Introduction
The expression of soluble adenylyl cyclase (sAC) was
recently documented in insulin-secreting INS-1E cells by reverse
transcription polymerase chain reaction, western blot analysis and
immunocytochemistry (1). It was
also hypothesized that an increase in the extracellular D-glucose
concentration from 2.5 to 16.0 mM in INS-1E cells induces a delayed
increase in levels of the secondary messenger, adenosine
3′,5′-cyclic monophosphate (cAMP), mediated by bicarbonate, calcium
and ATP-sensitive sAC, with subsequent phosphorylation of
mitogen-activated protein (MAP) kinase. This hypothesis is not
consistent with that formulated over 3 decades ago, stating that
the glucose-induced increase in the cAMP content of
insulin-producing cells is accounted for by the activation of
membrane-associated adenylate cyclase by calcium-calmodulin
(2).
Bicarbonate regulation of sAC was previously
revealed to be physiologically relevant in sperm (3,4),
bronchii (5) and the epididymis
(6). In light of these
observations, the possibility that a comparable mechanism exists in
insulin-producing cells should be explored. Firstly, the knowledge
that the omission of extracellular NaHCO3 inhibits
glucose-stimulated insulin release (7–9) was
recently extended to the secretory response of rat isolated
pancreatic islets to non-nutrient secretagogues (10). Secondly, the major fraction of
CO2 generated by the oxidative catabolism of D-glucose
in rat pancreatic islets is converted to
HCO3− by the intervention of a mitochondrial
type V carbonic anhydrase (11).
Thirdly, it has been identified that the efflux of
HCO3− from pancreatic islet cells may be
mediated through volume-regulated anion channels (VRACs) gated in
response to an increase in cell volume, itself provoked by a rise
in D-glucose concentration or extracellular hypotonicity, at least
following the initial ‘phosphate flush’ also attributable to the
gating of VRAC (12). Finally, it
was documented that insulin-producing cells express the
electrogenic sodium bicarbonate
(Na+-HCO3− ) cotransporter, NBCe1
(13). Inhibition of this
cotransporter by
5-chloro-2-hydroxy-3-[thiophene-2-carbonyl]indole-1-carboxamide
(tenidap) has been reported to result in alterations of
22Na+ net uptake and insulin secretion in rat
pancreatic islets (13,14). Therefore, NBCe1 may also play a
role in the regulation of HCO3− fluxes in
islet cells.
With this information in mind, the major aim of the
present study, conducted in rat isolated pancreatic islets and
tumoral insulin-producing cells of the BRIN-BD11 line (15), was to explore the effect of agents
targeting carbonic anhydrase activity, VRAC, NBCe1 or MAP kinase
activity, as well as the effect of changes in
HCO3−, Cl− and Na+
extracellular concentration, upon the insulin secretory response to
nutrient secretagogues (D-glucose, 2-ketoisocaproate [KIC]) or
extracellular hypoosmolarity, as measured in the absence or
presence of cAMP analogs and the phosphodiesterase inhibitor,
3-isobutyl-1-methylxanthine (IBMX).
Materials and methods
Drugs
Tenidap was a gift from Pfizer (Groton, CT, USA).
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA)
