Introduction
The presence of specific human gene single
nucleotide polymorphisms (SNP) has been associated with
susceptibility to several diseases (1). Evidence has demonstrated that
polymorphisms have substantial clinical effects on the prevalence,
incidence and prognosis of various major diseases, including asthma
in children which is associated with IL-13 (1,2) and
TNF-α-308A/G (3), Alzheimer’s
disease (4,5), human mental diseases (6,7),
chronic obstructive pulmonary disease (8,9) and
cardiovascular disease (10).
These diseases are often associated with the deoxyribonucleic acid
(DNA) sequences of the genes, in particular with the SNPs. A very
accurate prediction of diseases present may be obtained by
genotyping the relevant SNPs. Large-scale samples are required to
analyze and reveal the relationships between disease and SNPs.
At present, the following methods are typically
used: i) polymerase chain reaction-restriction fragment length
polymorphism (PCR-RFLP), a method which demands highly pure PCR
products, large quantities and restriction enzyme sites for the SNP
to be detected. Detection using this method would be very costly
and time consuming for large samples. ii) Sequencing, a method that
has high costs and is unsuitable for the quick analysis of many
clinical samples. iii) PCR using sequence-specific primers
(PCR-SSP), a method that is based on a critical PCR process with
detection of the amplified product by agarose gel electrophoresis.
The rapid and simple PCR amplification makes the technique suitable
for screening SNPs that are related to disease, treatments and drug
choice, particularly by large-sample polygenic genotyping. Thus, it
is a more favorable prospect for the clinical testing of gene
polymorphisms. However, multiple factors affect the PCR process.
During PCR-SSP, the sequence primers are required to be combined
and controlled with specific primers in the same reaction mixture
in the test tube to ensure that if amplification failure occurs, it
results from mismatched primers rather than from a failure of the
amplification system. Therefore, the reaction parameters have a
significant effect which may limit the applications of the method
in the detection of SNPs (11).
For this purpose, the parameters for the cycling and
operation systems were optimized to establish a protocol suitable
for the convenient, rapid and accurate polygenic genotyping of a
large number of samples.
Materials and methods
DNA
Genomic DNA from human peripheral blood donated by
researchers was prepared and used by our laboratory. The animal
genomic DNA was from healthy Kunming mice provided by The Animal
Experimental Center of Xinxiang Medical University. The DNA
extraction kit used was a universal genomic DNA extraction kit
SK1341 (Sangon Co. Ltd., Shanghai, China).
Primers
The PCR-SSP method optimized in our laboratory was
tested to analyze polymorphisms of the following genes: -308A/G
mutations in TNF-α, COMT 472 coding sequence G/A, the C/T mutation
at exon 188 of CYP2D6 *10B, mouse PPARγ and mouse
β-actin. The primers (synthesized by Sangon Co. Ltd., Shanghai,
China) used in the experiment are shown in Table I.
 | Table IPrimer sequences, amplification size
for the single locus PCR-SSP and sequencing. |
Table I
Primer sequences, amplification size
for the single locus PCR-SSP and sequencing.
| Gene name | Primer sequences (5′
to 3′) | Product size
(bp) |
|---|
| Human β-globin gene
(12) | F: GAA GAG CCA AGG
ACA GGT AC
R: CAA CTT CAT CCA CGT TCA CC | 268 |
| TNFα-308A/G (13) | F: TCC TCC CTG CTC
CGA TTC CG
Ra: CAA TAA GTT TTG AGG GGC ATG A
Rb: CAA TAA GTT TTG AGG GGC ATG G | 104 |
| CYP2D6
*10B exon 188 C/T (7) | F: ACC AGG CCC CTC
CAC CGG
Ra: AGG GGG CCT GGT GG
Ra: AGG GGG CCT GGT GA | 397 |
| COMT 472 coding
sequence G/A | F: ACT GTG GCT ACT
CAG CTG TG
Ra: TGC ACA CCT TGT CCT TCA C
Rb: TGC ACA CCT TGT CCT TCA T | 138 |
| Mouse β actin
(14) | F:
CCAGGGTGTGATGGTGGGAATG
R: CGCACGATTTCCCTCTCAGCTG | 510 |
| Mouse PPARγ (15) | F:
GACCACTCGCATTCCTTT
R: CCACAGACTCGGCACTCA | 265 |
Method optimization
The amplification system parameters, including the
concentrations of Mg2+, dNTPs, pfu Taq, primers and
control primers, were optimized using designed PCR-SSP reactions.
