Introduction
Renal cell carcinoma (RCC) is the most common
primary renal malignant neoplasm in adults. It accounts for ~3% of
adult malignancies and 90–95% of renal neoplasms. The gold standard
for RCC treatment is surgery, where nephron-sparing surgery,
laparoscopic and robotic surgery and minimally invasive procedures
have all decreased the morbidity of RCC (1). However, advanced or metastatic RCC
may develop resistance to chemotherapy or radiotherapy,
contributing to a poor prognosis (2). In order to develop effective
therapeutic strategies for RCC, further investigations are required
to understand the molecular pathogenesis of aggressive RCC.
Clusterin (CLU), also known as testosterone
repressed prostate message-2 or sulfated glycoprotein-2, is a
glycoprotein crucial to various pathophysiological processes
(3), such as tumor pathogenesis
and progression. CLU is overexpressed in a variety of tumors,
including in liver, pancreatic, colorectal, ovarian, prostate,
bladder and kidney cancer (4).
Furthermore, CLU expression levels correlate with the metastasis of
melanoma, gastric cancer, ovarian cancer and RCC (5–8).
However, the molecular mechanism by which CLU plays an oncogenic
role in RCC remains unclear.
Global expression analysis using microarrays may be
able to monitor the expression of thousands of genes in a
high-throughput manner to provide novel insights into the
mechanisms of cancer initiation, progression, resistance to
treatment and response to cellular microenvironments (9). Therefore, in the present study, we
used the human renal cancer cell line 786-O as an experimental
model. We knocked down CLU expression in 786-O cells using
lentiviral vector-mediated delivery of RNAi, and then compared the
gene expression profiles of knocked down CLU 786-O cells and
control cells. We demonstrated that CLU knockdown induces apoptosis
and inhibits the proliferation and migration of 786-O cells.
Furthermore, we identified differentially expressed genes after CLU
knockdown and analyzed the related pathways in which these genes
are involved.
Materials and methods
Cell culture
The 786-O cell line was purchased from the American
Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in
RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented with 10%
Gibco™ FBS (Life Technologies, Grand Island, NY, USA) at 37°C in a
standard humidified incubator containing 5% CO2 and 95%
O2. The study was approved by the Ethics Committee of
The First People's Hospital Affiliated to Guangzhou Medical
University (Guangzhou, China).
Lentivirus RNAi construct and
transfection
The following three siRNA sequences targeting human
CLU were provided by GeneChem Co., Ltd. (Shanghai, China): 1,
5′-CAGGGAAGTAAGTACGTCAATCTCGAGATTGACGTACTTACTTCCCTGTTTTT-3′; 2,
5′-GCTAAAGTCCTACCAGTGGAACTCGAGTTCCACTGGTAGGACTTTAGCTTTTT-3′; and 3,
5′-AGGGAAGTAAGTACGTCAATACTCGAGTATTGACGTACTTACTTCCCTTTTTT-3′. A
control siRNA with non-specific sequences was also produced. These
siRNAs were cloned into pGCSIL-GFP plasmids. Lentiviruses were
generated by the transfection of 80% confluent HEK293T cells with
recombinant pGCSIL-GFP plasmids and pHelper 1.0 and pHelper 2.0
helper plasmids (GeneChem Co., Ltd.) using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA). Lentiviruses were harvested in
serum-free medium after 2 days, filtered and concentrated in primed
Centricon Plus-20 filter devices (Millipore, Billerica, MA, USA),
and the titers of recombinant lentiviruses were determined.
The 786-O cells were seeded in 6-well plates and
grown to 60% confluence on the day of transfection. Four hours
prior to transfection, cells were placed in serum-free media. The
cells were transfected with a titrated siRNA vector diluted in
RPMI-1640, with the addition of 1 μl polybrene. Successful
knockdown of CLU was analyzed by real-time PCR and western blot
analysis.
Real-time PCR assay
Total RNA was extracted from the 786-O cells using
TRIzol (Invitrogen), according to the manufacturer's instructions.
