Introduction
Post-traumatic stress disorder (PTSD) is an anxiety
disorder that may develop following exposure to the threat of death
or serious injury and may cause affected invididuals to
continuously re-experience the traumatic event (1,2) and
react with intense fear, helplessness or horror for years. Patients
with this disorder persistently re-experience their traumatic
events in various ways, including intrusive and disturbing
recollections, nightmares, flashbacks and distress and
physiological reactivity on exposure to reminders of the event
(3). These individuals often avoid
reminders of the traumatic event and experience a restricted range
of effects.
One of the core neuroendocrine abnormalities related
to PTSD is the dysfunction of the hypothalamic-pituitary-adrenal
(HPA) axis, characterized by low levels of adrenocorticotropic
hormone (ACTH), plasma cortisol and urinary cortisol and enhanced
suppression of cortisol in response to low-dose dexamethasone
administration (4,5). These neuroendocrine findings specific
to PTSD have served as the basis for animal models and are useful
for elucidating the pathophysiology of PTSD. Single-prolonged
stress (SPS) is a reliable animal model of PTSD based on the
time-dependent dysregulation of the HPA axis which has been
developed and employed for PTSD studies (6,7). SPS
has been shown to induce enhanced inhibition of the HPA axis, which
is a putative neuroendocrinological hallmark of PTSD (8). In addition, SPS rats also exhibit
behavioral abnormalities (enhanced anxiety) that mimic the symptoms
of PTSD. SPS paradigms have been extensively applied in the
investigation of PTSD (9).
A convergent body of human and non-human studies
suggests that the amygdala mediates the acquisition and expression
of conditioned fear and the enhancement of emotional memory
(10), whereas the medial
prefrontal cortex (mPFC) mediates the extinction of conditioned
fear and the volitional regulation of negative emotion (11). It has been theorized that the mPFC
exerts an inhibitory effect on the amygdala and that a defect in
this inhibition could account for the symptoms of PTSD (12). A study of brain-injured and
trauma-exposed combat veterans confirmed that amygdala damage
reduces the likelihood of developing PTSD (13). However, contrary to the prediction
of the top-down inhibition model, mPFC damage also reduces the
likelihood of developing PTSD. The mPFC contributes significantly
to the modulation of memory consolidation with the storage of
emotionally relevant information and is critical for the formation
of long-term aversive memory, particularly for the modulation of
anxiety, fear and aggression (14). In addition, the mPFC may inhibit
the effect of the amygdala following SPS in PTSD rats and may also
be related to impaired fear extinction. It has been confirmed by
computed tomography and functional magnetic resonance imaging that
the mPFC of patients with PTSD are notably smaller than normal.
Calcium (Ca2+) is an influential
intracellular secondary messenger. Elevated Ca2+ binds
to numerous proteins, including low-affinity/high-capacity buffer
proteins. The influx of Ca2+ ions results in calmodulin
(CaM) activation. A number of Ca2+/CaM targets modulate
cellular signaling pathways. CaM kinase II (CaMKII) is a major
mediator of calcium signaling and is of particular importance in
the brain, contributing significantly to the regulation of nerve
functions (15,16).
In this study, our team briefly examined the changes
in Ca2+-CaM-CaMKIIα levels in the mPFC in order to
ascertain how Ca2+-CaM-CaMKIIα cascades participate in
PTSD.
Materials and methods
Animal model preparation and
grouping
A total of 21 male Wistar rats were randomly divided
into a control and SPS groups of 1 (1-day) and 7 days (7-day). The
control rats remained in their home cages with no handling for 7
days and were sacrificed at the same time as the SPS groups. The
SPS rats underwent the SPS procedure on the first day. The SPS
protocol (7,9) consisted of: a 2-h immobilization
(compression with plastic bags), a 20-min forced swim (25°C), and a
15-min rest, followed by ether anesthesia (until loss of
consciousness). Following SPS, the rats were fed routinely. The
study was approved by the ethics committee of China Medical
University.