with the exception of IBMX,
1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene
ethanolate (U0126),
2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98,059;
Calbiochem, Merck Biosciences, Darmstadt, Germany), cAMP
acetoxymethyl ester (cAMP-AM), adenosine-3′,5′-cyclic
monophosphorothioate (Sp-isomer) acetoxymethyl ester (Sp-cAMPS-AM),
Rp-isomer of 8-bromo-cAMPS (Rp-8-Br-cAMPS), Sp-8-Br-cAMPS (Biolog,
Bremen, Germany) and buffer compounds (Merck, Darmstadt, Germany).
Tissue culture materials were purchased from Starstedt (Nümbrecht,
Germany) and culture media from Invitrogen Life Technologies
(Carlstadt, Germany).
Insulin secretion
The methods used to measure insulin secretion from
the rat isolated pancreatic islets (16) or BRIN-BD11 cells (17) were performed as described
previously.
Statistical analysis
All results are presented as mean ± SEM with the
number of individual observations (n) or degrees of freedom (df).
The statistical significance of differences between mean values was
assessed using the Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Experiments in rat isolated pancreatic
islets
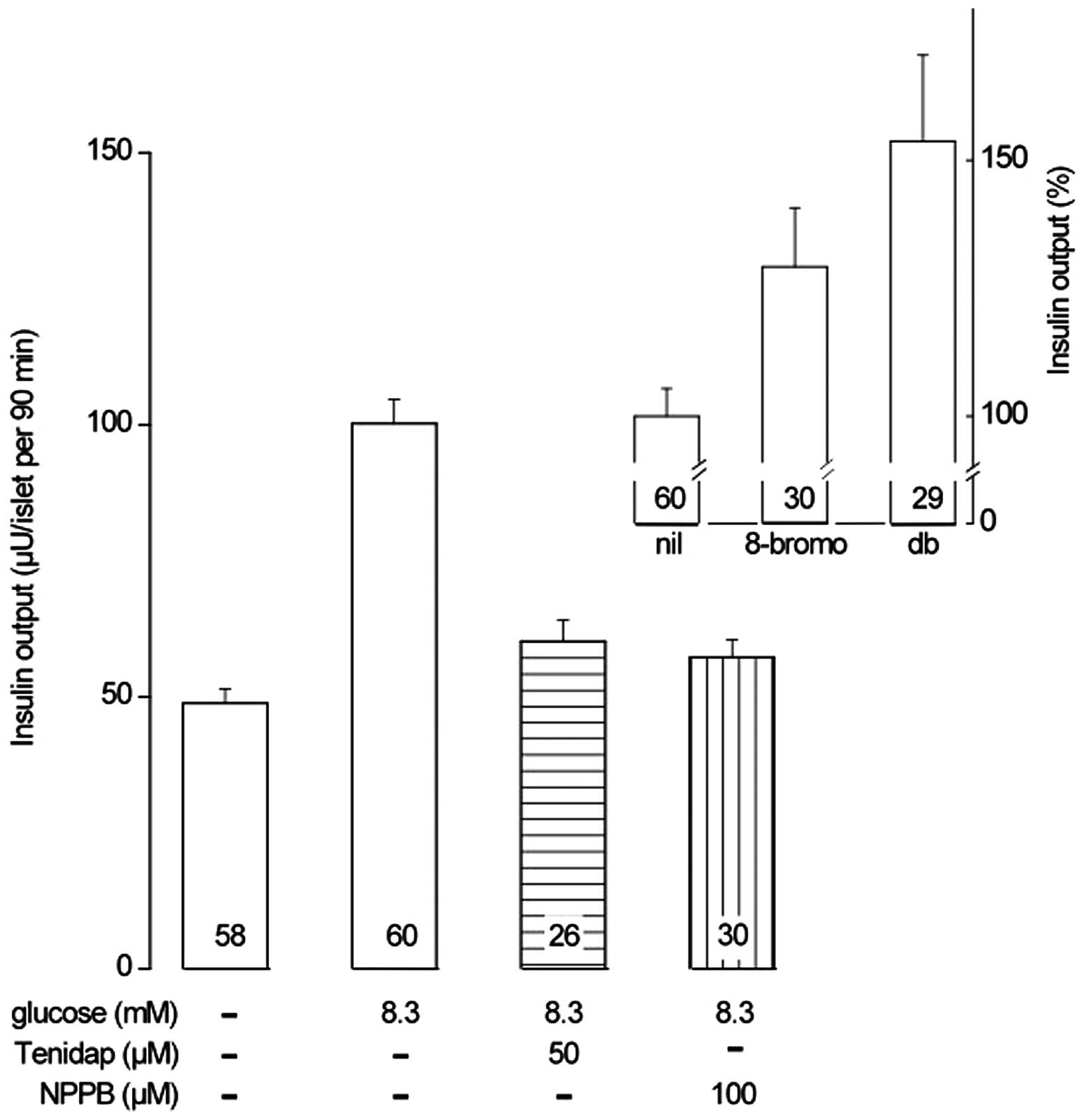
When incubated for 90 min, the insulin output of
islets exposed to 8.3 mM D-glucose averaged 100.2±4.4 μU/islet
(n=60), as compared with a basal value of 48.8±2.7 μU/islet (n=58)
in the absence of hexose (P<0.001; Fig. 1). Glucose-stimulated insulin
release was significantly increased by 8-Br-cAMP and dibutyryl-cAMP
(db-cAMP; 1.0 mM each; P<0.04; Table I). The relative magnitude of the
increase was not identified to be significantly different
(P>0.22) between these cAMP analogs, with an overall mean value
of 20.5±5.7% (df=115; P<0.001). As demonstrated in Fig. 1 (inset), the relative magnitude of
the enhancing action of the cAMP analogs upon the secretory
response to D-glucose was ~2-fold higher when corrected for basal
values, with an overall mean enhancing action of 41.1±11.4%
(df=115; P<0.001).
 | Table IRelative increment (%) of insulin
release from islets incubated with D-glucose as provoked by cAMP
analogs in the absence or presence of tenidap or NPPB. |
Table I
Relative increment (%) of insulin
release from islets incubated with D-glucose as provoked by cAMP
analogs in the absence or presence of tenidap or NPPB.
| cAMP analog | Inhibitor | Increment |
|---|
| 8-Bromo-cAMP | - | +13.76±6.53a (df=59) |
| db-cAMP | - | +27.64±9.46c (df=56) |
| Both | - | +20.52±5.71e (df=115) |
| 8-Bromo-cAMP | Tenidap | +28.49±8.39d (df=23) |
| db-cAMP | Tenidap | +14.23±12.26
(df=26) |
| Both | Tenidap | +20.94±7.59b (df=49) |
| 8-Bromo-cAMP | NPPB | −7.32±5.63
(df=29) |
| db-cAMP | NPPB | +14.46±12.99
(df=26) |
| Both | NPPB | +3.00±6.92
(df=55) |
Tenidap (50 μM) reduced the glucose-stimulated
insulin release by 42.5±4.8% (df=54; P<0.001). Similarly, NPPB
(100 μM) reduced the glucose-stimulated insulin release by
32.1±6.0% (df=58; P<0.001). The relative magnitudes of the
reductions induced by tenidap and NPPB were not observed to be
significantly different (df=112; P>0.18).
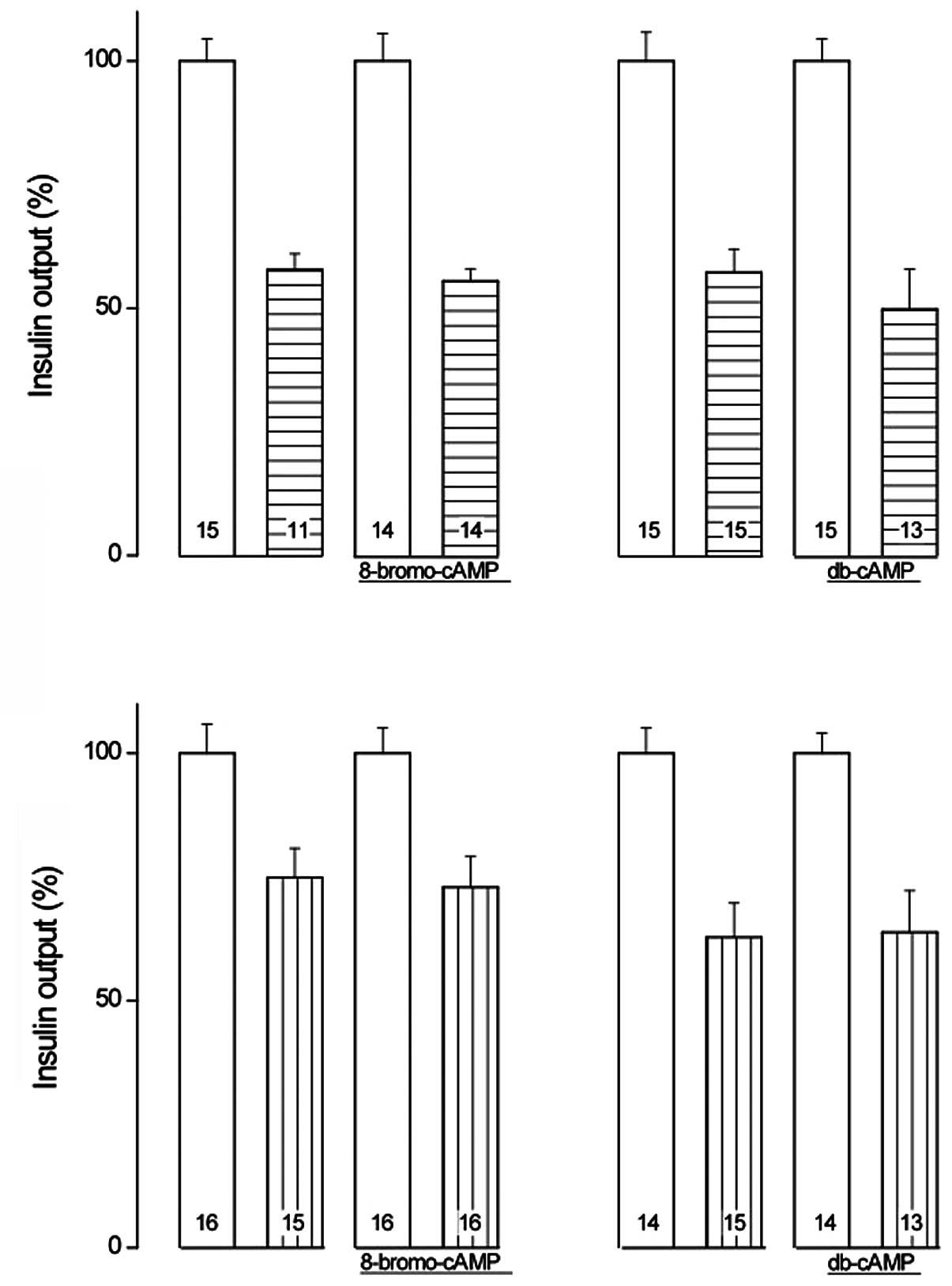
As demonstrated in Fig.
