP inhibitor,
curcumin, inhibited the expression of acetylated H3K56 and c-Myc
induced by ASF1B (Fig. 4D). The
effects of ASF1B on the transcriptional activity of c-Myc were
analyzed using ChIP. In siNC transfected PANC-1 and SW1990 cells,
33.9 and 81.4-fold enrichment of the promoter amplification of
c-Myc was observed using anti-H3K56ac antibodies compared with
anti-IgG (Fig. 4E). However, no
significant enrichment of the c-Myc promoter was observed in
ASF1B-silenced PANC-1 (P=0.847) and SW1990 (P=0.522) cells. These
data revealed that ASF1B promoted CBP-mediated H3K56ac to enhance
the transcriptional activity of c-Myc in pancreatic cancer cells
(Fig. 4F).
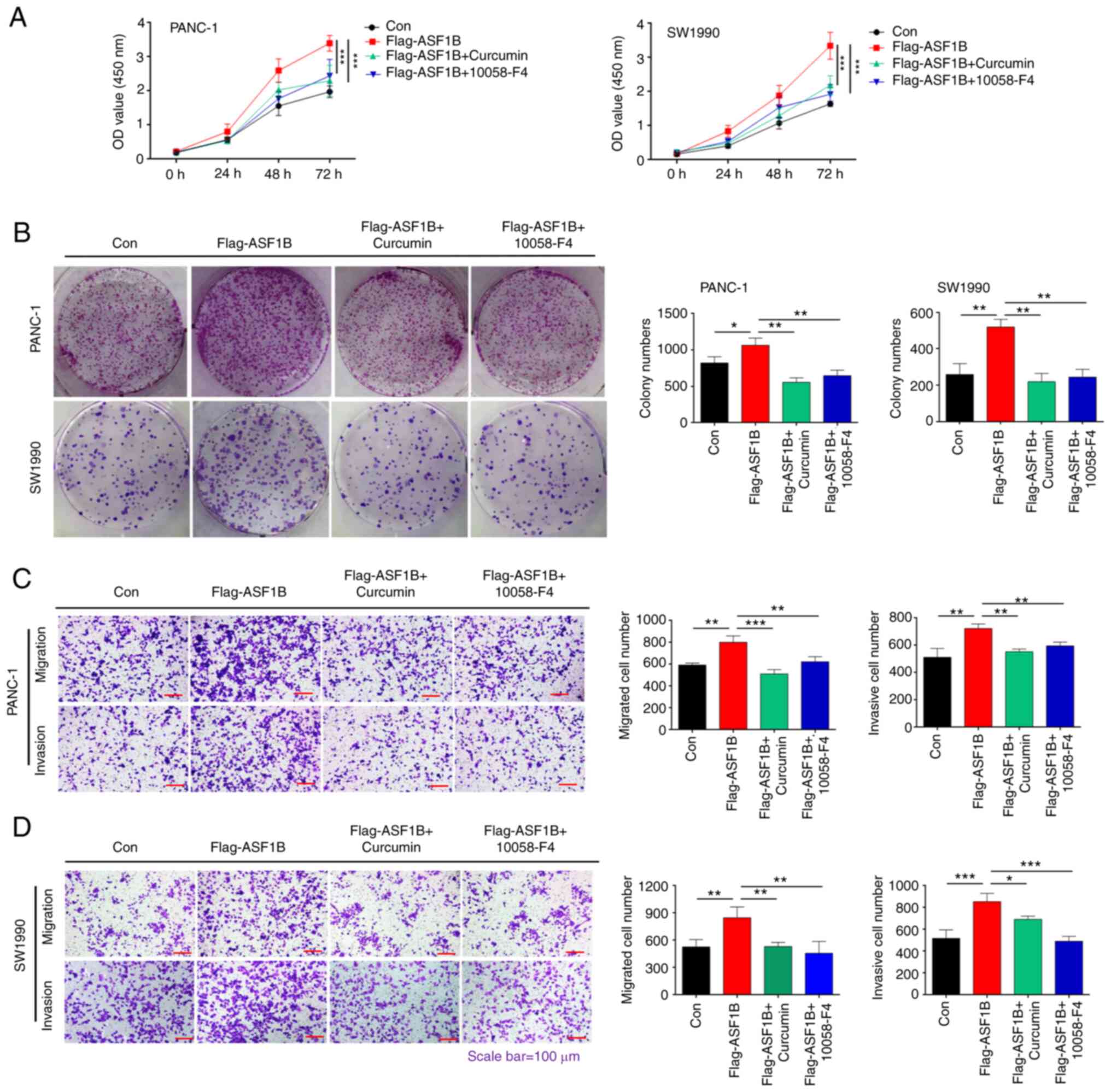
ASF1B promotes pancreatic cancer cell
proliferation, migration and invasion by activating
CBP/H3K56ac/c-Myc signaling
Subsequently, the present study evaluated whether
ASF1B contributes to the progression of pancreatic cancer by
modulating CBP/H3K56ac/c-Myc signaling in vitro. CCK-8
assays revealed that the overexpression of ASF1B enhanced the
proliferation of PANC-1 and SW1990 cells, and the CBP inhibitor
curcumin and the c-Myc inhibitor 10058-F4 blocked this enhancement
(Fig. 5A). In addition, treatment
with curcumin and 10058-F4 significantly inhibited the colony
formation of PANC-1 and SW1990 cells induced by ASF1B
overexpression (Fig. 5B).