The optimized reaction system was used to determine the melting
temperature of the sample DNA and the cycling parameters.
The PCR products were examined on a 2% agarose gel
(Shanghai Sangon Biological Engineering Technology & Service
Co. Ltd., Shanghai, China). The amplified products were confirmed
under UV light following ethidium bromide staining to determine the
optimum parameters. The molecular standard used was
GeneRuler™ 100 bp DNA Ladder (Sangon Co. Ltd.). Pfu Taq
DNA (Promega Corporation, Madison, WI, USA) and PTC-100 PCR machine
(Biogene, Richardson, TX, USA) were used.
Results
Reaction system
The amplification was performed in a 20 μl reaction
volume and the effect of varying each of the parameters was studied
while the others were kept constant. Firstly, the volume of 25
mmol/l Mg2+ was varied from 1 to 5 μl. Other parameters
were DNA from 50 to 100 ng, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 0.5
mM of each dNTP, 0.7 μM of each primer (Table I) and 3 units Pfu Taq DNA
polymerase. As shown in Fig. 1a, 3
μl was the optimal amount of Mg2+. Secondly, after the
optimal Mg2+ concentration was determined, the effects
of different final dNTP concentrations on the amplification were
compared in the range 0.25–1.0 mM, added in increments of 0.25M.
Fig. 1b reveals that 0.5 mM dNTPs
was optimal. The optimal quantity of pfu Taq DNA polymerase was 2.5
units; it was evaluated in the range 1.0–3.5 units, added in
increments of 0.5 units. The optimal common primer concentration
was 0.5 μM; it was evaluated in the range 0.3–0.9 μM and added in
increments of 0.2 μM (Fig. 1c and
d). Finally, the ratio of the internal control primer
concentration to the specific primer concentration was optimized
after the other optimal concentrations had been determined. As
shown in Fig. 1e, the optimum
ratio was 2/5.
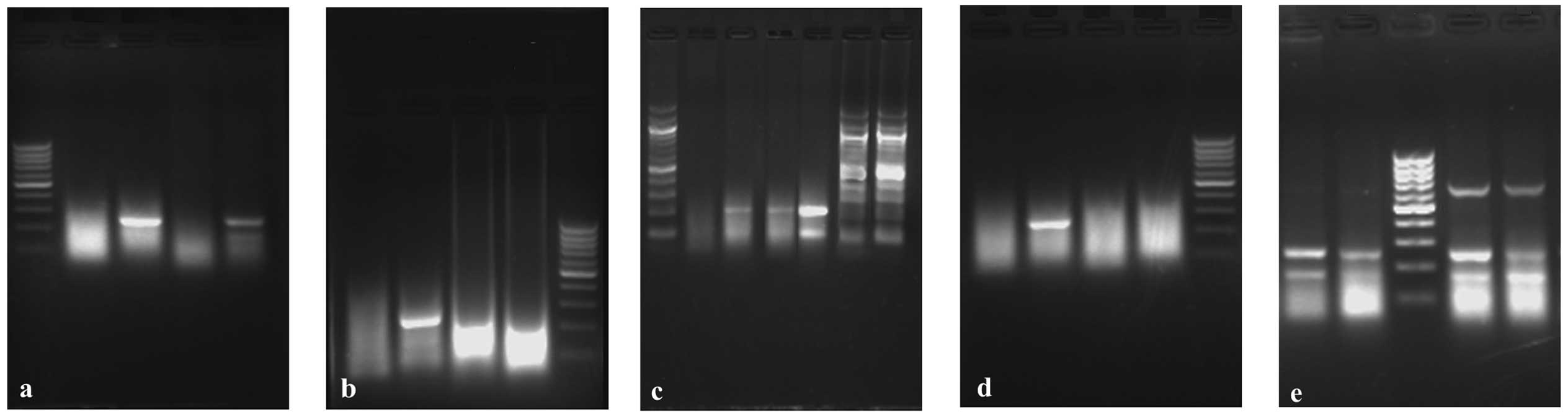 | Figure 1PCR-SSP system parameters
optimization. (a) Variation of Mg2+ concentration in
PCR. Lane 1: GeneRuler™ 100 bp DNA Ladder; lanes 2, 3, 4 and 5: 5,
3, 2 and 1 μl 25 mM Mg2+ in buffer, respectively. (b)
Effect of different dNTP concentrations on PCR. Lanes 1, 2, 3 and
4: 0.25, 0.5, 0.75 and 1.0 mmol/l dNTPs in the PCR mixture,
respectively; lane 5: GeneRuler 100 bp DNA Ladder. (c) Effect of
different concentrations of pfu Taq in PCR. Lane 1: GeneRuler 100
