Introduction
Several epidemiological studies have found that
diabetic patients using metformin have a lower risk of cancer in
comparison to those using other anti-diabetic drugs. A case-control
study by Li et al (1)
reported that the risk of pancreatic cancer was 62% lower in
diabetic patients who had been treated with metformin than those
who had never received the drug. Other observational cohort studies
demonstrated a decrease of 25–37% in cancer cases in diabetic
patients treated with metformin (2,3). A
study by Zhou et al (4)
suggested that most of the beneficial effects of metformin are
mediated through its ability to activate the AMP-activated protein
kinase (AMPK). AMPK is a key sensor of the cellular AMP/ATP ratio.
AMPK is activated by an increase in this proportion as a
consequence of the partial inhibition of the mitochondrial
respiratory chain by metformin (5). Various biological effects have been
attributed to the activation of AMPK by metformin. It interferes
with the action of the mammalian target of rapamycin (mTOR) that
functions as part of the cellular signaling processes regulating
cell growth, cell proliferation, cell motility, transcription and
protein synthesis (6,7). Furthermore, the upstream regulator of
AMPK is a protein kinase identified as LKB1 (8,9)
which is a well-known tumor suppressor. It has been suggested that
LKB1 is a critical barrier to pulmonary tumorigenesis,
differentiation and metastasis (10). This fact further highlights the
possible role of AMPK activation in the anticancer effects of
metformin. Angiogenesis, an essential component of tumor
progression, is primarily achieved through the proliferation,
survival, and migration of endothelial cells (11). Angiogenesis is believed to begin
with matrix metalloproteinase (MMP)-mediated degradation of the
blood vessel basement membrane which contains various extracellular
matrix (ECM) proteins. Subsequently, it is followed by sequential
changes in vascular endothelial cells (12). MMP-2 and -9, predominately
expressed in the endothelial cells, are directly involved in
endothelial cell migration and vascular remodeling during
angiogenesis (13,14).
Tan et al (15) reported that metformin decreases
angiogenesis in women suffering from polycystic ovary syndrome
(PCOS) by increasing the anti-angiogenic thrombospondin-1. In
addition, metformin in a murine sponge model was found to inhibit
angiogenesis by decreasing vascularization, macrophage recruitment,
collagen deposition and levels of the transforming growth factor β1
(16). It can be proposed that
metformin controls and reduces the progression of cancer through
its anti-angiogenic effects. The effect of metformin on human
umbilical vein endothelial cells (HUVECs), an established model for
angiogenesis study, has not been elucidated to date. The present
study seeks to address whether metformin interferes with
endothelial cell functions in terms of proliferation, migration and
MMP expression. In addition, we also speculated whether these
effects are mediated by AMPK.
Materials and methods
Materials
HUVECs were purchased from the National Cell Bank,
Pasteur Institute of Iran. Metformin was provided by the Osveh
Pharmaceutical Laboratory (Tehran, Iran). Fetal bovine serum (FBS),
Dulbecco's modified Eagle's medium (DMEM), TRIzol, and trypsin/EDTA
0.25% were obtained from Invitrogen (USA). Compound C, DMSO and MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
were obtained from Sigma-Aldrich (USA). The Quantitect reverse
transcription kit and Quantifast probe PCR+Rox vial kit were
obtained from Qiagen (USA). The LDH cytotoxicity assay kit was
purchased from Roche (Germany). All the other reagents used in the
experiments were of analytical grade.
Cell culture
HUVECs were cultured in DMEM medium supplemented
with 10% FBS. The culture was carried out at 37˚C in 5%
CO2. After the cells reached a confluence of 80%, they
were detached using 0.25% trypsin-EDTA. Subsequently, the cells
were subcultured once again.
Endothelial cell cytotoxicity assay
The experimental procedure was conducted according
to the method of Linford and Dorsa (17) for measuring the cytotoxicity and
cell lysis by detecting lactate dehydrogenase (LDH) activity
released from the damaged cells. HUVECs were cultured in a 96-well
culture plate at a density of 1×104 cells/well in DMEM
medium. After 24 h, metformin at different concentrations, compound
C (10 μM), and compound C plus 3 mM metformin were added to the
wells and the cells were incubated for an additional 72 h. The
plates were centrifuged at 200 × g. Then, 100 μl of the LDH assay
mixture was added, and the plates were incubated at 37˚C for 30
min. The LDH release was estimated at 490 nm, using ELISA (Behring
ELISA Processor) and expressed as a percentage of the control. All
of the experiments were performed in triplicate.
Endothelial cell proliferation assay
This assay aimed to determine whether metformin
affects cell proliferation. HUVECs were seeded at a density of
1×104 cells/well in a 12-well culture plate and allowed
to attach for 24 h. Next, the cells were washed twice with PBS and
treated with different concentrations of metformin. HUVECs were
treated with 10 μM compound C for 30 min alone or before adding
metformin at the 3 mM concentration [in the present series of
experiments DMSO (0.8%) was used as a vehicle]. After a 72-h
incubation, the cells were washed with PBS and harvested using
trypsin-EDTA. The cell count and viability were determined by
trypan blue dye exclusion assay. All the experiments were performed
in triplicate.
MTT proliferation assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) proliferation assay is an index of cell
viability and proliferation. The cells were cultured in a 96-well
culture plate at a density of 3×103 cells/well for 24 h.