Transwell assay results demonstrated that migration and invasion
induced by ASF1B overexpression in PANC-1 and SW1990 cells were
significantly inhibited by treatment with curcumin and 10058-F4
(Fig. 5C and D). In summary, these
data indicated that ASF1B promoted the progression of pancreatic
cancer by activating CBP/H3K56ac/c-Myc signaling in
vitro.
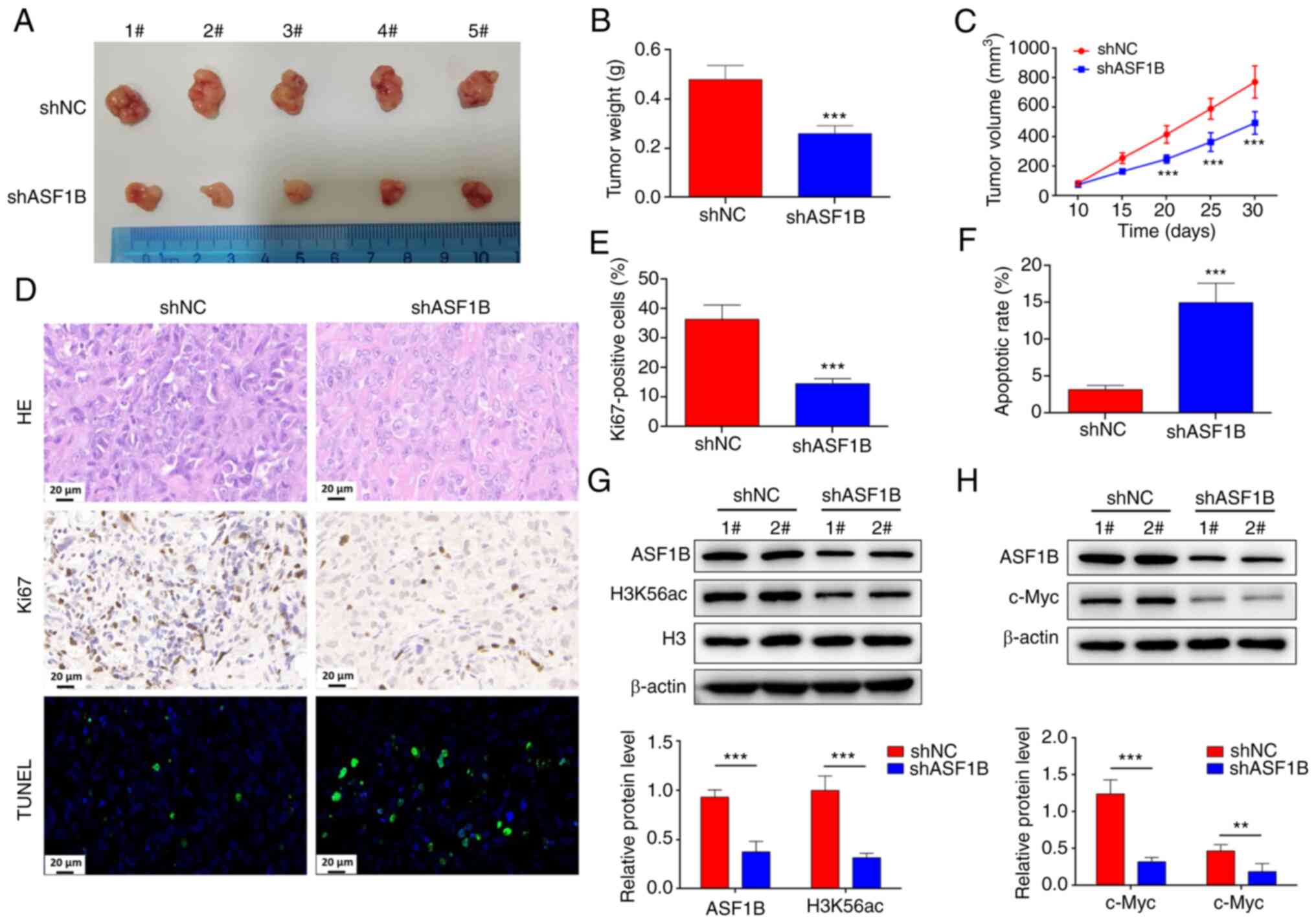
Silencing of ASF1B reduces c-Myc
expression and pancreatic cancer growth in vivo
The present study further determined the role of
ASF1B in pancreatic cancer development in vivo. Therefore,
pancreatic cancer xenografts were established by injecting BALB/c
nude mice with PANC-1 cells that were pre-transfected with shNC or
shASF1B. The proof of successful transfection of shASF1B is shown
in Fig. S2. Compared with those
in the shNC group, the mice in the shASF1B group had a lower tumor
weight and volume at day 20, 25 and 30 (Fig. 6A-C). HE staining results showed
that, compared with the shNC group, the shASF1B group tumor tissues
exhibited focal necrosis (Fig.
6D). The percentage of Ki67-positive cells was reduced, and the
TUNEL-positive cell rate was increased significantly in the tumor
tissues of the shASF1B group (Fig.
6D-F). Furthermore, ASF1B was weakly expressed in the shASF1B
group, with low expression of H3K56ac and c-Myc (Fig. 6G and H), indicating that silencing
of ASF1B inhibited c-Myc expression and tumor growth in
vivo.
Discussion
Pancreatic cancer is one of the deadliest
malignancies of the digestive system worldwide (32). It has been previously reported that
the histone chaperone ASF1B is a biomarker for predicting the
outcome of human cancers, including pancreatic cancer (15,33).
Silencing of ASF1B influences prostate cancer cell apoptosis by
inhibiting PI3K/AKT signaling (11). ASF1B increases migration and growth
of clear cell renal cell carcinoma by activating AKT signaling
(34). Silencing of ASF1B induces
pancreatic cancer cell death and inhibits cancer cell
micrometastasis via inactivation of the PI3K/AKT signaling pathway