2, the relative extent of the inhibitory action of tenidap upon
insulin release was not observed to differ significantly
(P>0.78) in the presence (44.5±6.0%; df=26) or absence
(42.2±6.0%; df=24) of 8-Br-cAMP, being significant (P<0.001) in
both cases. The relative inhibitory action of tenidap upon insulin
release did not differ significantly (P>0.52) in the presence
(50.2±9.0%; df=26) or absence of db-cAMP (42.7±7.4%; df=28), and
was significant (P<0.001) in both cases.
When compared within the same experiments, the
relative extent of the inhibitory action of NPPB also failed to
differ significantly (P>0.86) in the presence (27.1±8.1%; df=30)
or absence (25.1±8.4%; df=29) of 8-Br-cAMP, which was significant
(P<0.006) in both cases. In addition, the inhibitory action of
NPPB was not identified to be significantly different (P>0.93)
in the presence (36.2±9.2%; df=25) or absence (27.2±8.7%; df=27) of
db-cAMP, and was significant (P<0.001) in both cases (Fig. 2).
In the presence of tenidap, the two cAMP analogs
augmented the mean levels of insulin release. The overall mean
relative magnitude of this increase (20.9±7.6%; df=49; P<0.009)
was almost identical to that recorded in the absence of inhibitors
of insulin release (Table I). In
the islets exposed to NPPB, however, the enhancing action of the
cAMP analogs failed to achieve statistical significance, indicating
that, under the present experimental conditions, NPPB suppressed an
essential component of the secretory response to D-glucose.
Experiments in BRIN-BD11 cells
Reference data
Basal insulin output from BRIN-BD11 cells incubated
in an isotonic medium (1.1 mM D-glucose) was 61.5±4.1 μU/ml/30 min
(n=39). Output increased by 70.0±5.8 μU/ml/30 min (paired
comparison within each experiment; n=33; P<0.001) when the
BRIN-BD11 cells were exposed to a hypotonic medium [50 mM reduction
in NaCl concentration (n=30) or a one-third dilution of the usual
medium (n=3)]. No significant difference was observed (P>0.8) in
insulin output under the latter two experimental conditions.
Addition of KIC (10.0 mM) to the isotonic medium increased insulin
output by 30.5±2.8 μU/ml/30 min (paired comparison; n=8;
P<0.001).
Effect of cAMP analogs and
phosphodiesterase inhibitors
cAMP-AM (0.1–0.2 mM) and IBMX (0.5 mM) increased the
insulin output of BRIN-BD11 cells incubated in an isotonic medium
to 254.8±15.1% (n=3; P<0.01) of the control value, compared with
139.3±12.8% (n=3; P<0.005) when BRIN-BD11 cells incubated in
hypotonic medium were also exposed to cAMP-AM and IBMX. The
hypotonicity-induced increment in insulin output, expressed as
μU/ml/30 min, was ~2-fold lower (P<0.05) in the presence of
cAMP-AM and IBMX (47.1±6.3; n=3) than in their absence (81.6±7.7;
n=2). In addition, the insulin output of the BRIN-BD11 cells
incubated in isotonic medium in the presence of cAMP-AM (0.1 mM)
and IBMX (0.5 mM) was 201.8±15.4% (n=12; P<0.001) of the paired
control value. The latter percentage was not noted to differ
significantly (P>0.13) from that recorded in the first set of
experiments.
Further experiments, all conducted in the isotonic
medium, indicated that IBMX (0.5 mM) increased insulin output to
191.5±14.3% (n=12) of the paired value determined in its absence.
When compared within the same experiments, the insulin output in
the presence of IBMX (0.5 mM) was ≥97.3±3.4% (n=8) of that recorded
in the presence of IBMX (0.5 mM) and cAMP-AM (0.1 mM).
Insulin output in the presence of other cAMP analogs
in isotonic medium in the absence of IBMX was 105.4±4.4% (n=4) for
8-Br-cAMP (1.0 mM), 115.3±3.6% (n=4; P<0.025) for Sp-8-Br-cAMPS
(0.1 mM) and 337.4±10.7% (n=4; P<0.001) for Sp-cAMPS-AM (0.1
mM).
For the purpose of comparison, the ratio for insulin
output in the hypotonic/isotonic medium in the absence of any cAMP
analog or phosphodiesterase inhibitor was 242.7±11.5% (n=16;
P<0.001).
Effect of NPPB
The VRAC inhibitor, NPPB (0.1 mM), eliminated the
secretory response to KIC (10 mM). Thus, instead of an increase in
insulin output of 40.8±2.7% (n=2; P<0.05), as recorded in the
presence of KIC but absence of NPPB, a mean decrease of 19.9±1.1%
(n=2; P<0.05) was observed in the presence of KIC and NPPB. In
the presence of NPPB, the cAMP analogs did not fully restore the
insulinotropic action of KIC. Compared with a KIC-induced increment
of 36.9±5.6 μU/ml/30 min, as observed in the absence of NPPB and a
decrement of 17.9±2.6 μU/ml/30 min, as recorded in the presence of
KIC and NPPB, values obtained in the presence of KIC and NPPB did
not exceed +5.9±1.7 μU/ml/30 min in the presence of 1.0 mM
8-Br-cAMP, −2.5±0.4 μU/ml/30 min in the presence of 1.0 mM db-cAMP
and −2.7±0.1 μU/ml/30 min in the presence of 1.0 mM dioctanoyl-cAMP
or 0.1 mM 2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate
tyrosyl methyl ester (n=2 in all cases). The paired difference from
basal values obtained in the presence of KIC, NPBB and a cAMP
analog yielded an overall mean value of +0.2±1.9 μU/ml/30 min
(n=6), significantly higher (P<0.004) than the reduced insulin
output recorded in the presence of KIC and NPPB and significantly
lower (P<0.001) than the increased insulin output recorded in
the presence of KIC.