After being treated with different concentrations of metformin,
compound C (10 μM), and compound C plus 3 mM of metformin for a
further 72 h, the cells were washed twice with PBS and subjected to
the MTT assay. The cells were incubated with the MTT solution at a
final concentration of 0.5 mg/ml for 3 h. Subsequently, the cells
were lysed in DMSO. The optical density was measured at 540 nm
using an ELISA reader (Behring ELISA Processor). All of the samples
were assayed in triplicate, and the mean value for each experiment
was calculated. The obtained results are expressed as a percentage
of the control, which is considered to be 100%.
Endothelial cell migration assay
HUVECs were cultured in a 6-well culture plate. A
wound was made in the cell area using a sterile yellow tip when the
cells achieved 80–90% confluence. The variation in the wound width
within the experiments was approximately 5%. First, the cells were
washed with PBS. Then, the cells were treated with a medium
containing different concentrations of metformin and 2% FBS (2% FBS
allows cell survival but not cell proliferation). After a 72-h
incubation, the cells were washed twice with PBS, fixed with
methanol and stained with Giemsa. The cell migration into the
scratched area was photographed at a magnification of ×40.
Subsequently, the cell migration was quantified by calculating the
difference in the denuded area using a computerized planimetry
package (Landcalc, UK). The obtained data were expressed as a
percentage of the migration in the untreated endothelial cells.
RNA isolation and real-time quantitative
PCR
The total cellular RNA was extracted from the
cultured cells (~1×105) using TRIzol. The cells were
lysed in 1 ml TRIzol and incubated at room temperature for 5 min.
Then, 200 ml chloroform was added to the lysate, incubated for 3
min, and centrifuged for 15 min at 12,000 × g at 4˚C. The aqueous
layer was removed, mixed with an equal volume of isopropanol and
incubated for 1 h at 4˚C. The purified RNA was precipitated by
centrifugation at 12,000 × g for 15 min and was finally dissolved
in 50 μl diethylpyrocarbonate (DEPC)-treated water. One microgram
of the total-RNA was converted to cDNA using the Quantitect reverse
transcription kit (Qiagen). Real-time PCR was performed by the
Quantifast Probe PCR+Rox vial kit (Qiagen) using the ABI Step One
Plus Detection system (Applied Biosystems, USA). The cycling
conditions were 45 cycles in two steps. An initial denaturation
step at 95˚C for 3 min was followed by denaturation at 95˚C for 3
sec, and annealing-extension at 60˚C for 30 sec. For
quantification, the target gene was normalized to the internal
standard gene 18S. The primers were designed for detection of MMP-2
and -9 gene expression: MMP-2, forward, 5′-TTGATGGCATCGCTCAGATC-3′
and reverse, 5′-TTGTCACGTGGCGTCACAGT-3′; MMP-9, forward,
5′-GACGCAGACATCGTCATCCA-3′ and reverse,
5′-CACAACTCGTCATCGTCGAAA-3′; 18S rRNA, forward,
5′-CGGCTACCACATCCAAGGAA-3′ and reverse, 5′-GCT
GGAATTACCGCGGCT-3′.
Statistics
Data are presented as the mean ± SD. One-way ANOVA
was used to make comparisons between groups. A Student-Newman-Keuls
post test was performed to compare the mean values between the
treatment groups and the control in case the ANOVA analysis
indicated significant differences. Differences between the groups
were considered significant at P<0.05.
Results
Effects of metformin on LDH release from
endothelial cells
HUVECs were incubated with different concentrations
of the drugs for 72 h to determine whether metformin, compound C,
and DMSO (as a vehicle) are cytotoxic against endothelial cells.
Subsequently, the lactate dehydrogenase (LDH) release was measured.
The LDH activity of the control and treated groups is shown in
Table I. The LDH values among the
groups were almost identical with no significant differences.
 | Table IEffects of metformin, compound C, and
DMSO on lactate dehydrogenase (LDH) activity in HUVECs. |
Table I
Effects of metformin, compound C, and
DMSO on lactate dehydrogenase (LDH) activity in HUVECs.