(14,15). By analyzing TCGA, ASF1B was
identified to be highly expressed in PAAD samples, and its
expression was positively associated with poor survival rate. In
the present study, high ASF1B expression was also demonstrated in
31 patients with pancreatic cancer. In PANC-1 and SW1990 cells,
ASF1B silencing repressed cell proliferation, migration and
invasion, indicating the oncogenic role of ASF1B in pancreatic
cancer. In addition, to the best of our knowledge, the present
study was the first to demonstrate that ASF1B induced c-Myc
expression through CBP-mediated H3K56ac. ASF1B exerted oncogenic
effects in pancreatic cancer, possibly through the activation of
CBP/H3K56ac/c-Myc signaling.
c-Myc has been recognized as a critical regulator of
pancreatic cancer progression (19). Various factors that target c-Myc
expression have been demonstrated to be effective in regulating
tumor progression. For instance, pescadillo ribosomal biogenesis
factor 1 enhances resistance to extra-terminal inhibitors and cell
proliferation by increasing c-Myc expression in pancreatic cancer
(35). F-box and WD repeat domain
containing 7 is a negative modulator of glycolytic metabolism that
regulates c-Myc/thioredoxin interacting protein signaling in
pancreatic cancer (36).
Bromodomain PHD finger transcription factor is critical for
transcriptional activation of c-Myc in tumorigenesis (37). Proteasome activator subunit 3
increases pancreatic cancer progression via c-Myc/glycolysis
signaling (38). The cooperation
of dysregulated KRAS and Myc via inhibition of the type I
interferon pathway achieves immune evasion in pancreatic cancer
cells (39). The present study
revealed that ASF1B was positively associated with c-Myc and that
ASF1B promoted c-Myc expression in pancreatic cancer cells. To the
best of our knowledge, the present study was the first to
demonstrate an association between ASF1B and c-Myc in the
development of pancreatic cancer, and presented a novel mechanism
of ASF1B-mediated pancreatic cancer involving c-Myc activation.