bp DNA Ladder; lanes 2, 3, 4, 5, 6 and 7: 1.0, 1.5, 2.0, 2.5, 3.0
and 3.5 units of pfu Taq DNA in PCR mixture, respectively. (d)
Different concentrations of common primers in the PCR system. Lanes
1, 2, 3 and 4: 0.3, 0.5, 0.7 and 0.9 μmol/l primers, respectively;
lane 5: GeneRuler 100 bp DNA Ladder. (e) Different ratios of
internal control primers to specific primers in the PCR system.
Lane 3: GeneRuler 100 bp DNA Ladder; lanes 1, 2, 4 and 5: the ratio
of internal control primers to common primers of 2/5, 4/5, 1 and
1.5. PCR-SSP, polymerase chain reaction using sequence-specific
primers; DNA, deoxyribonucleic acid. |
Cycling and validation
The primers, PCR mixture and electrophoresis
conditions were as previously described. The optimal thermocycler
conditions for the amplification were evaluated, including 95°C for
1, 3, 5, 10, 15 and 20 min, 95°C for 50 sec, 56°C for 50 sec and
72°C for 60 sec, followed by 72°C for 10 min. The denaturation
times were evaluated and revealed an optimal duration time of 15
min (Fig. 2a). For certain SNPs
that were difficult to amplify by conventional PCR, evaluation was
performed by touchdown and modified touchdown PCR (Figs. 2b and c).
 | Figure 2Optimized parameters of the PCR-SSP
cycling. (a) Different denaturation times in PCR cycling. Lane 4:
GeneRuler™ 100 bp DNA Ladder; lanes 1, 2, 3, 5, 6 and 7:
denaturation times of 1, 3, 5, 10, 15 and 20 min, respectively. (b)
Different touchdown programs used in PCR. Lanes 1 and 2:
conventional PCR; lane 3: GeneRuler 100 bp DNA Ladder; lane 4: a
common touchdown program; lane 5: optimized touchdown PCR. (c)
Different SNPs tested by the optimized touchdown program. PCR-SSP,
polymerase chain reaction using sequence-specific primers. |
The same primers were used in the optimized PCR: 20
μl reaction system contained 2 μl 1X PCR buffer, 3 μl 25 mmol/l
MgCl2, positive control primer at a final concentration
of 0.2 μmol/l, the common primer and primers a or b at final
concentrations of 0.5 μmol/l, dNTP at a final concentration of 0.5
mmol/l and 2.5 units pfu Taq DNA polymerase. The PCR programs used
the touchdown cycling method were: 95°C for 15 min, 95°C for 30
sec, 70°C for 1 min and 72°C for 10 min repeated for 5 cycles; 95°C
for 30 sec, 69.5°C for 1 min, then decreased by 0.5°C every cycle,
72°C for 1.5 min, repeated for 30 cycles; and 72°C for 10 min.