| Groups (n=4) | LDH (%
control) |
|---|
| Control | 1 |
| Metformin (500
μM) | 0.6±0.5 |
| Metformin (1
mM) | 1.0±0.9 |
| Metformin (2
mM) | 1.4±0.8 |
| Metformin (3
mM) | 2.3±1.5 |
| Metformin (4
mM) | 3.8±1.6 |
| Metformin (5
mM) | 1.3±0.6 |
| Compound C (10
μM) | 0±0 |
| Metformin (3 mM) +
compound C (10 μM) | 2.9±2 |
| DMSO (0.8%;
vehicle) | 0±0 |
Effects of metformin on endothelial cell
proliferation
Incubation of the unstimulated human umbilical vein
endothelial cells with different concentrations of metformin
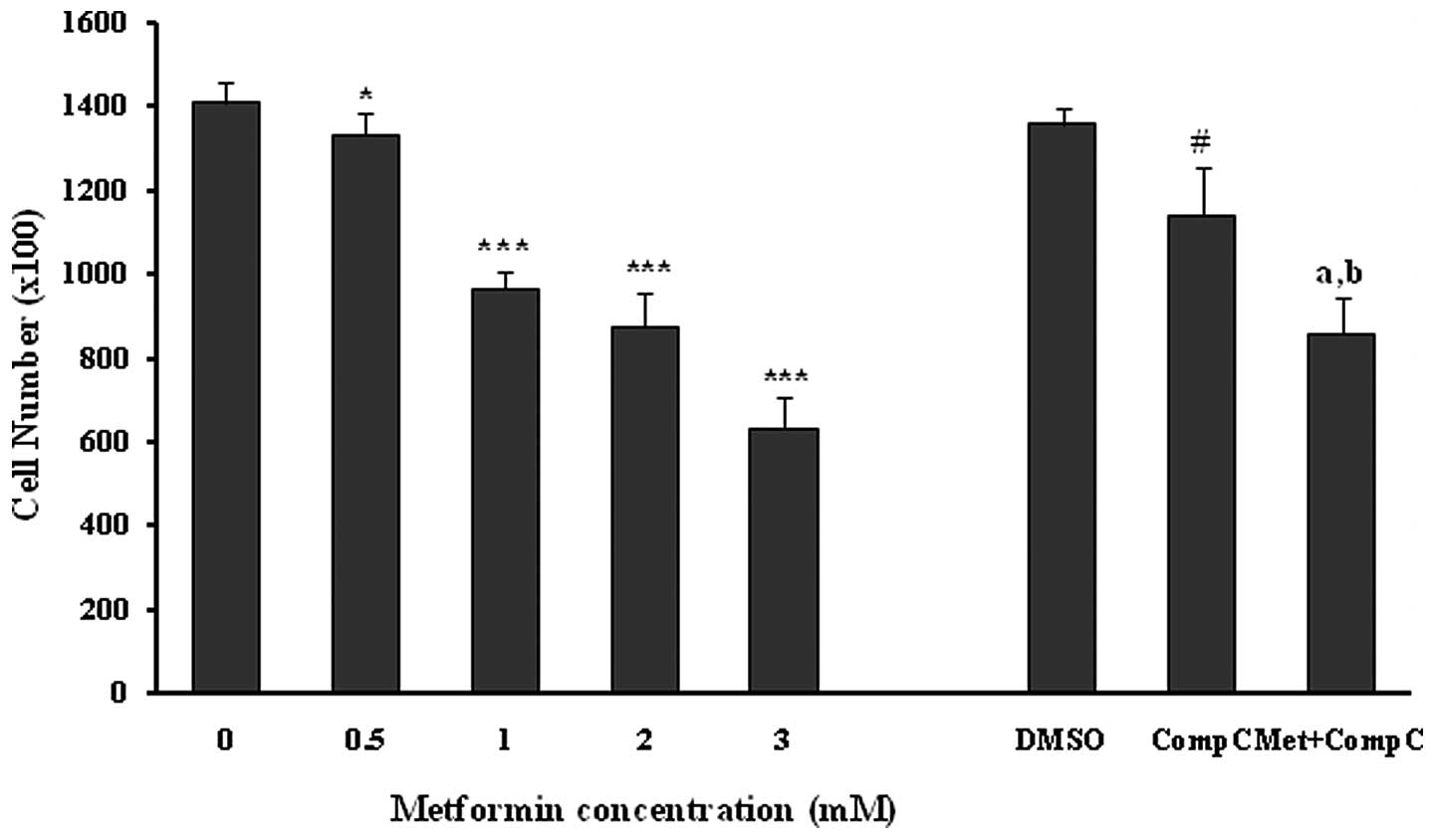
(0.5–3.0 mM) for 72 h induced a marked (P<0.05; P<0.001) and
dose-dependent reduction in the number of cells (Fig. 1). Compound C was used as a
pharmacological inhibitor of AMPK for evaluating the role of the
AMPK pathway in the metformin anti-proliferation effects of HUVECs.
Compound C, at a concentration of 10 μM, caused a 16% reduction
(P<0.001) in the cell proliferation by itself. Metformin at the
concentration of 3 mM produced a strong (P<0.001) inhibition of
HUVEC proliferation both in the presence (37% inhibition) and
absence (55% inhibition) of compound C in comparison to the related
controls. However, the anti-proliferation effect of metformin (3
mM) was significantly (18%; P<0.01), but not completely reversed
by compound C (Fig. 1). This
inhibition did not result from a cytotoxic effect, as assessed by
the LDH release from the control and the treated groups (Table I).