NPPB (0.1 mM) also eliminated the secretory response
evoked by extracellular hypotonicity. In a first series of 4
experiments, the output of insulin (μU/ml/30 min) was increased
(P<0.01) from a basal value of 82.0±8.0 in an isotonic medium to
165.1±21.8 in the hypotonic medium. In the presence of NPPB, the
release of insulin recorded in the hypotonic medium did not exceed
55.4±7.8 μU/ml/30 min (P<0.06 vs. basal value). Again, the cAMP
analogs failed to restore the secretory response evoked by the
exposure of the BRIN-BD11 cells to the hypotonic medium. Thus, in
the presence of NPPB, the output of insulin (μU/ml/30 min) from
cells exposed to the hypotonic medium averaged no more than
64.1±20.9 (n=3) in the presence of 1.0 mM 8-Br-cAMP, 24.5±5.7 (n=2)
in the presence of 1.0 mM db-cAMP, 51.8 (n=1) in the presence of
1.0 mM dioctanoyl-cAMP and 28.3 (n=1) in the presence of 0.1 mM
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester. These observations indicate that the insulin output
from the BRIN-BD11 cells exposed to hypotonic medium in the
presence of NPPB and a cAMP analog was only 27.6±4.5% (n=7) of the
value from cells exposed to hypotonic medium in the absence of
NPPB.
Even in the presence of 0.5 mM IBMX, the release of
insulin from BRIN-BD11 cells exposed to NPPB (0.1 mM) in the
hypotonic medium did not exceed 51.3±4.8% (n=3) of the value
determined in the hypotonic medium and absence of NPPB, despite the
presence of 0.1 mM cAMP-AM (n=2) or
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester (n=1). However, the latter percentage was
significantly higher (P<0.02) than that recorded in the absence
of IBMX but presence of a cAMP analog (27.6±4.5%; n=7). The results
demonstrate that the enhancing action of the phosphodiesterase
inhibitor on insulin secretion remained operative in the cells
exposed to NPPB.
These observations were confirmed in a further
series of four experiments in which the release of insulin
(μU/ml/30 min) from the BRIN-BD11 cells averaged 110.2±4.7 in the
hypotonic medium, compared with (P<0.001) 47.8±3.6 in the
isotonic medium and 48.1±3.5 in the hypotonic medium containing
NPPB (0.1 mM). Fig. 3 presents the
major observations relevant to analyses conducted in the presence
of NPPB.
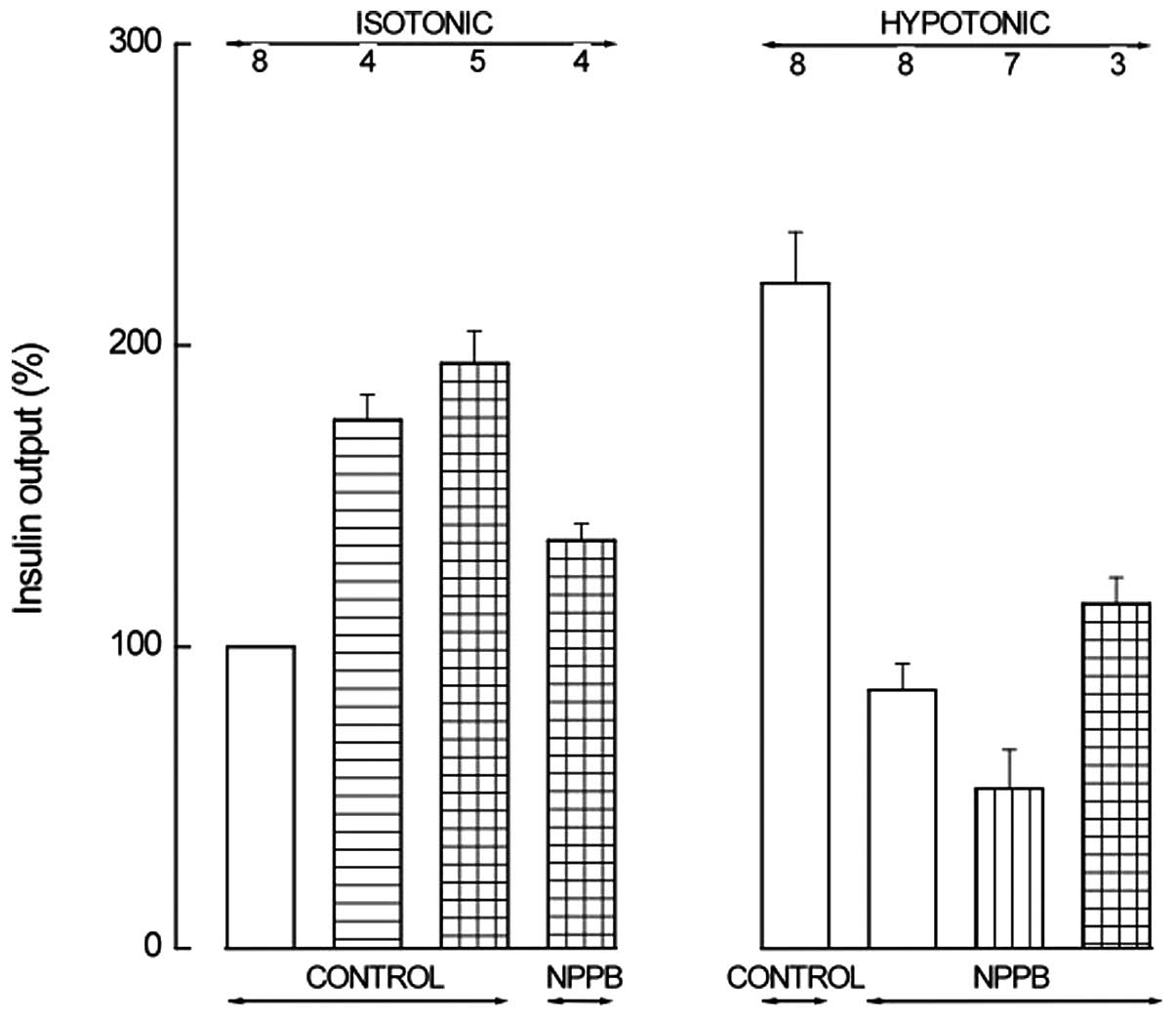 | Figure 3Insulin output from BRIN-BD11 cells
incubated for 30 min in isotonic (left) or hypotonic (right) medium
in the absence (control) or presence of NPPB. The incubation medium
also contained IBMX (0.5 mM; horizontally hatched column) alone or
together with cAMP-AM (0.1 mM; horizontally and vertically hatched
columns) in the left panel; in the right panel, the incubation
medium also contained a cAMP analog (0.1 mM 8-Br-cAMP, 1.0 mM
db-cAMP, 1.0 mM dioctanoyl-cAMP or 0.1 mM
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester; vertically hatched column) or IBMX (0.5 mM) and a
cAMP analog (0.1 mM cAMP-AM or 0.1 mM
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester; horizontally and vertically hatched column). Means ±
SEM were collected in a series of 8 experiments, are expressed
relative to the paired basal value in the isotonic medium and refer
to the number of determinations indicated above each column.