The modified PCR-SSP reaction system and touchdown
cycling were used to evaluate human SNPs and mouse genes.
TNF-α-308A/G, C/T at exon 188 of CYP2D6 *10B, COMT 472
G/A, mouse PPARγ and mouse β-actin were analyzed. Fig. 2c clearly shows the amplified
bands.
Discussion
To optimize the protocol for the simultaneous
polymorphism analysis of SNPs in large samples of multiple genes,
the reaction was performed in 20 μl liquid mixture. The
Mg2+ concentration is more important to the polymerase,
hence it was optimized first. For other parameters, conventional
concentrations were utilized for smooth optimization.
PCR-SSP is based on the principle that recombinant
Taq DNA polymerase is more specific for the oligonucleotide primers
that completely match the target gene. If a primer that completely
matches one genotype of the allele is designed and the PCR process
is strictly controlled, then the matching primer will be amplified
(positive results), whereas the mismatched primer will not
(negative results). The results of PCR-SSP are interpreted based on
whether an amplification band is visible, and the amplified
products of two lanes are used to determine the genotype of a
sample. Considering that numerous factors affect a PCR procedure,
an internal reference must be used for each reaction. The internal
reference generally used is the human globin gene, which is
universally present in the conserved region of the human genome. In
reactions showing positive bands, the amplification band of the
internal reference may be present, attenuated or absent depending
on the concentration of the specific primer pair and internal
reference primer pair (16–18).
Primer concentrations are very significant in
PCR-SSP reactions (11) to avoid
false negative results. The optimized common primer concentration
is 0.5 μM and the ratio of internal control primer to specific
primer is 2/5.
If the amplification reaction occurs smoothly, the
DNA template must be a single band. Human genes are extremely
complicated. Thus, the denaturation time was optimized. The results
indicated that 15 min is the optimum duration for human and mouse
genes. To avoid Taq polymerase inactivation, 2.5 units pfu Taq were
used, which is slightly higher than the usual concentration.
The proposed optimized method is similar to the
touchdown method and standard PCR protocols, with the exception of
a special cycling program in which a series of gradually decreasing
annealing temperatures was set. Furthermore, a variety of primers
were added to the reaction system so that different target gene
specific sequences were amplified satisfactorily. The basic
principle of this approach was to choose an annealing temperature
that was 15°C higher than the calculated annealing temperature.
This temperature was gradually decreased back to the annealing
temperature and then finally reduced further to 5°C lower than the
annealing temperature. Such a strategy ensures that the first
hybridization between the primer and the template occurs under the
most complementary conditions and it may be cycled at different
denaturing and annealing temperatures.
In conclusion, a PCR-SSP protocol modified to
optimize the diagnosis of diseases associated with multiple gene
polymorphisms has been developed. This PCR-SSP protocol is faster
and less expensive than the currently used procedures, particularly
in the primary screening of large samples and various point
mutations.
Acknowledgements
This study was supported by the Natural Science
Foundation of Henan Province, China [No: 12A310005]; National
Natural Science Foundation of China [No: 30970055]; and Xinxiang
Medical University Projects [No: ZD200936, ZD2011-17].
References
|
1
|
Knight JC: Functional implications of
genetic variation in non-coding DNA for disease susceptibility and
gene regulation. Clin Sci (London). 104:493–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He JQ, Chan-Yeung M, Becker AB, et al:
Genetic variants of the IL13 and IL4 genes and atopic diseases in
at-risk children. Genes Immun. 4:385–389. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Y, Zhang J, Tian C, et al: The -308
G/A polymorphism in TNF-α gene is associated with asthma risk: an
update by meta-analysis. J Clin Immunol. 31:174–185. 2011.
|
|
4
|
Faltraco F, Bürger K, Zill P, et al:
Interleukin-6–174 G/C promoter gene polymorphism C allele reduces
Alzheimer’s disease risk. J Am Geriatr Soc. 51:578–579. 2003.
|
|
5
|
Licastro F, Grimaldi LM, Bonafé M, et al:
Interleukin-6 gene alleles affect the risk of Alzheimer’s disease
and levels of the cytokine in blood and brain. Neurobiol Aging.