Intracellular free calcium in mPFC
cells
Rats of the control and SPS groups were decapitated
rapidly and the brains were removed and immediately placed in a
dish standing on crushed ice. The mPFC was then dissected out
according to the atlas of rats (17), snap-frozen in liquid nitrogen and
prepared for cell suspension using a routine method. Furthermore,
the cell suspension was loaded with 1 mmol/l fura-2-acetoxymethyl
ester (Fura-2/AM) (Beyotime Institute of Biotechnology, Haimen,
China) for 35 min and then detected with a spectrofluorometer.
Immunohistochemistry
Rats of the control and SPS groups were prepared by
left ventricle perfusion fixation (18) with 4% buffered paraformaldehyde and
the mPFCs were post-fixed in the same fixative at 4°C for 24 h and
then embedded in paraffin. Paraffin sections (5-μm) were prepared
for the morphological studies. The mPFC sections were treated with
5% bovine serum albumin (BSA) and 0.3% Triton X-100 in PBS for 30
min at room temperature for blocking of non-specific staining,
followed by incubation with mouse monoclonal antibody against CaM
(Sigma, St. Louis, MO, USA; 1:100) or CaMKIIα (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA; 1:200) overnight at 4°C.
Following incubation with goat anti-mouse IgG (Boster, Wuhan,
China; 1:100) for 2 h, sections were treated with the
streptomycin-avidin-biotin-peroxidase complex (SABC) for 1 h at
room temperature. Moreover, they were washed three times with PBS
following each incubation and subsequently incubated with
3,3′-diaminobenzidine (DAB) and H2O2. In
order to assess non-specific staining, a few sections in every
experiment were processed with the omission of the antibody.
Western blotting
Fresh mPFC of the control and SPS rats were
respectively homogenized with sample buffer containing 200 mM TBS,
pH 7.5, 4% sodium dodecyl sulfate (SDS), 20% glycerol and 10%
2-mercaptoethanol and were denatured by boiling for 3 min. The
protein fraction (30 μg/lane) extracted from each sample was
separated by 12% (w/v) gradient SDS-polyacrylamide gel
electrophoresis (PAGE) and transferred to a PVDF membrane
(Millipore, Bedford, MA, USA). After blocking with 5% dried skimmed
milk in 0.05% Tween-20-containing TBST at room temperature for 2 h
and incubating with a primary antibody comprising a mouse
monoclonal antibody against CaM (1:1,000) or CaMKIIα (1:5,000)
overnight at 4°C, respectively, the membrane was incubated with
anti-mouse IgG-HRP (Santa Cruz Biotechnology, Inc.; 1:5000)
secondary antibodies for another 2 h at room temperature. Finally,
the PVDF membrane was washed three times with TBST prior to
visualization using enhanced chemiluminescence (ECL; Amersham
Pharmacia Biotech, Buckinghamshire, UK).
Reverse transcription-polymerase chain
reaction
The total mRNA from the mPFC was extracted using the
TRIzol kit according to the manufacturer’s instructions. The
forward and reverse sequences of the primers (synthesized by
Shenggong Biotech Co., Shanghai, China) were according to the
serial numbers from GenBank and are listed in Table I(19). The cycling reaction for CaM was as
follows: 94°C for 4 min, followed by amplification for 32 cycles of
30 sec at 94°C, 30 sec at 58°C and 40 sec at 72°C and a final 7-min
extension at 72°C. For CaMKIIα, the reaction was started at 95°C
for 2 min, followed by amplification for 33 cycles of 30 sec at
95°C, 30 sec at 55°C and 40 sec at 72°C and a final 5-min extension
at 95°C. β-actin mRNA used as an internal control was co-amplified
with CaM and CaMKIIα. The products were observed following
electrophoresis on a 1.2% agarose gel and the density of each band
was analyzed on the Gel Image Analysis System. The levels of CaM
and CaMKIIα mRNA were determined by calculating the density ratios
of CaM mRNA/β-actin mRNA and CaMKIIα mRNA/β-actin mRNA.
 | Table IPrimer sequences for CaM and
CaMKIIα. |
Table I
Primer sequences for CaM and
CaMKIIα.
| Name | Upstream primer
(5′-3′) | Downstream primer
(5′-3′) | Product size
(bp) |
|---|
| CaM | ggcatcctgctt
tagcctgag | acatgctatccc
tctcgtgtga | 328 |
| CaMKIIα |
catcctcaccactatgctg |
atcgatgaaagtccaggccg | 284 |
| β-actin |
atcacccacactgtgcccatc |
acagagtacttgcgctcagga | 542 |
Statistical analysis
All data were expressed as the mean ± standard
deviation (SD). Data analysis among groups was performed using
one-way analysis of variance (ANOVA) with SPSS 13.0 software.