Reference basal value in the isotonic medium was 64.9±7.6 μU/ml/30
min (n=8). NPPB, 5-nitro-2-(3-phenylpropylamino)benzoic acid; IBMX,
3-isobutyl-1-methylxanthine; cAMP, adenosine 3′,5′-cyclic
monophosphate; AM, acetoxymethyl ester; db, dibutyryl. |
Effect of tenidap
In the absence of tenidap, which is postulated to be
an inhibitor of the
Na+-HCO3−-cotransporter, NBCe1,
KIC (10.0 mM) increased insulin output by 25.7±5.8 μU/ml/30 min
(n=3). The paired difference between insulin output in the presence
of KIC and tenidap (50 μM) and the basal value in the absence of
KIC yielded a mean negative value of −7.9±6.6 μU/ml/30 min (n=3).
Tenidap decreased the KIC-stimulated insulin output to 61.3±2.0%
(n=3; P<0.005) of its paired control value. In the presence of
KIC and tenidap, 8-Br-cAMP or db-cAMP (both 1.0 mM) did not
significantly increase insulin output whereby average output in the
presence of the cAMP analogs was 109.8±7.4% (n=6) of the paired
value recorded in their absence. Only dioctanoyl-cAMP (1.0 mM) and
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester (0.1 mM) augmented the insulin output evoked by KIC in
the presence of tenidap from 53.0±11.5 to 97.5±11.6 μU/ml/30 min
(n=3 in both cases; P<0.06). The latter mean value was not
significantly different (P>0.65) from that recorded in the
presence of KIC only (86.6±19.1 μU/ml/30 min; n=3).
In a series of eight experiments, the
hypotonicity/isotonicity ratio for insulin release was 254.2±22.2%
(n=8; P<0.001). When tenidap (50 μM) was present in the
hypotonic medium, insulin release was decreased to 29.6±3.3% (n=8;
P<0.001) of the paired value recorded in its absence and
represented no more than 70.5±8.5% (n=8; P<0.01) of that
recorded in the isotonic medium in the absence of tenidap. When
8-Br-cAMP (1.0 mM; n=4), db-cAMP (1.0 mM; n=3) or
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester (0.1 mM; n=1) was added to the hypotonic medium
containing tenidap, the release of insulin was only 113.2±10.4%
(n=8; p>0.25) of the paired value in their absence. Even in the
presence of 0.5 mM IBMX and 0.1 mM cAMP-AM or 0.1 mM
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester, the output of insulin from the BRIN-BD11 cells
incubated in the hypotonic medium containing tenidap did not exceed
127.6±19.2% (n=2; P>0.38) of the paired value in the hypotonic
medium in the presence of tenidap only. In the presence of tenidap,
only dioctanoyl-cAMP (1.0 mM) was observed to significantly
increase the insulin output from the BRIN-BD11 cells exposed to the
hypotonic medium from 33.3±7.8 to 101.1±15.0 μU/ml/30 min (n=2 in
both cases; P<0.06). Even in BRIN-BD11 cells incubated in the
isotonic medium in the presence of IBMX (0.5 mM) and cAMP-AM (0.1
mM), tenidap (50 μM) decreased insulin output from 86.9±7.0
μU/ml/30 min (n=4) recorded in the absence of tenidap to 54.3±5.8
μU/ml/30 min (n=4; P<0.02). The tenidap-induced decrease in
insulin release was 32.6±4.8 μU/ml/30 min (n=4; P<0.008).
Fig. 4 presents the major
observations relevant to the analyses conducted in the presence of
tenidap.
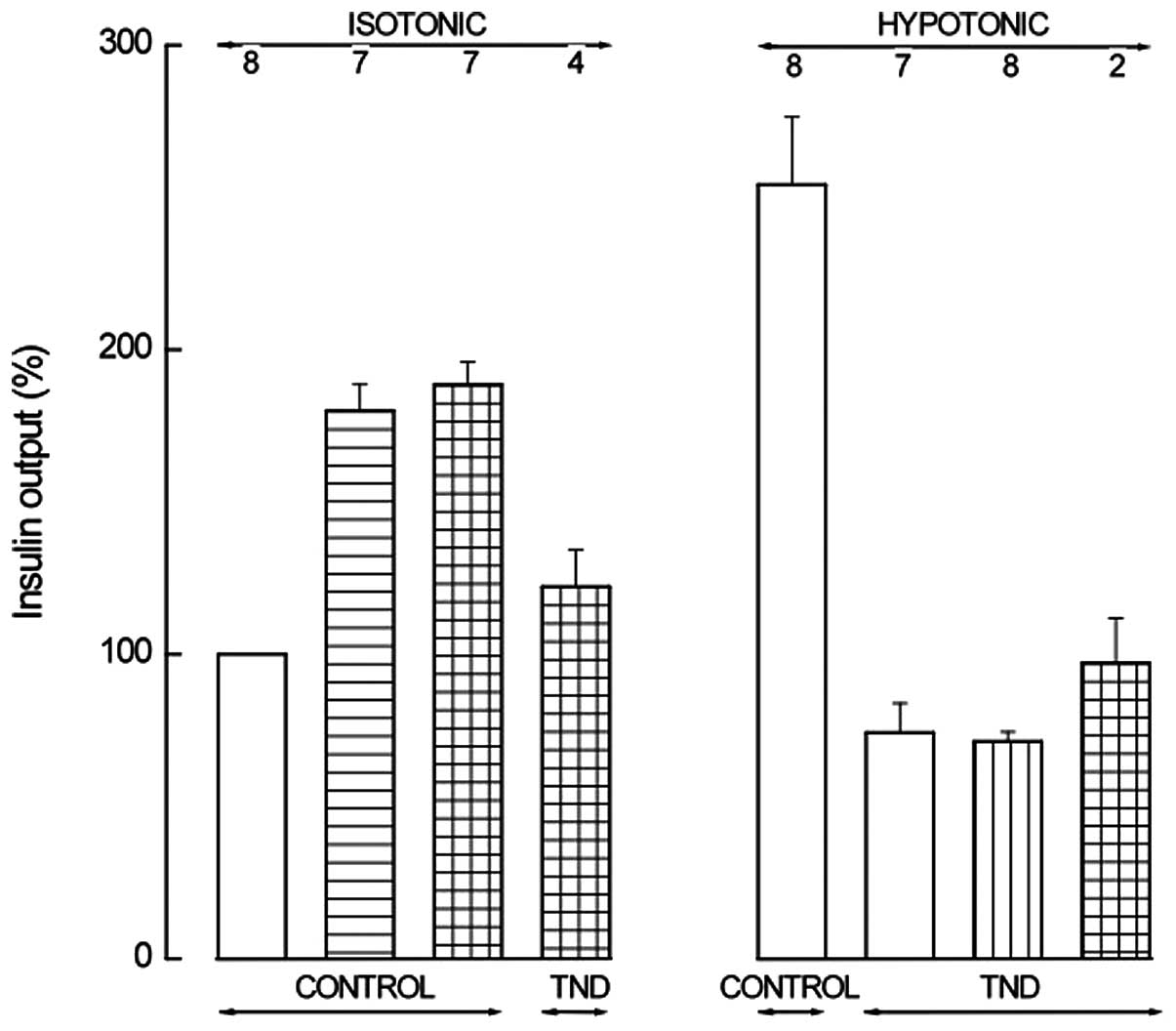 | Figure 4Insulin output from BRIN-BD11 cells
incubated for 30 min in an isotonic (left) or hypotonic (right)
medium in the absence (control) or presence (TND) of tenidap. The
incubation medium also contained IBMX (0.5 mM; horizontally hatched
column) alone or together with cAMP-AM (0.1 mM; horizontally and
vertically hatched columns) in the left panel; in the right panel,
the incubation medium contained a cAMP analog (1.0 mM 8-Br-cAMP,
1.0 mM db-cAMP or 0.1 mM 2′-O-monosuccinyladenosine-3′,5′-cyclic