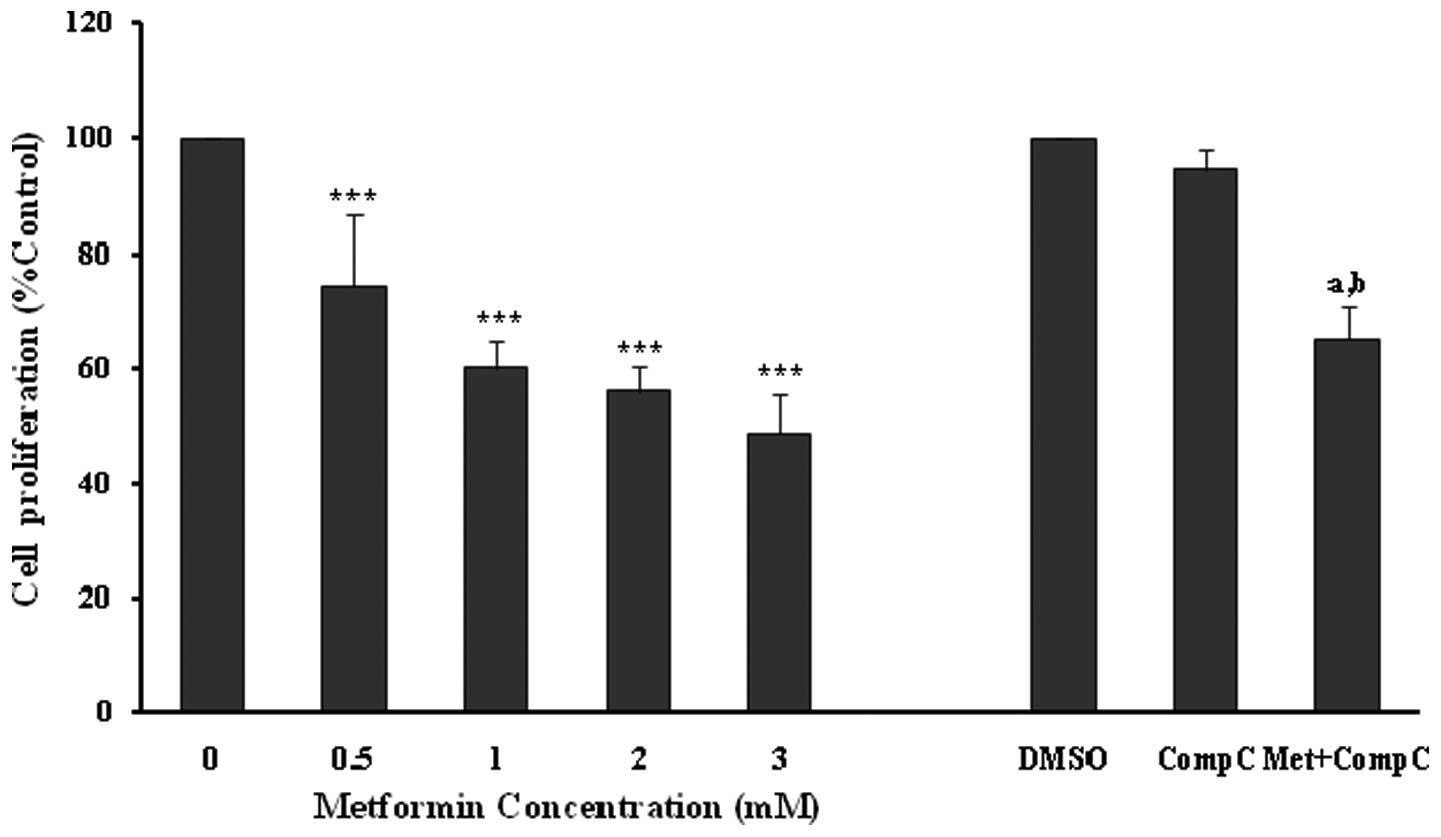
The anti-proliferative effects of metformin were
confirmed using an MTT proliferation assay (Fig. 2), with similar significant
(P<0.001) and concentration-dependent decreases noted in
endothelial cell proliferation. However, compound C did not affect
the cell viability in this set of experiments (Fig. 2). Metformin at 3 mM produced a
significant (P<0.001; 35%) inhibition of endothelial cell
proliferation in the presence of compound C. However, the
inhibitory effect was much lower in comparison to the cells treated
with metformin (3 mM) alone (P<0.001; 52%). The results of the
MTT assay also showed that compound C partially blocked the
anti-proliferative action of metformin (Fig. 2), and this was comparable with that
of the cell counting experiments (Fig.
1).
Effects of metformin on endothelial cell
migration
The ‘wound’ repair model of migration was used to
evaluate the effect of metformin on endothelial cell migration.
Confluent scrape-wounded HUVEC monolayers were incubated for 72 h
with metformin in the presence or absence of compound C.
Subsequently, the degree of closure of the ‘wound’ was assessed. It
was observed that metformin at concentrations of 0.5–3.0 mM induced
a strong and significant (P<0.001) concentration-dependent
inhibition of ‘wound’ repair in HUVECs from 31 to 80%, respectively
(Fig. 3). Compound C significantly
inhibited the migration (P<0.001), which was consistent with its
effect on endothelial cell numbers. However, in comparison to the
metformin alone-treated cells (3 mM), compound C partially, but
significantly (P<0.001) reversed the anti-migratory effect of
metformin.