P<0.05 was considered to indicate a statistically significant
difference.
Results
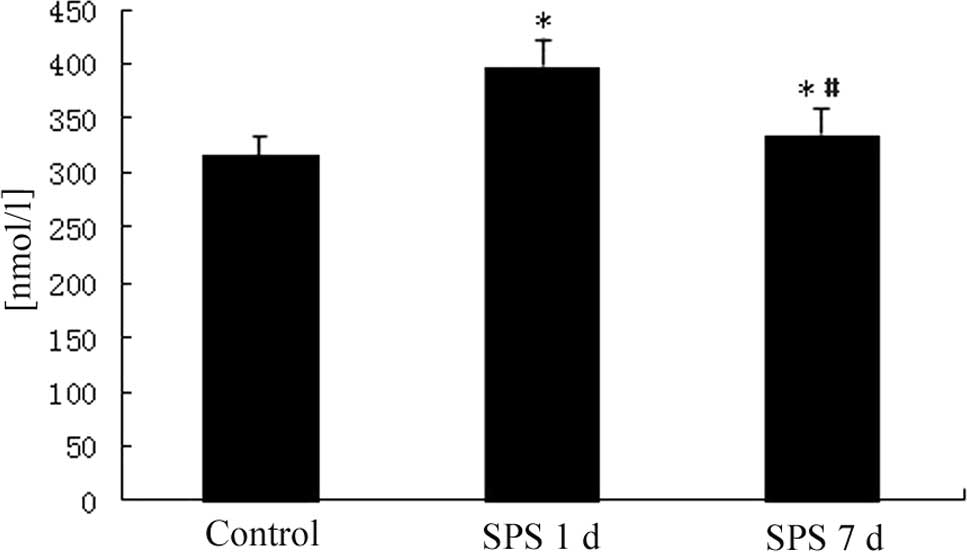
Free Ca2+ concentration in
mPFC
The intracellular free Ca2+ levels in the
mPFC neurons were notably higher than in the control group 1 day
after SPS and had returned to normal levels 7 days after SPS
(Fig. 1).
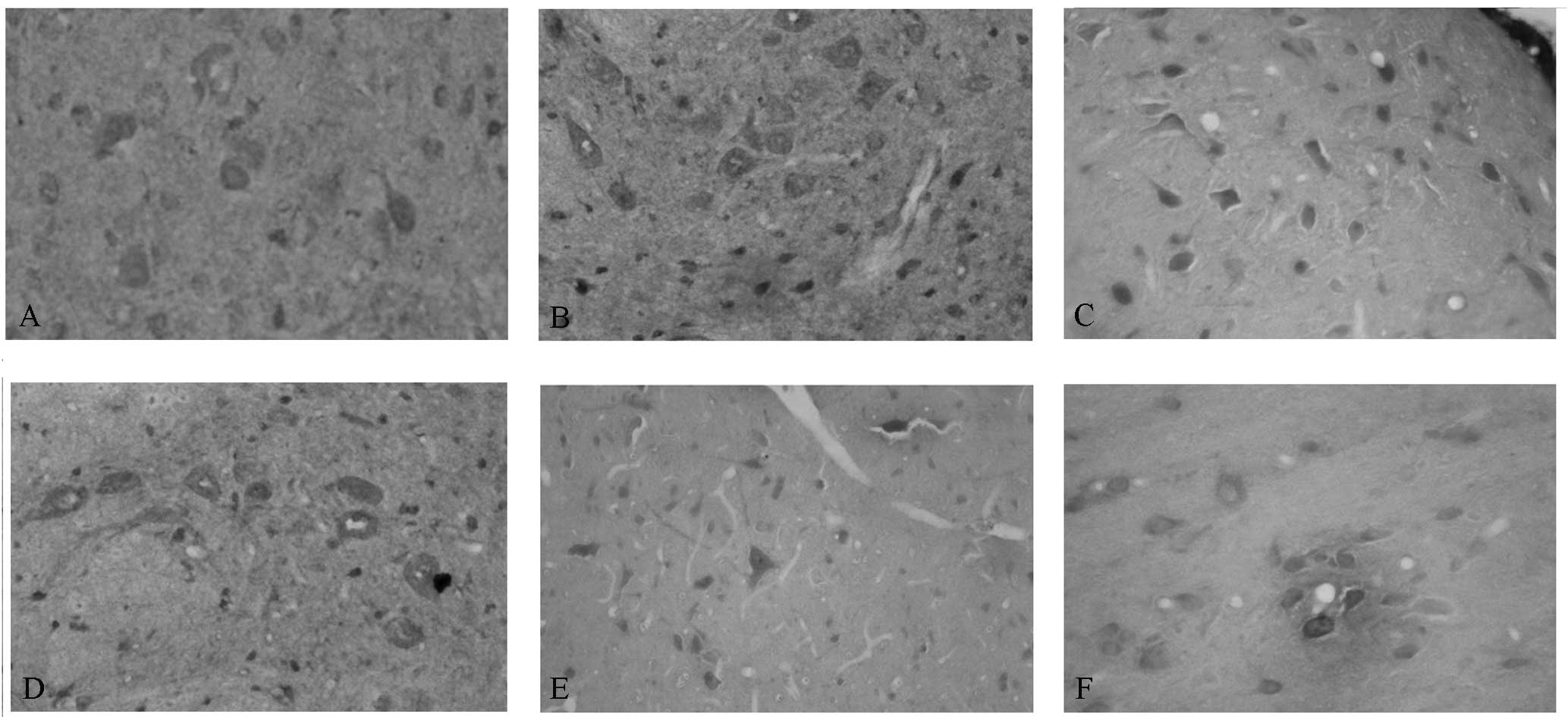
Immunohistochemical observation of CaM
and CaMKIIα
Our team observed the CaM and CaMKIIα levels in the
mPFC of the control and SPS rats. The sites of expression of CaM
and CaMKIIα were distributed mainly in the cytoplasm and appeared
as buffy particles (Fig. 2A and
D). In SPS rats, increased CaM levels were observed; the
highest expression levels were identified 1 day after SPS
stimulation (Fig. 2B and C); by
contrast, decreased CaMKIIα levels were observed (Fig. 2E and F).
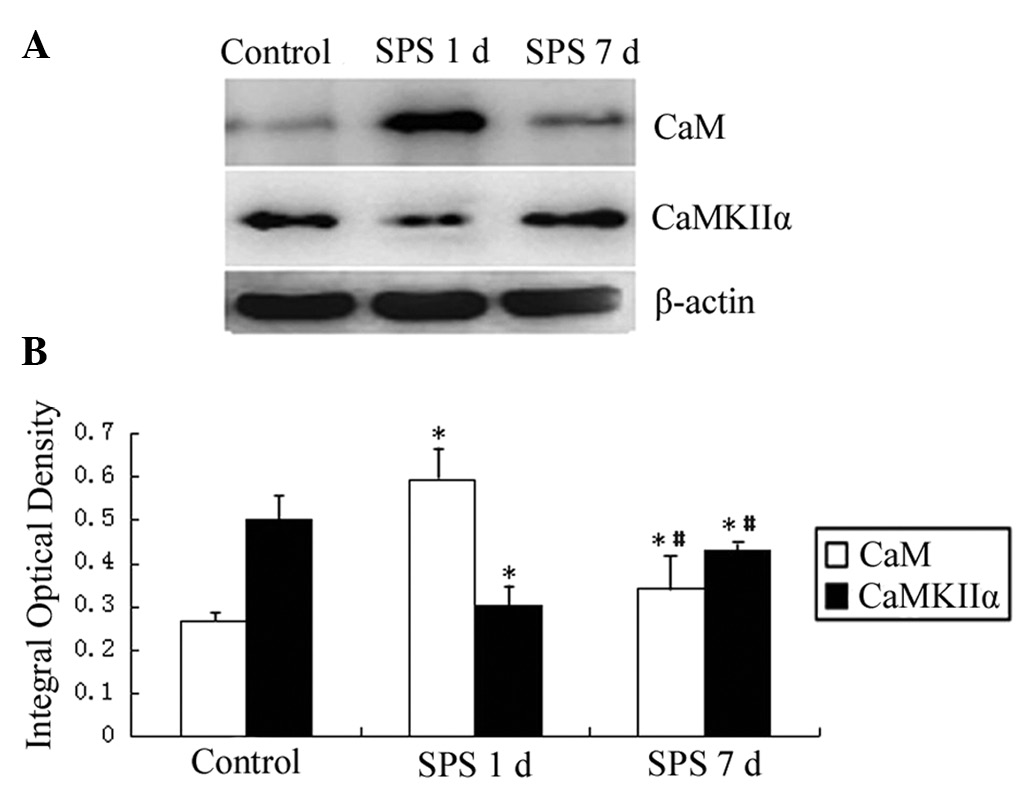
Western blotting of CaM and CaMKIIα
The CaM, CaMKIIα and β-actin immunoreactive signals
appeared at 16.7, 50 and 42 kDa, respectively (data not shown), and
the band density mean value of the control group was set as 100%.
Data were expressed as normalized optical density.
In the SPS group, the expression of CaM protein in
the mPFC was highest on day 1 and was downregulated a little on day
7, but remained higher than the control. Significantly lower