monophosphate tyrosyl methyl ester; vertically hatched column) or
both IBMX (0.5 mM) and a cAMP analog (0.1 mM cAMP-AM or 0.1 mM
2′-O-monosuccinyladenosine-3′,5′-cyclic monophosphate tyrosyl
methyl ester; horizontally and vertically hatched column). Means ±
SEM were collected in a series of 8 experiments, are expressed
relative to the paired basal value in the isotonic medium and refer
to the number of determinations indicated above each column. The
reference basal value in the isotonic medium was 55.5±9.5 μU/ml/30
min (n=8; P>0.45, vs. the 8 experiments in Fig. 3). Control values of the hypotonic
medium were 254.2±22.2% (n=8) of the paired basal value in the
isotonic medium (P>0.25, vs. the 8 experiments in Fig. 3). Tenidap,
5-chloro-2-hydroxy-3-(thiophene-2-carbonyl)indole-1-carboxamide;
IBMX, 3-isobutyl-1-methylxanthine; cAMP, adenosine 3′,5′-cyclic
monophosphate; AM, acetoxymethyl ester. |
Effect of MAP kinase inhibitors
In the isotonic medium, the MAP kinase inhibitors,
U0126 (10 μM) and PD98,059 (50 μM), reduced insulin output to
81.3±6.1% (n 4; P<0.06) of the paired control value. The
relative extent of the reduction was not noted to be significantly
different between the isotonic medium and either the isotonic
medium containing 10 mM KIC or the hypotonic medium (df = 6;
P>0.5).
Effect of 2-hydroxyestriol
The effects of the sAC inhibitor, 2-hydroxyestriol,
upon the secretory response to extracellular hypotonicity were
analyzed. Insulin output in the absence of 2-hydroxyestriol was
60.9±4.7 μU/ml/30 min (n=6; P<0.001) higher in the hypotonic
medium than the isotonic medium. In addition, 2-hydroestriol (50
μM) was not observed to significantly affect insulin output in
BRIN-BD11 cells incubated in isotonic or hypotonic medium. Insulin
output in the presence of 2-hydroxyestriol (50 μM) was 94.0±8.2%
(n=4; P>0.5) of the paired control value.
At a concentration of 100 μM, 2-hydroxyestriol
exhibited a modest inhibitory effect, with an insulin output of
84.2±3.1% (n=8; P<0.002) of the paired control value. This
reduction was not determined to differ significantly between the
isotonic and hypotonic medium (P>0.36). Insulin release in
BRIN-BD11 cells exposed to the hypotonic medium was inhibited by
2-hydroxyestriol (100 μM; P>0.5) or the membrane permeant,
metabolically stable inhibitor of cAMP-dependent protein kinase,
Rp-8-Br-cAMPS (100 μM), with an overall mean value of 82.8±5.6%
(n=8; P<0.025) of the representative control.
Effect of HCO3−
and/or Cl− omission
The omission of NaHCO3 severely decreased
the secretory response to KIC (10 mM), the KIC-induced increment in
insulin output not exceeding 5.0 μU/ml/30 min. Similarly, the
paired ratio between insulin output in the hypotonic/isotonic
medium was no more than 121.1±13.6% (n=3) in the absence of
NaHCO3, compared with 188.6±14.3% (n=12) in the presence
of NaHCO3 (P<0.05). In the absence of
NaHCO3, insulin output evoked by KIC or extracellular
hypotonicity was decreased by 7.1±0.1 μU/ml/30 min (n=2; P<0.01)
in the presence of 50 μM tenidap. In the presence of tenidap,
neither 8-Br-cAMP (1.0 mM) nor db-cAMP (1.0 mM) were observed to
significantly affect the insulin output of BRIN-BD11 cells exposed
to KIC or extracellular hypotonicity in the absence of
NaHCO3, with a mean increment not exceeding 2.3±1.1
μU/ml/30 min (n=4). However, dioctanoyl-cAMP (1.0 mM), markedly
increased insulin release from the BRIN-BD11 cells exposed, in the
presence of tenidap and absence of NaHCO3, to KIC or
hypotonic medium. The increased insulin output attributable to
dioctanoyl-cAMP was 63.9±1.6 μU/ml/30 min (n=2).