First-strand cDNA was generated from 2 μg total RNA using the
PrimeScript RT reagent kit (Takara, Dalian, China) with random
primers. Real-time PCR was performed on an ABI Prism 7300 (Applied
Biosystems, Foster City, CA, USA). The specific primers were as
follows: CLU, 5′-TCCGCGGCATTCTTTGGGCG-3′ and
5′-GCACTGGGAGGCGCCGTATT-3′; and β-actin,
5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and 5′-CCTTGCACATGCCGGAGCCGT-3′.
Thermal cycling was initiated with a denaturation step for 1 min at
95°C followed by 40 cycles performed in two steps; 5 sec at 95°C
and 30 sec at 60°C. The relative mRNA level of CLU was compared to
that of β-actin and was calculated by the 2−ΔΔCt method.
Each Ct value used for these calculations was the mean of
triplicate results obtained for each reaction.
Western blot analysis
The 786-O cells were lysed in RIPA buffer
supplemented with protease inhibitors. The supernatant was
collected for the protein concentration assay. Equal amounts of
protein (30 μg) were separated in 10% SDS-polyacrylamide gels and
transferred to PVDF membranes. The membranes were blocked using
non-fat milk in TBST and probed with either a CLU (Abcam,
Cambridge, MA, USA) or GAPDH antibody (Santa Cruz Biotech, Santa
Cruz, CA, USA), followed by incubation with a secondary antibody.
Immunoreactivity signals were developed using an enhanced
chemiluminescence (ECL) kit (GE Healthcare, Piscataway, NJ,
USA).
Cell proliferation assay
The proliferation of 786-O cells was assessed using
a WST-1 kit (Beyotime, Haimen, China), according to the
manufacturer's instructions. After lentivirus infection, the cells
were seeded in 6-well plates and incubated at 37°C. Cell
proliferation was assessed based on the absorbance measured at 450
nm using a multiwell spectrophotometer (Eppendorff, Hamburg,
Germany).
Wound healing assay
After lentivirus infection, 786-O cells were seeded
at 5×105 cells/well in 6-well plates and cultured in
RPMI-1640 medium supplemented with 10% FBS for ~24 h to near
confluence. The cell monolayer was scraped in a straight line using
a 20 μl pipette tip to create a scratch, and the medium was changed
to remove detached cells. Images were captured at 0, 6, 12 and 24 h
after scratching and analyzed using the Image J program to
calculate cell migration distance. Cells from six representative
fields were counted.
Flow cytometry analysis of apoptosis
Apoptosis was evaluated using annexin V/propidium
iodide (PI; BD-Biosciences, San Jose, CA, USA) staining followed by
flow cytometry analysis. After lentivirus infection, the cells were
plated in 6-well plates at a density of 1×106
cells/well, cultured at 37°C in a 5% CO2 incubator for
three days, then gently trypsinized and washed with ice-cold PBS.
The cells were resuspended in 500 μl 1X binding buffer and stained
with annexin V and PI. The samples were subjected to flow cytometry
analysis within 1 h using a flow cytometer (BD LSRII;
BD-Biosciences) and the data were analyzed using BD FACS Diva
software (BD-Biosciences).
Microarray analysis
Total RNA was extracted from 786-O cells using
TRIzol (Invitrogen), according to the manufacturer's instructions,
and purified with an RNeasy Mini kit (Qiagen, Mississauga, Ontario,
ON, Canada). The integrity of the purified RNA was examined using
agarose gel electrophoresis, and the quality and quantity of
purified RNA were assessed using an Agilent 2100 Bioanalyzer RNA
6000 NanoChip (Agilent, Palo Alto, CA, USA). Only RNA samples with
an A260/A280 between 1.7 and 2.2 were used in further experiments.
Total RNA (5 μg) was used to generate cDNA, which was labeled with
the NimbleGen one-color DNA labeling kit (Roche NimbleGen, Madison,
WI, USA). The labeled cDNA was hybridized in a NimbleGen human gene
expression 12×135 K microarray (Roche NimbleGen, Madison, WI, USA),
according to the manufacturer's instructions. The washed arrays
were spin-dried and scanned using the Genepix 4000B scanner (Axon
Instruments, Union City, CA, USA).