protein expression of CaMKIIα was identified in the SPS group
compared with the control on days 1 and 7 (Fig. 3).
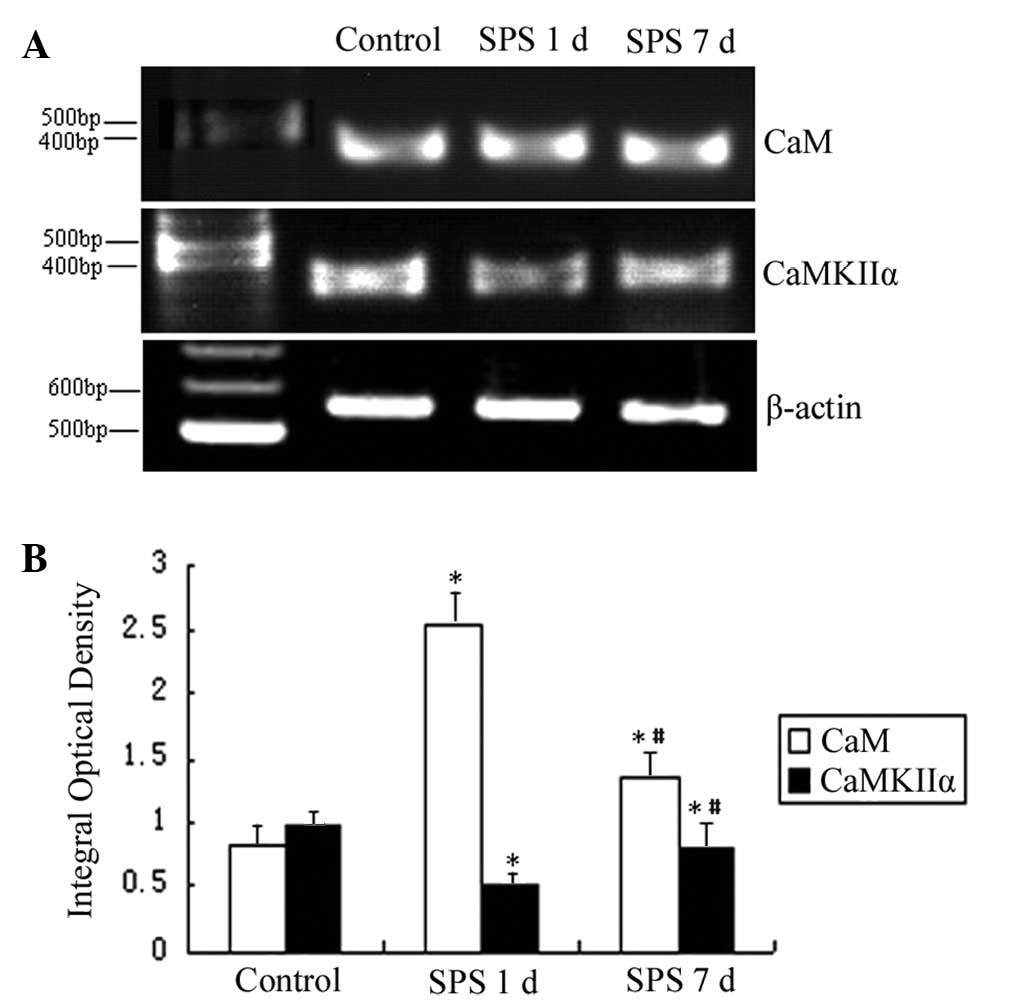
mRNA expression of CaM and CaMKIIα
The mRNA levels of CaM and CaMKIIα were normalized
to the β-actin mRNA level. The mRNA expression levels of CaM
significantly increased in the mPFC of the SPS group compared with
the control rats. However, the mRNA expression levels of CaMKIIα
markedly decreased in the SPS group rats (Fig. 4).
Discussion
Over the past decade, basic animal research and
human neuroimaging studies have begun to outline the specific
neural circuitry dedicated to emotional functioning (20). These studies have partially
inspired hypotheses with regard to the dysfunction in this
circuitry that leads to the development and maintenance of PTSD.
While they have provided useful initial information and guided the
initial functional neuroanatomical and neurophysiological studies
into PTSD, it is becoming increasingly clear that the scope of
these studies does not fully capture the complexity of the changes
occurring during trauma exposure and PTSD development.
Evidence that the mPFC is involved in a neural
mechanism that modulates a differential susceptibility of multiple
memory systems to emotional arousal during acquisition, memory
consolidation (21) and/or memory