In the absence of Cl− or Cl−
and HCO3− and in the absence or presence of
the carbonic anhydrase inhibitor, ethoxyzolamide
(6-ethoxy-2-benzothiazolsulfonamide; 0.5 mM), the paired ratio
between insulin ouput in the hypotonic/isotonic medium was
extremely low (P<0.04), averaging no more than 115.2±18.6%
(n=3). Under these experimental conditions, the addition of
8-Br-cAMP (1.0 mM) to the incubation medium was not observed to
significantly increase insulin output, which averaged 110.4±20.1%
(n=3) of the paired control. Ethoxyzolamide (0.5 mM) increased
insulin output from the BRIN-BD11 cells incubated in the absence of
Cl− and HCO3− to 187.2±4.1% (n=3;
P<0.005) of the paired control value determined in the presence
of Cl− and HCO3− in the isotonic,
hypotonic or hypotonic medium containing 8-Br-cAMP (1.0 mM).
Effect of Na+ omission
Insulin release from BRIN-BD11 cells incubated in an
isotonic medium deprived of Na+ (substitution of NaCl
(115 mM) by an equiosmolar mixture of
2-amino-2-hydroxymethyl-1,3-propanediol, N-methyl-D-glucosamine and
sucrose and of NaHCO3 (24 mM) by an equimolar amount of
choline bicarbonate) was observed to be 2–3-fold higher than that
found in the control isotonic medium. The hypotonicity/isotonicity
ratio for insulin output, which exceeded 200% under the usual
experimental conditions, did not exceed 103.6±3.3% (n=2; P>0.48)
in the Na+-free medium. When the BRIN-BD11 cells were
exposed to the Na+-free hypotonic medium, addition of
8-Br-cAMP (1.0 mM) or IBMX (0.5 mM) and cAMP-AM (0.1 mM) increased
insulin output by 17.9 and 53.7%, respectively.
Discussion
The present study aimed to evaluate several novel
observations associated with the interaction between cAMP, VRAC and
the Na+-HCO3−-cotransporter,
NBCe1, in the regulation of nutrient- and hypotonicity-induced
insulin release.
Studies in rat isolated pancreatic islets have
demonstrated that tenidap and NPPB inhibit glucose-stimulated
insulin release while 8-Br-cAMP and db-cAMP increase the
insulinotropic action of hexose (12,13,18).
cAMP analogs were observed to augment insulin output in the
presence or absence of tenidap to the same extent and NPPB
suppressed the enhancing action of the cAMP analogs on
glucose-stimulated insulin secretion (Table I). The latter observation is
consistent with the hypothesis that NPPB, by opposing the gating of
VRACs, suppresses an essential component of the secretory response
to D-glucose. It is well established that agents increasing the
cAMP content of non-tumoral insulin-producing cells fail to augment
insulin output from islets incubated at low D-glucose
concentrations (18). By contrast,
the maintenance of a marked positive response to the cAMP analogs
in the presence of tenidap indicates that the role of NBCe1 in
ionic fluxes does not represent an essential permissive process for
the expression of D-glucose insulinotropic action.
In the analyses conducted in BRIN-BD11 cells, KIC
was used as a nutrient secretagogue instead of D-glucose due to the
poor secretory responsiveness of these tumoral insulin-producing
cells to the hexose (19). The
effects of cAMP analogs on insulin output from the BRIN-BD11 cells
differed, in part, from those in isolated rat pancreatic islets. In
contrast to the situation observed in rat islets (18), the phosphodiesterase inhibitor,
theophylline, markedly enhanced insulin output from BRIN-BD11 cells
incubated in isotonic medium even when the cells were exposed to a
low D-glucose concentration (1.1 mM). In the absence of
theophylline, Sp-cAMPS-AM (0.1 mM) and to a significantly more
marked extent, Sp-8-Br-cAMPS (0.1 mM) also enhanced insulin output
from the BRIN-BD11 cells incubated in isotonic medium.
In the BRIN-BD11 cells, the secretory response to
KIC was eliminated in the presence of NPPB. Under these
experimental conditions, the cAMP analogs simply brought insulin
output to a level comparable to that obtained in the absence of KIC
and NPPB. Similarly, in BRIN-BD11 cells exposed to the hypotonic
medium in the presence of NPPB, the output of insulin recorded in
the presence of cAMP analogs was not identified to be significantly
different (df=9; P<0.33) from that recorded in their absence.
Even in the presence of IBMX (0.5 mM) and a cAMP analog, insulin
release from BRIN-BD11 cells exposed to hypotonic medium in the
presence of NPPB (93.4±14.3 μU/ml/30 min; n=3) was not identified
to be significantly different (P>0.53) from the corresponding
basal value (81.8±10.6 μU/ml/30 min; n=3).
As demonstrated in rat isolated pancreatic islets,
the effect of the NBCe1 inhibitor, tenidap, on insulin release from
the BRIN-BD11 cells differed, in part, from that of NPPB. Tenidap
was identified to decrease insulin output from BRIN-BD11 cells
exposed to KIC or the hypotonic medium to mean values lower than
the basal values recorded in the absence of tenidap. However,
selected cAMP analogs restored insulin output from BRIN-BD11 cells
exposed to KIC in the presence of tenidap to a level comparable to
that recorded in the presence of KIC but absence of tenidap.
Similarly, dioctanoyl-cAMP increased insulin output by ~3-fold in
BRIN-BD11 cells exposed to hypotonic medium and tenidap.
A role for cAMP-responsive MAP kinase in the
secretory activity of BRIN-BD11 cells was demonstrated by the
inhibitory effects of U0126 and PD98,059 on basal and KIC- or
hypotonicity-stimulated insulin release. Analyses conducted in the
presence of 2-hydroxyestriol indicated a limited role for sAC in
this secretory activity. It must be noted, however, that in both
cases, basal and stimulated insulin output from the BRIN-BD11 cells
was affected to a comparable extent by the tested inhibitors.
However, this does not exclude a role for sAC in glucose-induced
insulin release from isolated pancreatic islets.
Whilst the analyses conducted in BRIN-BD11 cells
exposed to media deprived of HCO3− extend to
the present experimental design the unfavourable effect of
HCO3− omission on insulin secretion (7,8), the
results revealed an unexpected enhancement of insulin secretion by
ethoxyzolamide.
Finally, observations in BRIN-BD11 cells exposed to
a Na+-deprived medium indicate a favourable effect of
Na+ omission on basal insulin output which may be
associated with a reduced consumption of ATP by Na+,
K+-ATPase.