Epigenetic regulation serves a critical role in
cancer development, and histone acetylation modulates
cancer-related gene expression (40,41).
Histone H3K56ac and histone acetyltransferase CBP are involved in
cancer progression. It has been reported that hypoxia-provoked
K63-polyubiquitinated herpes virus-associated ubiquitin-specific
protease affects CBP-regulated acetylation of H3K56 on the
promoters of hypoxia-inducible factor 1α-targeted genes to increase
metastasis in tumor progression (42). mTORC2 signaling positively supports
H3K56ac to regulate metabolic re-programming in gliomas (43). Chromatin marks on Notch-related
enhancers exhibit large-scale variations in H3K56ac upon activation
(44). In addition, ASF1B is
reportedly essential for regulating H3K56ac and this modification
in gene transcription and replication (45,46).
Genetic deletion of the H3K56 deacetylase enzyme sirtuin 6 leads to
increased c-Myc transcription (47,48).
The present study revealed that ASF1B enhanced c-Myc expression by
modulating H3K56ac and promoting H3K56ac at the c-Myc promoter.
These data revealed a novel mechanism involving
ASF1B/CBP/H3K56ac/c-Myc in pancreatic cancer progression.
In conclusion, ASF1B promoted pancreatic cancer
progression by activating c-Myc through CBP-mediated H3K56ac. The
present findings provide novel insights into the mechanism by which
ASF1B modulates pancreatic cancer development via CBP/H3K56ac/c-Myc
signaling. Therefore, ASF1B may serve as a potential target for
pancreatic cancer therapy.
Supplementary Data
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MZha conceived and designed the study. LZ, MZho, EW,
and BM performed experiments. QinL, XW, YW, and QioL analyzed and
interpreted the data. MZha wrote the manuscript. All authors read
and approved the final manuscript. MZha, YW and QioL confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Animal experiments were performed in accordance with
the Guide for the Care and Use of Laboratory Animals. Animal and
patient experiments were approved by the Ethics Committee of Cancer
Hospital Affiliated to Zhengzhou University (Ethics No.
2019-05-006; Zhengzhou, China). This research was conducted under
the guidance of the Declaration of Helsinki. All patients provided
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
ASF1B
|
anti-silencing function 1B
|
|
CBP
|
CREB-binding protein
|
|
H3K56ac
|
histone H3 lysine 56 acetylation
|
|
PAAD
|
pancreatic adenocarcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
Acknowledgments
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China joint fund project (grant no. u1404820),
Tackling Key Problems of Science and Technology in Henan Province
(grant no. 212102310137) and Henan Medical Science and Technology
Research plan project (joint construction) (grant no.
lhgj20190628).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019.
|
|
2
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016.
|
|
3
|
Kang MJ, Jang JY and Kim SW: Surgical
resection of pancreatic head cancer: What is the optimal extent of
surgery? Cancer Lett. 382:259–265. 2016.
|
|
4
|
Garrido-Laguna I and Hidalgo M: Pancreatic
cancer: From state-of-the-art treatments to promising novel
therapies. Nat Rev Clin Oncol. 12:319–334. 2015.
|
|
5
|
Giovannetti E, van der Borden CL, Frampton
AE, Ali A, Firuzi O and Peters GJ: Never let it go: Stopping key
mechanisms underlying metastasis to fight pancreatic cancer. Semin
Cancer Biol. 44:43–59. 2017.
|
|
6
|
Collisson EA, Bailey P, Chang DK and
Biankin AV: Molecular subtypes of pancreatic cancer. Nat Rev
Gastroenterol Hepatol. 16:207–220. 2019.
|
|
7
|
Ilic M and Ilic I: Epidemiology of
pancreatic cancer. World J Gastroenterol. 22:9694–9705. 2016.
|
|
8
|
Mousson F, Ochsenbein F and Mann C: The
histone chaperone Asf1 at the crossroads of chromatin and DNA
checkpoint pathways. Chromosoma. 116:79–93. 2007.
|
|
9
|
Cote JM, Kuo YM, Henry RA, Scherman H,
Krzizike DD and Andrews AJ: Two factor authentication: Asf1
mediates crosstalk between H3 K14 and K56 acetylation. Nucleic
Acids Res. 47:7380–7391. 2019.
|
|
10
|
Paul PK, Rabaglia ME, Wang CY, Stapleton
DS, Leng N, Kendziorski C, Lewis PW, Keller MP and Attie AD:
Histone chaperone ASF1B promotes human β-cell proliferation via
recruitment of histone H3.3. Cell Cycle. 15:3191–3202. 2016.