retrieval, may provide a novel approach to elucidating the range of
mnemonic dysfunctions in PTSD.
The mPFC is significantly involved in emotional
adjustment. The function of emotional adjustment was induced in
PTSD. With the extension of the SPS stimulation time, an increasing
number of neurons of the mPFC of rats became smaller (22) and the outline of the small neurons
was indistinct. In the future, this finding may help scientists to
understand how brain damage is likely to affect thought, action and
the ability to reflect.
The volitional control of negative emotion is
another affective function that is germane to PTSD and one in which
the mPFC and amygdala again are significant (23). That is, during emotional
regulation, an increase in the activity of the mPFC is associated
with a decrease in the activity of the amygdala as well as with the
experience of negative effects. Furthermore, this normative
mPFC-amygdala inverse coupling during emotional regulation is
disrupted in patients with major depressive disorder, which is
characterized by pathologically high levels of negative
effects.
The special role of the mPFC in the processing of
threat-related stimuli, particularly anger and fear, is well
documented (24). Abundant
evidence from animal and human investigations strongly suggests
that the mPFC is responsible for the enhancement of explicit memory
associated with emotional arousal (25,26).
In addition, numerous lines of evidence have implicated the mPFC as
a substrate for the stress-related modulation of memory.
CaM, as a ubiquitous Ca2+ sensor protein,
is involved in almost all intracellular events. CaMKIIα is the
molecular basis of learning and memory (27), but in the absence of bound
Ca2+/CAM, CaMKII is in an inactive conformation. The
influx of Ca2+ results in CaMKII activation.
Ca2+/CaMKII is a major mediator of Ca2+
signaling and is of particular importance in the brain,
contributing significantly to the regulation of nerve functions,
including learning and memory (28). It is speculated that CaMKIIα
responds to a strong and/or repeated stimulus in which the cellular
Ca2+ concentration is relatively high. CaMKIIα is highly
effective in synaptic plasticity and is considered as one of the
best candidates for a memory molecule (29).
In this study, the detection of free Ca2+
in the mPFC neurons revealed Ca2+ overload 1 day after
SPS stimulation. Further analysis of CaM, the main
Ca2+-conjugated protein in the CNS, revealed that the
expression of total CaM in the mPFC markedly increased 1 day after
SPS stimulation, suggesting that the CaM content changed
synchronously with changes in the Ca2+ concentration.