Effects of metformin on MMP-2 and -9
expression in HUVECs
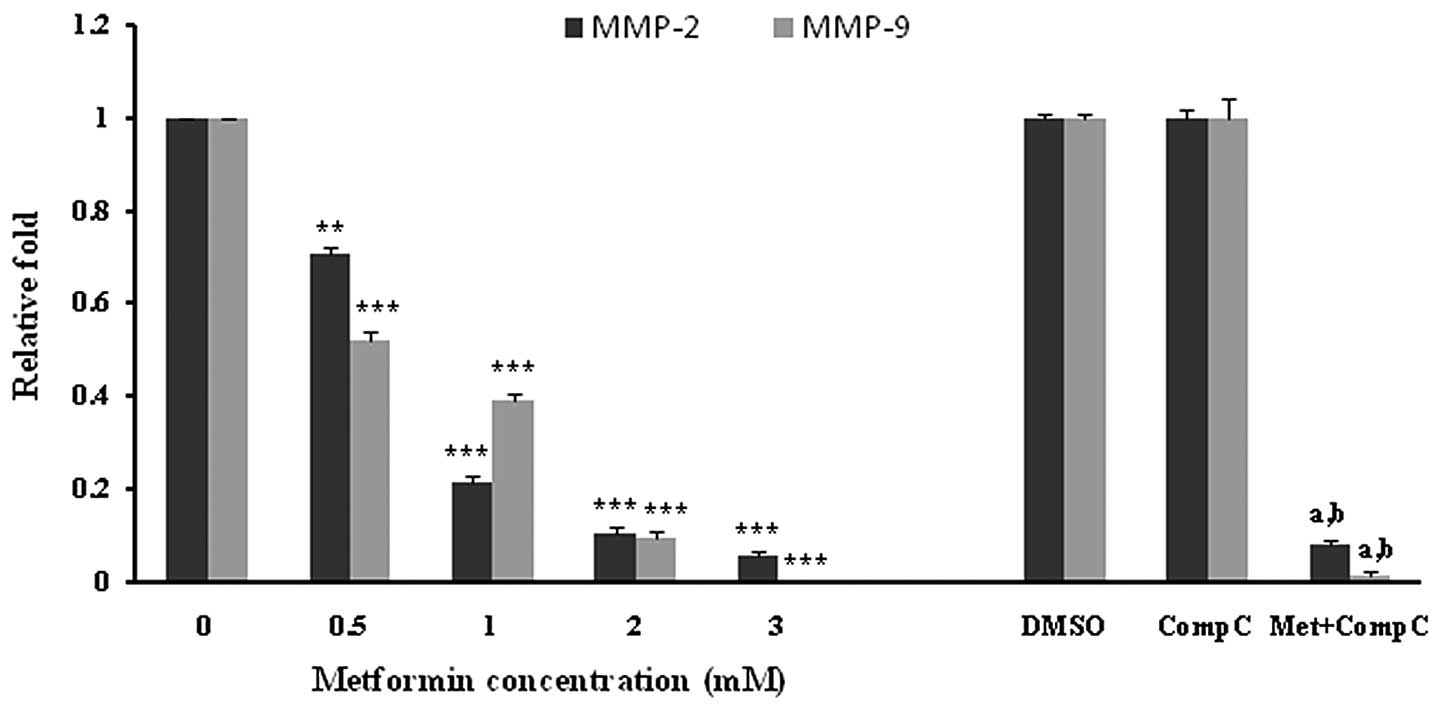
HUVECs were incubated with different concentrations
of metformin for 72 h. Subsequently, the mRNA expression of MMP-2
and -9 was examined. Metformin significantly (P<0.001) decreased
both the MMP-2 and -9 mRNA levels in a concentration-dependent
manner (Fig. 4). The most marked
decline in mRNA expression was noted with 3 mM of metformin. DMSO,
as a vehicle, or compound C, as an AMPK inhibitor, had no effect on
the mRNA expression of MMPs. However, it was observed that compound
C significantly (P<0.05), but not completely, reversed the
suppressive effect of metformin (3 mM) on the MMP-2 and -9 mRNA
expression (Fig. 4).
Discussion
Angiogenesis is an integral part of tumor growth and
metastasis that has gained increased interest as a core component
in cancer therapy. Several case-control and observational cohort
clinical trials have reported that systemic treatment with the
anti-diabetic drug metformin considerably decreased the risk of
different types of cancer in diabetic patients (1–3,18–20).
A recent study on mice exposed to tobacco carcinogenesis
demonstrated that metformin decreased the tumor burden by 72%,
which was correlated with decreased cellular proliferation and
marked inhibition of mTOR in tumors (21). However, it is necessary to clarify
whether metformin exerts the anticancer effect, at least in part,
through an anti-angiogenic effect. The present study found that
metformin produced a potent anti-proliferative and anti-angiogenic
effect in vitro on HUVECs, and that this effect was
associated with reduced mRNA expression of MMP-2 and -9.
It was clear from the experiments that metformin
produced a potent (P<0.001) and concentration-dependent
inhibitory effect on HUVEC proliferation. A concentration of 3 mM
of metformin reduced the endothelial cell numbers in culture by at
least 55%. The effect was also confirmed by an MTT proliferation
assay. The anti-proliferative effect of metformin was not due to
cytotoxicity due to the fact that treatment of endothelial cells
with different concentrations of metformin for extended periods of
time neither affected the integrity of the cell monolayer nor was
it associated with increased LDH release, an indicator of
cytotoxicity. Furthermore, the obtained data showed that the
migration of the cells into the denuded area 72 h after the
cultures were treated with metformin was significantly (P<0.001)
lower in comparison to the untreated control. To the best of our
knowledge, not many studies have dealt with the effects of
metformin on angiogenesis, in particular the effects of metformin
on the endothelial cell proliferation and migration. A study
conducted on PCOS women treated with metformin reported that
metformin decreased angiogenesis by increasing the serum
anti-angiogenic thrombospondin-1 (15). Also, a recent in vivo study
in a murine sponge model demonstrated that metformin inhibited
inflammatory angiogenesis by decreasing the levels of transforming
growth factor-β1 (TGF-β1) (16).
In addition, our findings revealed that the MMP-2 and -9 expression
in unstimulated HUVECs was markedly downregulated, following the
metformin treatment of endothelial cells in a
concentration-dependent manner. Inhibitory action of metformin, as
a pharmacological activator of AMPK, on the MMP expression has been
described in human fibrosarcoma cells (22). However, no study has evaluated the
effect of the drug on MMP expression in vascular endothelial cells.
MMPs are involved in many endothelial cell processes, such as cell
migration and angiogenesis, as well as in tumor invasion or
metastasis. These enzymes play an important role in physiological
tissue remodeling, and also in pathological remodeling associated
with conditions such as wound healing and tumor growth. In
particular, MMP-2 and -9, the two predominately expressed MMPs in
endothelial cells, have been directly implicated in the process of
endothelial cell migration. This is accomplished through
proteolysis of the components of the extracellular matrix (13,14).