|
|
11
|
Han G, Zhang X, Liu P, Yu Q, Li Z, Yu Q
and Wei X: Knockdown of anti-silencing function 1B histone
chaperone induces cell apoptosis via repressing PI3K/Akt pathway in
prostate cancer. Int J Oncol. 53:2056–2066. 2018.
|
|
12
|
Corpet A, De Koning L, Toedling J,
Savignoni A, Berger F, Lemaître C, O'Sullivan RJ, Karlseder J,
Barillot E, Asselain B, et al: Asf1b, the necessary Asf1 isoform
for proliferation, is predictive of outcome in breast cancer. EMBO
J. 30:480–493. 2011.
|
|
13
|
Rosty C, Sheffer M, Tsafrir D, Stransky N,
Tsafrir I, Peter M, de Crémoux P, de La RA, Salmon R, Dorval T, et
al: Identification of a proliferation gene cluster associated with
HPV E6/E7 expression level and viral DNA load in invasive cervical
carcinoma. Oncogene. 24:7094–7104. 2005.
|
|
14
|
Kim JH, Youn Y, Lee JC, Kim J, Ryu JK and
Hwang JH: Downregulation of ASF1B inhibits tumor progression and
enhances efficacy of cisplatin in pancreatic cancer. Cancer
Biomark. 34:647–659. 2022.
|
|
15
|
Wang K, Hao Z, Fu X, Li W, Jiao A and Hua
X: Involvement of elevated ASF1B in the poor prognosis and
tumorigenesis in pancreatic cancer. Mol Cell Biochem.
477:1947–1957. 2022.
|
|
16
|
Thomas LR, Foshage AM, Weissmiller AM and
Tansey WP: The MYC-WDR5 nexus and cancer. Cancer Res. 75:4012–4015.
2015.
|
|
17
|
Pelengaris S and Khan M: The c-MYC
oncoprotein as a treatment target in cancer and other disorders of
cell growth. Expert Opin Ther Targets. 7:623–642. 2003.
|
|
18
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012.
|
|
19
|
Hessmann E, Schneider G, Ellenrieder V and
Siveke JT: MYC in pancreatic cancer: Novel mechanistic insights and
their translation into therapeutic strategies. Oncogene.
35:1609–1618. 2016.
|
|
20
|
Wirth M, Mahboobi S, Kramer OH and
Schneider G: Concepts to target MYC in pancreatic cancer. Mol
Cancer Ther. 15:1792–1798. 2016.
|
|
21
|
Hogg SJ, Beavis PA, Dawson MA and
Johnstone RW: Targeting the epigenetic regulation of antitumour
immunity. Nat Rev Drug Discov. 19:776–800. 2020.
|
|
22
|
Chen Y, Hong T, Wang S, Mo J, Tian T and
Zhou X: Epigenetic modification of nucleic acids: From basic
studies to medical applications. Chem Soc Rev. 46:2844–2872.
2017.
|
|
23
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
|
|
24
|
Audia JE and Campbell RM: Histone
modifications and cancer. Cold Spring Harb Perspect Biol.
8:a0195212016.
|
|
25
|
Liu Z, Yang L, Sun Y, Xie X and Huang J:
ASF1a enhances antiviral immune response by associating with CBP to
mediate acetylation of H3K56 at the Ifnb promoter. Mol Immunol.
78:57–64. 2016.
|
|
26
|
Zhang L, Serra-Cardona A, Zhou H, Wang M,
Yang N, Zhang Z and Xu RM: Multisite substrate recognition in
asf1-dependent acetylation of histone H3 K56 by Rtt109. Cell.
174:818–830.e811. 2018.
|
|
27
|
Shaukat A, Khan MHF, Ahmad H, Umer Z and
Tariq M: Interplay between BALL and CREB binding protein maintains
H3K27 acetylation on active genes in drosophila. Front Cell Dev
Biol. 9:7408662021.
|
|
28
|
Das C, Lucia MS, Hansen KC and Tyler JK:
CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature.
459:113–117. 2009.
|
|
29
|
Tang Z, Kang B, Li C, Chen T and Zhang Z:
GEPIA2: An enhanced web server for large-scale expression profiling
and interactive analysis. Nucleic Acids Res. 47:W556–W560.
2019.
|
|
30
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012.
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
32
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017.
|
|
33
|
Hu X, Zhu H, Zhang X, He X and Xu X:
Comprehensive analysis of pan-cancer reveals potential of ASF1B as
a prognostic and immunological biomarker. Cancer Med. 10:6897–6916.