This occurred as a result of the SPS increasing the intracellular
free Ca2+ levels in the mPFC neurons and thereby
inducing the overexpression of CaM. The change in CaMKIIα from
inactive to active led to a decreased content of CaMKIIα in the
mPFC following SPS exposure. Due to the importance of the
Ca2+-CaM-CaMKIIα signaling pathway in the plasticity of
the central nervous system, learning and memory, mind, behavior and
other types of cognitive activities (30), the dysfunction of the
Ca2+-CaM-CaMKIIα pathway of the mPFC might be the
pathobiological basis for the abnormality of affect and behavior
induced by PTSD.
To date, the pathogenesis of PTSD has not been
entirely clarified. PTSD may result in a series of biochemical and
physiological abnormalities in the brain, which lead to dysfunction
of the mPFC. Thus, the pathogenesis of PTSD requires further
study.
In conclusion, the SPS rats exhibit behavioral
abnormalities that mimic the symptoms of PTSD. The dysfunction of
Ca2+-CaM-CaMKIIα in the SPS rats decreases the
inhibition of the amygdala, which might be the pathobiological
basis of the abnormality of affect and behavior induced by
PTSD.
Acknowledgements
The authors would like to thank all staff members of
the China Medical University Experiment Center for their technical
support. In addition, this study was supported by a grant from the
National Natural Science Foundation of China (no. 81171282).
References
|
1
|
Liberzon I and Martis B: Neuroimaging
studies of emotional responses in PTSD. Ann NY Acad Sci.
1071:87–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shin LM, Wright CI, Cannistraro PA, Wedig
MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR,
Krangel TS, et al: A functional magnetic resonance imaging study of
amygdala and medial prefrontal cortex responses to overtly
presented fearful faces in posttraumatic stress disorder. Arch Gen
Psychiatry. 62:273–281. 2005. View Article : Google Scholar
|
|
3
|
Liberzon I, King AP, Britton JC, Phan KL,
Abelson JL and Taylor SF: Paralimbic and medial prefrontal cortical
involvement in neuroendocrine responses to traumatic stimuli. Am J
Psychiatry. 164:1250–1258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eckart C, Stoppel C, Kaufmann J,
Tempelmann C, Hinrichs H, Elbert T, Heinze HJ and Kolassa IT:
Structural alterations in lateral prefrontal, parietal and
posterior midline regions of men with chronic posttraumatic stress
disorder. J Psychiatry Neurosci. 36:176–186. 2011. View Article : Google Scholar
|
|
5
|
Shin LM, Rauch SL and Pitman RK: Amygdala,
medial prefrontal cortex, and hippocampal function in PTSD. Ann NY
Acad Sci. 1071:67–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hughes KC and Shin LM: Functional
neuroimaging studies of post-traumatic stress disorder. Expert Rev
Neurother. 11:275–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koenigs M and Grafman J: Posttraumatic
stress disorder: the role of medial prefrontal cortex and amygdala.
Neuroscientist. 15:540–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adou E, Miller JS, Ratovoson F, Birkinshaw
C, Andriantsiferana R, Rasamison VE and Kingston DG:
Antiproliferative cardenolides from Pentopetia androsaemifolia
Decne from the Madagascar rain forest. Indian J Exp Biol.
48:248–257. 2010.
|
|
9
|
Weinberg MS, Johnson DC, Bhatt AP and
Spencer RL: Medial prefrontal cortex activity can disrupt the
expression of stress response habituation. Neuroscience.
168:744–756. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang CH, Berke JD and Maren S:
Single-unit activity in the medial prefrontal cortex during
immediate and delayed extinction of fear in rats. PLoS One.
5:e119712010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
St Jacques PL, Botzung A, Miles A and
Rubin DC: Functional neuroimaging of emotionally intense
autobiographical memories in post-traumatic stress disorder. J
Psychiatr Res. 45:630–637. 2011.PubMed/NCBI
|
|
12
|
King AP, Abelson JL, Britton JC, Phan KL,
Taylor SF and Liberzon I: Medial prefrontal cortex and right insula
activity predict plasma ACTH response to trauma recall. Neuroimage.