Metformin has been used as an anti-diabetic drug
since 1957 (23). The drug reduces
blood sugar levels mainly through three mechanisms: decreased
hepatic glucose production (24,25),
increased skeletal myocyte glucose uptake (26,27),
and reduction of hepatic lipids (28). AMP-activated protein kinase (AMPK)
provides a candidate target, which is capable of mediating the
beneficial metabolic effects of metformin (4). AMPK is an important intracellular
energy sensor, which activates the catabolic pathways that generate
ATP. In addition, AMPK also inactivates ATP-consuming anabolic
pathways when the cellular AMP/ATP ratio is increased (29). Proliferation and migration are
ATP-consuming processes. Thus, AMPK activity may be required for
optimal cell proliferation and survival, in particular under stress
conditions. It was observed that tumor xenografts prepared from
Ras-transformed mouse embryo fibroblasts lacking AMPK lose their
ability to grow in a hypoxic environment (30), and 5-amino-4-imidazole carboxamide
riboside (AICAR), an AMPK agonist, increases the angiogenesis of
endothelial progenitor cells by phosphorylation of acetyl-coenzyme
A carboxylase (ACC) and eNOS (31). Furthermore, it has been reported
that the activation of AMPK signaling in endothelial cells is
essential for angiogenesis under hypoxic conditions (32), but not in normoxia. On the
contrary, a growing body of evidence indicates that AMPK activation
inhibits the growth and/or survival of various cancer cell lines
(33–38). It is now obvious that AMPK is
regulated by a well-recognized tumor suppressor known as LKB1
(39), and that activation of AMPK
by metformin requires LKB1. Furthermore, AMPK activation by
metformin inhibits the mammalian target of rapamycin (mTOR), a
protein that plays a critical role in transcription, cell growth,
proliferation and migration (6,7).
It was observed that compound C, a cell-permeable
pyrazolopyrimidine derivative, acts as a potent and selective
ATP-competitive inhibitor of AMPK (4). In the present study, we demonstrated
that the anti-proliferative and anti-migratory effects of metformin
on endothelial cells as well as the inhibitory effect of metformin
on mRNA expression of MMP-1 and MMP-2 were significantly but not
completely blocked by compound C. This indicates that the AMPK
pathway is involved, at least in part, in the anti-angiogenic
action of metformin. Surprisingly, compound C alone showed a slight
but significant anti-proliferative and anti-migratory action. These
paradoxical effects in the present study probably imply the
involvement of AMPK-dependent and AMPK-independent mechanisms in
metformin anti-angiogenic actions. The other possibility is that
both the activation and inhibition of AMPK cause anti-proliferative
effects through different downstream pathways. Regarding the
energy-saving and ATP-producing roles of AMPK through enhancing
fatty acid oxidation, glycolysis and glucose uptake (30,40),
especially in ATP deprivation conditions, it is conceivable that
AMPK inhibition by compound C to some extent leads to the
inhibitory effects on HUVEC proliferation and migration. However,
AMPK activation has a wider role in reducing circulating levels of
insulin-like growth factor and inhibition of cell differentiation,
proliferation, and growth through the suppression of mTOR (41), elongation factor-2 (42), and the cyclin (43) pathways. In this study, comparison
of the strong inhibitory effects of metformin, as an AMPK
activator, with the weak suppressive effects of compound C, as an
AMPK antagonist, on the proliferation and migration of human
umbilical vein endothelial cells indicates the potentially
beneficial effects of AMPK activation in preventing angiogenesis
and related diseases.
Many factors have been identified as stimulators of
the MMP expression in endothelial cells (44) but little is known about the
inhibitors of these elements in normal cells under physiological
conditions. Using an AMPKα-knockout mouse, Morizane et al
(45) reported that total AMPKα
deletion significantly elevated MMP-9 expression in embryonic
fibroblast cells. The authors also demonstrated that AMPK
activation by AICAR or by A769662 in wild-type fibroblasts
suppressed MMP-9 expression. Thus, it was concluded that both the
activity and the presence of AMPKα contribute as a regulator of
MMP-9 expression. Similarly, the present study demonstrated that
the expression of MMP-2 and -9 mRNA was decreased in HUVECs
incubated with metformin and this decrease was reversed partially
by compound C as an inhibitor of AMPK.
Collectively, the results of this study suggest that
metformin may have potential effects in arresting the progression
of tumors by inhibiting endothelial cell proliferation and
migration through the suppression of MMP-2 and -9 mRNA expression.
In addition, AMPK activity, at least in part, is required for the
above-mentioned effects. In conclusion, the results may clarify the
beneficial effect of metformin in reducing cancer incidence in
diabetic patients receiving the drug.
Acknowledgements
The present study was supported by grants from the
Research Vice Chancellors of the Tabriz University of Medical
Sciences, Tabriz, Iran and from the Research Vice Chancellors of
the Tehran University of Medical Sciences, Tehran, Iran.