2021.
|
|
34
|
Jiangqiao Z, Tao Q, Zhongbao C, Xiaoxiong
M, Long Z, Jilin Z and Tianyu W: Anti-silencing function 1B histone
chaperone promotes cell proliferation and migration via activation
of the AKT pathway in clear cell renal cell carcinoma. Biochem
Biophys Res Commun. 511:165–172. 2019.
|
|
35
|
Jin X, Fang R, Fan P, Zeng L, Zhang B, Lu
X and Liu T: PES1 promotes BET inhibitors resistance and cells
proliferation through increasing c-Myc expression in pancreatic
cancer. J Exp Clin Cancer Res. 38:4632019.
|
|
36
|
Jensen MB, Lawaetz JG, Andersson AM,
Petersen JH, Nordkap L, Bang AK, Ekbom P, Joensen UN, Prætorius L,
Lundstrøm P, et al: Vitamin D deficiency and low ionized calcium
are linked with semen quality and sex steroid levels in infertile
men. Hum Reprod. 31:1875–1885. 2016.
|
|
37
|
Richart L, Carrillo-de Santa Pau E,
Rio-Machin A, de Andrés MP, Cigudosa JC, Lobo VJ and Real FX: BPTF
is required for c-MYC transcriptional activity and in vivo
tumorigenesis. Nat Commun. 7:101532016.
|
|
38
|
Guo J, Hao J, Jiang H, Jin J, Wu H, Jin Z
and Li Z: Proteasome activator subunit 3 promotes pancreatic cancer
growth via c-Myc-glycolysis signaling axis. Cancer Lett.
386:161–167. 2017.
|
|
39
|
Muthalagu N, Monteverde T,
Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, Laing S,
Kruspig B, Upstill-Goddard R, Shaw R, et al: Repression of the type
I interferon pathway underlies MYC- and KRAS-dependent evasion of
NK and B cells in pancreatic ductal adenocarcinoma. Cancer Discov.
10:872–887. 2020.
|
|
40
|
Okugawa Y, Grady WM and Goel A: Epigenetic
alterations in colorectal cancer: Emerging biomarkers.
Gastroenterology. 149:1204–1225.e1212. 2015.
|
|
41
|
Benton CB, Fiskus W and Bhalla KN:
Targeting histone acetylation: Readers and writers in leukemia and
cancer. Cancer J. 23:286–291. 2017.
|
|
42
|
Wu HT, Kuo YC, Hung JJ, Huang CH, Chen WY,
Chou TY, Chen Y, Chen YJ, Chen YJ, Cheng WC, et al:
K63-polyubiquitinated HAUSP deubiquitinates HIF-1alpha and dictates
H3K56 acetylation promoting hypoxia-induced tumour progression. Nat
Commun. 7:136442016.
|
|
43
|
Vadla R and Haldar D: Mammalian target of
rapamycin complex 2 (mTORC2) controls glycolytic gene expression by
regulating histone H3 Lysine 56 acetylation. Cell Cycle.
17:110–123. 2018.
|
|
44
|
Skalska L, Stojnic R, Li J, Fischer B,
Cerda-Moya G, Sakai H, Tajbakhsh S, Russell S, Adryan B and Bray
SJ: Chromatin signatures at Notch-regulated enhancers reveal
large-scale changes in H3K56ac upon activation. EMBO J.
34:1889–1904. 2015.
|
|
45
|
Weng M, Yang Y, Feng H, Pan Z, Shen WH,
Zhu Y and Dong A: Histone chaperone ASF1 is involved in gene
transcription activation in response to heat stress in Arabidopsis
thaliana. Plant Cell Environ. 37:2128–2138. 2014.
|
|
46
|
Han J, Zhou H, Li Z, Xu RM and Zhang Z:
Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and
regulated by ASF1 is required for replisome integrity. J Biol Chem.
282:28587–28596. 2007.
|
|
47
|
Sebastián C, Zwaans BM, Silberman DM,
Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber
D, et al: The histone deacetylase SIRT6 is a tumor suppressor that
controls cancer metabolism. Cell. 151:1185–1199. 2012.
|
|
48
|
Cai J, Zuo Y, Wang T, Cao Y, Cai R, Chen
FL, Cheng J and Mu J: A crucial role of SUMOylation in modulating
Sirt6 deacetylation of H3 at lysine 56 and its tumor suppressive
activity. Oncogene. 35:4949–4956. 2016.
|