47:872–880. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaw ME, Moores KA, Clark RC, McFarlane
AC, Strother SC, Bryant RA, Brown GC and Taylor JD: Functional
connectivity reveals inefficient working memory systems in
post-traumatic stress disorder. Psychiatry Res. 172:235–241. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sailer U, Robinson S, Fischmeister FP,
König D, Oppenauer C, Lueger-Schuster B, Moser E, Kryspin-Exner I
and Bauer H: Altered reward processing in the nucleus accumbens and
medial prefrontal cortex of patients with posttraumatic stress
disorder. Neuropsychologia. 46:2836–2844. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Geuze E, Westenberg HG, Heinecke A, de
Kloet CS, Goebel R and Vermetten E: Thinner prefrontal cortex in
veterans with posttraumatic stress disorder. Neuroimage.
41:675–681. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bremner JD, Elzinga B, Schmahl C and
Vermetten E: Structural and functional plasticity of the human
brain in posttraumatic stress disorder. Prog Brain Res.
167:171–186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paxinos G and Watson C: The Rat Brain in
Stereotaxic Coordinates. 4th edition. Academic Press; San Diego,
CA: 1998
|
|
18
|
Liberzon I and Sripada CS: The functional
neuroanatomy of PTSD: a critical review. Prog Brain Res.
167:151–169. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao B, Han F and Shi YX: Dysfunction of
Ca2+/CaM kinase IIalpha cascades in the amygdala in
post-traumatic stress disorder. Int J Mol Med. 24:795–799.
2009.
|
|
20
|
Williams LM, Kemp AH, Felmingham K, Barton
M, Olivieri G, Peduto A, Gordon E and Bryant RA: Trauma modulates
amygdala and medial prefrontal responses to consciously attended
fear. Neuroimage. 29:347–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hull AM: Neuroimaging findings in
post-traumatic stress disorder. Systematic review. Br J Psychiatry.
181:102–110. 2002.PubMed/NCBI
|
|
22
|
Rauch SL, Shin LM and Phelps EA:
Neurocircuitry models of posttraumatic stress disorder and
extinction: human neuroimaging research-past, present, and future.
Biol Psychiatry. 60:376–382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corbo V, Clément MH, Armony JL, Pruessner
JC and Brunet A: Size versus shape differences: contrasting
voxel-based and volumetric analyses of the anterior cingulate
cortex in individuals with acute posttraumatic stress disorder.
Biol Psychiatry. 58:119–124. 2005.
|
|
24
|
Anderson MC and Green C: Suppressing
unwanted memories by executive control. Nature. 410:366–369. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cabeza R, Ciaramelli E, Olson IR and
Moscovitch M: The parietal cortex and episodic memory: an
attentional account. Nat Rev Neurosci. 9:613–625. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cardinal RN, Parkinson JA, Hall J and
Everitt BJ: Emotion and motivation: the role of the amygdala,
ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev.
26:321–352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Muigg P, Hetzenauer A, Hauer G, Hauschild
M, Gaburro S, Frank E, Landgraf R and Singewald N: Impaired
extinction of learned fear in rats selectively bred for high
anxiety-evidence of altered neuronal processing in
prefrontal-amygdala pathways. Eur J Neurosci. 28:2299–2309. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Milad MR, Pitman RK, Ellis CB, Gold AL,
Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP and Rauch SL:
Neurobiological basis of failure to recall extinction memory in
posttraumatic stress disorder. Biol Psychiatry. 66:1075–1082. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Akirav I and Maroun M: The role of the
medial prefrontal cortex-amygdala circuit in stress effects on the
extinction of fear. Neural Plast. 2007:308732007.PubMed/NCBI
|
|
30
|
Milad MR, Vidal-Gonzalez I and Quirk GJ:
Electrical stimulation of medial prefrontal cortex reduces
conditioned fear in a temporally specific manner. Behav Neurosci.
118:389–394. 2004. View Article : Google Scholar : PubMed/NCBI
|