References
|
1
|
D LiSC YeungMM HassanM KonoplevaJL
AbbruzzeseAntidiabetic therapies affect risk of pancreatic
cancerGastroenterology137482488200910.1053/j.gastro.2009.04.01319375425
|
|
2
|
JM EvansLA DonnellyAM Emslie-SmithDR
AlessiAD MorrisMetformin and reduced risk of cancer in diabetic
patientsBMJ33013041305200510.1136/bmj.38415.708634.F715849206
|
|
3
|
G LibbyLA DonnellyPT DonnanDR AlessiAD
MorrisJM EvansNew users of metformin are at low risk of incident
cancer: a cohort study among people with type 2 diabetesDiabetes
Care3216201625200910.2337/dc08-217519564453
|
|
4
|
G ZhouR MyersY LiY ChenX ShenJ
Fenyk-MelodyM WuJ VentreT DoebberN FujiiRole of AMP-activated
protein kinase in mechanism of metformin actionJ Clin
Invest10811671174200110.1172/JCI1350511602624
|
|
5
|
MR OwenE DoranAP HalestrapEvidence that
metformin exerts its anti-diabetic effects through inhibition of
complex 1 of the mitochondrial respiratory chainBiochem
J15348200010839993
|
|
6
|
N HayN SonenbergUpstream and downstream of
mTORGenes Dev1819261945200410.1101/gad.1212704
|
|
7
|
CS BeeversF LiL LiuS HuangCurcumin
inhibits the mammalian target of rapamycin-mediated signaling
pathways in cancer cellsInt J
Cancer119757764200610.1002/ijc.2193216550606
|
|
8
|
SA HawleyJ BoudeauJL ReidKJ MustardL UddTP
MakelaDR AlessiDG HardieComplexes between the LKB1 tumor
suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream
kinases in the AMP-activated protein kinase cascadeJ
Biol228200310.1186/1475-4924-2-2814511394
|
|
9
|
JM LizcanoO GoranssonR TothM DeakNA
MorriceJ BoudeauSA HawleyL UddTP MakelaDG HardieLKB1 is a master
kinase that activates 13 kinases of the AMPK subfamily, including
MARK/PAR-1EMBO J23833843200410.1038/sj.emboj.760011014976552
|
|
10
|
H JiMR RamseyDN HayesLKB1 modulates lung
cancer differentiation and
metastasisNature448807810200710.1038/nature0603017676035
|
|
11
|
J FolkmanPA D'AmoreBlood vessel formation:
what is its molecular
basis?Cell8711531155199610.1016/S0092-8674(00)81810-38980221
|
|
12
|
R KalluriBasement membranes: structure,
assembly and role in tumour angiogenesisNat Rev
Cancer3422433200310.1038/nrc109412778132
|
|
13
|
TA HaasEndothelial cell regulation of
matrix metalloproteinasesCan J Physiol
Pharmacol8317200510.1139/y04-120
|
|
14
|
JP SluijterDP de KleijinG
PasterkampVascular remodeling and protease inhibition: bench to
bedsideCardiovas
Res69595603200610.1016/j.cardiores.2005.11.02616387286
|
|
15
|
BK TanR AdyaJ ChenS FarhatullahD HeutlingD
MitchellH LehnertHS RandevaMetformin decreases angiogenesis via
NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic
thrombospondin-1Cardiovasc
Res83566574200910.1093/cvr/cvp13119414528
|
|
16
|
DO XavierLS AmaralMA GomesMetformin
inhibits inflammatory angiogenesis in a murine sponge modelBiomed
Pharmacother64220225201010.1016/j.biopha.2009.08.00420053525
|
|
17
|
NJ LinfordDM Dorsa17beta-estradiol and the
phytoestrogen genistein attenuate neuronal apoptosis induced by the
endoplasmic reticulum calcium-ATPase inhibitor
thapsigarginSteroids6710291040200210.1016/S0039-128X(02)00062-4
|
|
18
|
SL BowkerSR MajumdarP VeugelersJA
JohnsonIncreased cancer-related mortality for patients with type 2
diabetes who use sulfonylureas or insulinDiabetes
Care29254258200610.2337/diacare.29.02.06.dc05-1558
|
|
19
|
CJ CurrieCD PooleEA GaleThe influence of
glucose-lowering therapies on cancer risk in type 2
diabetesDiabetologia5217661777200910.1007/s00125-009-1440-619572116
|
|
20
|
JL WrightJL StanfordMetformin use and
prostate cancer in Caucasian men: results from a population-based
case-control studyCancer Causes
Control2016171622200910.1007/s10552-009-9407-y19653109
|
|
21
|
RM MemmottJR MercadoCR MaierS KawabataSD
FoxPA DennisMetformin prevents tobacco carcinogen - induced lung
tumorigenesisCancer Prev Res
(Phila)310661076201010.1158/1940-6207.CAPR-10-005520810672
|
|
22
|
YP HwangHG JeongMetformin blocks migration
and invasion of tumour cells by inhibition of matrix
metalloproteinase-9 activation through a calcium and protein kinase
Ca-dependent pathway:
phorbol-12-myristate-13-acetate-induced/extracellular
signal-regulated kinase/activator protein-1Br J
Pharmacol160119512112010
|
|
23
|
G SchaferBiguanides. A review of history,
pharmacodynamics and therapyDiabete Metab914816319836352352
|
|
24
|
M StumvollN NurjhanG PerrielloG DaileyJE
GerichMetabolic effects of metformin in non-insulin-dependent
diabetes mellitusN Engl J
Med333550554199510.1056/NEJM1995083133309037623903
|
|
25
|