In conclusion, the present study demonstrates marked
differences in the responsiveness to cAMP analogs depending on the
conditions and agents used to stimulate or inhibit insulin
secretion from rat isolated pancreatic islets or insulin-producing
tumoral BRIN-BD11 cells. However, these observations do not appear
to be to associated with the activation or inactivation of sAC. In
addition, the inhibition of sAC (by 2-hydroxyestriol) or MAP kinase
(by U0126 or PD98,059) did not have a significant effect on insulin
output and insulin secretion recorded in the presence of KIC or in
hypotonic medium, thus indicating that sAC and MAP kinase are not
involved in the secretory response to a nutrient secretagogue or
extracellular hypoosmolarity. In BRIN-BD11 cells, cAMP and its
analogs consistently increased insulin output, whilst in isolated
pancreatic islets these agents only amplified the secretory
response to D-glucose. However, in both cases, VRACs appear to be
involved in the effect of intracellular cAMP.
Acknowledgements
The authors thank C. Demesmaeker for secretarial
help. The present study was supported by a grant from the Belgian
Foundation for Scientific Medical Research (no. 3.452007).
Abbreviations:
|
cAMP
|
adenosine 3′,5′-cyclic
monophosphate
|
|
8-Br-cAMP
|
8-bromo-cAMP
|
|
cAMP-AM
|
cAMP acetoxymethyl ester
|
|
db-cAMP
|
dibutyryl-cAMP
|
|
IBMX
|
3-isobutyl-1-methylxanthine
|
|
KIC
|
2-ketoisocaprate
|
|
MAP kinase
|
mitogen-activated protein kinase
|
|
NPPB
|
5-nitro-2-(3-phenylpropylamino)benzoic
acid
|
|
Rp/Sp-8-Br-cAMPS
|
8-bromoadenosine-3′,5′-cyclic
monosphosphorothioate, Rp/Sp isomer
|
|
VRAC
|
volume-regulated anion channel
|
References
|
1
|
Ramos LS, Zippin JH, Kamenetsky M, Bruck J
and Levin LR: Glucose and GLP-1 stimulate cAMP production via
distinct adenylyl cyclases in INS-1E insulinoma cells. Gen Physiol.
132:329–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valverde I, Vandermeers A, Anjaneyulu R
and Malaisse WJ: Calmodulin activation of adenylate cyclase in
pancreatic islets. Science. 206:225–227. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esposito G, Jaiswal BS, Xie F,
Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, Van
Duin M, Conti M and Gossen JA: Mice deficient for soluble adenylyl
cyclase are infertile because of a severe sperm-motility defect.
Proc Natl Acad Sci USA. 105:2993–2998. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hess KC, Jones BH, Marquez B, Chen Y, Ord
TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, et al:
The ‘soluble’ adenylyl cyclase in sperm mediates multiple signaling
events required for fertilization. Dev Cell. 9:249–259. 2005.
|
|
5
|
Schmid A, Sutto Z, Nlend MC, Horvath G,
Schmid N, Buch J, Levin LR, Conner GE, Fregien N and Salathe M:
Soluble adenylyl cyclase is localized to cilia and contributes to
ciliary-beat frequency regulation via production of cAMP. J Gen
Physiol. 130:99–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pastor-Soler N, Beaulieu V, Litvin TN, Da
Silva N, Chen Y, Brown P, Buck J, Levin LR and Breton S:
Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that
regulates pH-dependent V-ATPase recycling. J Biol Chem.
278:49523–49529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Henquin J-C and Lambert AE: Extracellular
bicarbonate ions and insulin secretion. Biochim Biophys Acta.
381:437–442. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Henquin J-C and Lambert AE: Bicarbonate
modulation of glucose-induced biphasic insulin release by rat
islets. Am J Physiol. 231:713–721. 1976.PubMed/NCBI
|
|
9
|
Malaisse WJ, Hutton JC, Kawazu S,
Herchuelz A, Valverde I and Sener A: The stimulus-secretion
coupling of glucose-induced insulin release. XXXV The links between
metabolic and cationic events. Diabetologia. 16:331–341. 1979.
View Article : Google Scholar
|
|
10
|
Sener A and Malaisse WJ: Secretory, ionic
and metabolic events in rat pancreatic islets deprived of
extracellular NaHCO3. Metab Funct Res Diab. 5:1–3.
2012.
|
|
11
|
Sener A, Jijakli H, Zahedi Asl S, Courtois
P, Yates AP, Meuris S, Best LC and Malaisse WJ: Possible role of
carbonic anhydrase in rat pancreatic islets: enzymatic, secretory,
metabolic, ionic and electrical aspects. Am J Physiol.
292:E1624–E1630. 2007.PubMed/NCBI
|
|
12
|
Louchami K, Zhang Y, Beauwens R, Malaisse
WJ and Sener A: Is the glucose-induced phosphate flush in
pancreatic islets attributable to gating of volume-sensitive anion
channels? Endocrine. 31:1–4. 2007.PubMed/NCBI
|
|
13
|
Soyfoo MS, Bulur N, Virreira M, Louchami
K, Lybaert P, Crutzen R, Perret J, Delporte C, Roussa E, Thevenod
F, Best L, Yates AP, Malaisse WJ, Sener A and Beauwens R:
Expression of the electrogenic
Na+-HCO3−-cotransporters NBCe1-A
and NBCe1-B in rat pancreatic islet cells. Endocrine. 35:449–458.
2009.PubMed/NCBI
|
|
14
|
Bulur N, Louchami K, Sener A, Beauwens R
and Malaisse WJ: Effect of tenidap on sodium fluxes in pancreatic
islet cells: methodological aspects. Metab Funct Res Diab. 2:1–4.
2009.
|
|
15
|
McClenaghan NH, Barnett CR, Am-Sing E,
Abdel-Wahab YH, O’Harte FP, Yoon TW, Swanston-Flatt SK and Flatt
PR: Characterisation of a novel glucose-responsive
insulin-secreting cell line, BRIN-BD11, producted by electrofusion.
Diabetes. 45:1132–1140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Malaisse-Lagae F and Malaisse WJ: Insulin
release by pancreatic islets. Methods in Diabetes Research. I(Part
B)Larner J and Pohl S: John Wiley & Sons; New York, NY: pp.
147–152. 1984
|
|
17
|
Beauwens R, Best L, Markadieu N, Crutzen
R, Louchami K, Brown P, Yates AP, Malaisse WJ and Sener A:
Stimulus-secretion coupling of hypotonicity-induced insulin release
in BRIN-BD11 cells. Endocrine. 30:353–363. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brisson GR, Malaisse-Lagae F and Malaisse
WJ: The stimulus-secretion coupling of glucose-induced insulin
release. VII A proposed site for adenosine-3′,5′-cyclic
monophosphate. J Clin Invest. 51:232–241. 1972.PubMed/NCBI
|
|
19
|
Bulur N, Zhang Y, Malaisse WJ and Sener A:
Insulin release from isolated pancreatic islets, dispersed islet
cells and tumoral insulin-producing cells: a re-examination. Metab
Funct Res Diab. 3:20–24. 2010.
|