24:921–926. 2003.
|
|
6
|
Eley TC, Tahir E, Angleitner A, et al:
Association analysis of MAOA and COMT with neuroticism assessed by
peers. Am J Med Genet B Neuropsychiatr Genet. 120B:90–96. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roh HK, Kim CE, Chung WG, Park CS,
Svensson JO and Bertilsson L: Risperidone metabolism in relation to
CYP2D6*10 allele in Korean schizophrenic patients. Eur J Clin
Pharmacol. 57:671–675. 2001.
|
|
8
|
He JQ, Foreman MG, Shumansky K, et al:
Associations of IL6 polymorphisms with lung function decline and
COPD. Thorax. 64:698–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Córdoba-Lanús E, de-Torres JP,
López-Aguilar C, et al: Association of IL-6 gene polymorphisms and
COPD in a Spanish population. Resp Med. 102:1805–1811. 2008.
|
|
10
|
Elmas E, Bugert P, Popp T, et al: The
P-selectin gene polymorphism Val168Met: a novel risk marker for the
occurrence of primary ventricular fibrillation during acute
myocardial infarction. J Cardiovasc Electrophysiol. 21:1260–1265.
2010. View Article : Google Scholar
|
|
11
|
Fugger L, Morling N, Ryder LP, Odum N and
Svejgaard A: Technical aspects of typing for HLA-DP alleles using
allele-specific DNA in vitro amplification and sequence-specific
oligonucleotide probes. Detection of single base mismatches. J
Immunol Methods. 129:175–185. 1990. View Article : Google Scholar
|
|
12
|
Bon MA, van Oeveren-Dybicz A and van den
Bergh FA: Genotyping of HLA-B27 by real-time PCR without
hybridization probes. Clin Chem. 46:1000–1002. 2000.PubMed/NCBI
|
|
13
|
Yang R, Yang X, Qian C and Liu J: Rapid
detection of gene polymorphism of clinical pediatrics diseases by
PCR-SSP. J Appl Clin Pediatrics. 21:687–688. 2006.(In Chinese).
|
|
14
|
Schäffler A, Weigert J, Neumeier M,
Schölmerich J and Buechler C: Regulation and function of
collagenous repeat containing sequence of 26-kDa protein gene
product ‘cartonectin’. Obesity (Silver Spring). 15:303–313.
2007.
|
|
15
|
Grottkau BE, Purudappa PP and Lin YF:
Multilineage differentiation of dental pulp stem cells from green
fluorescent protein transgenic mice. Int J Oral Sci. 2:21–27. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aldener-Cannavá A and Olerup O: HLA-DOB1
“low-resolution” typing by PCR amplification with sequence-specific
primers (PCR-SSP). Eur J Immunogenet. 21:447–455. 1994.
|
|
17
|
Zetterquist H and Olerup O: Identification
of the HLA-DRB1*04, -DRB1*07, and -DRB1*09 alleles by PCR
amplification with sequence-specific primers (PCR-SSP) in 2 hours.
Hum Immunol. 34:64–74. 1992.
|
|
18
|
Olerup O and Zetterquist H: HLA-DR typing
by PCR amplification with sequence-specific primers (PCR-SSP) in 2
hours: an alternative to serological DR typing in clinical practice
including donor-recipient matching in cadaveric transplantation.
Tissue Antigens. 39:225–235. 1992. View Article : Google Scholar
|