RS HundalM KrssakS DufourMechanism by
which metformin reduces glucose production in type 2
diabetesDiabetes4920632069200010.2337/diabetes.49.12.206311118008
|
|
26
|
HS HundalT RamlalR ReyesLA LeiterA
KlipCellular mechanism of metformin action involves glucose
transporter translocation from an intracellular pool to the plasma
membrane in L6 muscle cellsEndocrinology131116511731992
|
|
27
|
D GaluskaLA NolteJR ZierathH
Wallberg-HenrikssonEffect of metformin on insulin-stimulated
glucose transport in isolated skeletal muscle obtained from
patients with
NIDDMDiabetologia37826832199410.1007/BF004043407988785
|
|
28
|
HZ LinSQ YangC ChuckareeF KuhajdaG
RonnetAM DiehlMetformin reverses fatty liver disease in obese,
leptin-deficient miceNat Med69981003200010.1038/7969710973319
|
|
29
|
DG HardieSA HawleyJW ScottAMP-activated
protein kinase - development of the energy sensor conceptJ
Physiol574715200610.1113/jphysiol.2006.10894416644800
|
|
30
|
KR LaderouteK AminJM CalaoaganM KnappT LeJ
OrdunaM ForetzB Viollet5′-AMP-activated protein kinase (AMPK) is
induced by low-oxygen and glucose deprivation conditions found in
solid-tumor microenvironmentsMol Cell Biol26533653472006
|
|
31
|
X LiY HanW PangC LiX XieJY ShyyY
ZhuAMP-activated protein kinase promotes the differentiation of
endothelial progenitor cellsArterioscler Thromb Vasc
Biol2817891795200810.1161/ATVBAHA.108.17245218599796
|
|
32
|
D NagataM MogiK WalshAMP-activated protein
kinase (AMPK) signaling in endothelial cells is essential for
angiogenesis in response to hypoxic stressJ Biol
Chem2783100031006200310.1074/jbc.M30064320012788940
|
|
33
|
BA KefasY CaiK KerckhofsZ LingG MartensH
HeimbergD PipeleersM Van de CasteeleMetformin-induced stimulation
of AMP-activated protein kinase in beta-cells impairs their glucose
responsiveness and can lead to apoptosisBiochem
Pharmacol68409416200410.1016/j.bcp.2004.04.00315242807
|
|
34
|
M SaitohK NagaiK NakagawaT YamamuraS
YamamotoT NishizakiAdenosine induces apoptosis in the human gastric
cancer cells via an intrinsic pathway relevant to activation of
AMP-activated protein kinaseBiochem
Pharmacol6720052011200410.1016/j.bcp.2004.01.020
|
|
35
|
R RattanS GiriAK SinghI
Singh5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside
inhibits cancer cell proliferation in vitro and in vivo via
AMP-activated protein kinaseJ Biol
Chem2803958239593200510.1074/jbc.M50744320016176927
|
|
36
|
A IsakovicL HarhajiD StevanovicZ MarkovicM
Sumarac-DumanovicV StarcevicD MicicV TrajkovicDual antiglioma
action of metformin: cell cycle arrest and mitochondria-dependent
apoptosisCell Mol Life
Sci6412901302200710.1007/s00018-007-7080-417447005
|
|
37
|
W ZhouWF HanLE LandreeFatty acid synthase
inhibition activates AMP-activated protein kinase in SKOV3 human
ovarian cancer cellsCancer
Res6729642971200710.1158/0008-5472.CAN-06-343917409402
|
|
38
|
R OkoshiT OzakiH YamamotoK AndoN KoidaS
OnoT KodaT KamijoA NakagawaraH KizakiActivation of AMP-activated
protein kinase induces p53-dependent apoptotic cell death in
response to energetic stressJ Biol
Chem28339793987200810.1074/jbc.M70523220018056705
|
|
39
|
DR AlessiK SakamotoJR
BayascasLKB1-dependent signaling pathwaysAnnu Rev
Biochem75137163200610.1146/annurev.biochem.75.103004.14270216756488
|
|
40
|
MM ShawWK GurrRJ McCrimmonDF SchorderetRS
Sherwin5'AMP-activated protein kinase alpha deficiency enhances
stress-induced apoptosis in BHK and PC12 cellsJ Cell Mol
Med112862982007
|
|
41
|
MB AntonoffJ D'CunhaTeaching an old drug
new tricks: metformin as a targeted therapy for lung cancerSemin
Thorac Cardiovasc
Surg22195196201010.1053/j.semtcvs.2010.10.01521167452
|
|
42
|
LQ Hong-BrownCR BrownDS HuberCH
LangLopinavir impairs protein synthesis and induces eEF2
phosphorylation via the activation of AMP-activated protein kinaseJ
Cell Biochem105814823200810.1002/jcb.2188218712774
|
|
43
|
JE KimHC ChoiLosartan inhibits vascular
smooth muscle cell proliferation through activation of
AMP-activated protein kinaseKorean J Physiol
Pharmacol14299304201010.4196/kjpp.2010.14.5.29921165328
|
|
44
|
R HanemaaijerP KoolwijkL le ClercqWJ de
VreeVW van HinsberghRegulation of matrix metalloproteinase
expression in human vein and microvascular endothelial cells.
Effects of tumour necrosis factor alpha, interleukin 1 and phorbol
esterBiochem J158038091993
|
|
45
|
Y MorizaneA ThanosK TakeuchiAMP-activated
protein kinase suppresses matix metalloproteinase-9 expression in
mouse embryonic fibroblastsJ Biol
Chem2861603016038201110.1074/jbc.M110.19939821402702
|