Microarray data analysis
Images were extracted and processed using NimbleScan
v2.4 software and analyzed using the NimbleGen software (both Roche
NimbleGen). The lognormal-normal model was employed in order to
estimate the expression of each gene. Genes were ranked according
to this value and the results were filtered further according to
the magnitude of the change in expression; only genes that were at
least 2-fold upregulated or downregulated were considered. Gene
Ontology (GO) analysis for the differentially expressed genes was
performed by using DAVID (http://david.abcc.ncifcrf.gov/. Accessed April 18,
2013), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
were analyzed using the Ingenuity Pathway Analysis software package
(IPA; Ingenuity Systems, Redwood City, CA, USA).
Statistical analysis
Values were represented as the means ± SD for at
least triplicate determination, and analyzed using one-way ANOVA
and an LSD test. All statistical analyses were performed using SPSS
13.0, where P<0.05 was considered to indicate a statistically
significant difference.
Results
Evaluation of CLU knockdown in 786-O
cells
To verify that the CLU-RNAi lentivirus efficiently
knocked down CLU in 786-O cells, we performed real-time PCR
analysis to detect CLU mRNA levels in 786-O cells transduced by a
different CLU-RNAi lentivirus and a negative control lentivirus.
The results demonstrated that no. 3 CLU RNAi was most the efficient
at reducing the CLU mRNA levels (data not shown). We performed
western blot analysis to detect the CLU protein levels in the 786-O
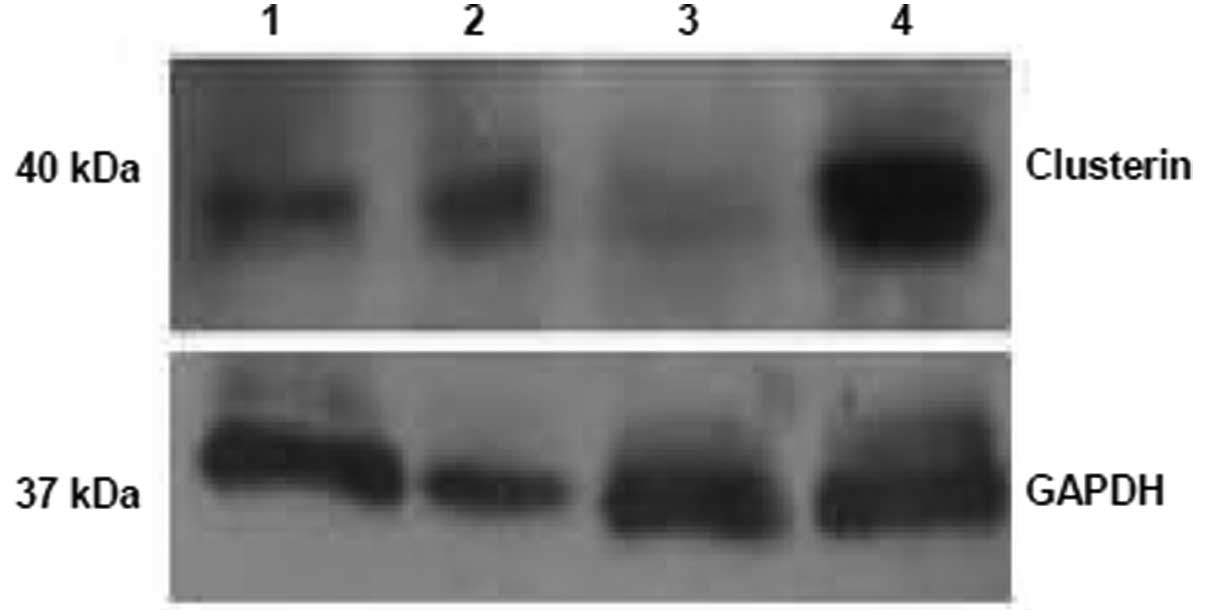
cells transduced by a different lentivirus. The results
demonstrated that no. 3 CLU RNAi was the most efficient at reducing
the CLU protein levels (Fig. 1),
consistent with real-time PCR results. Therefore, we chose no. 3
CLU RNAi to knockdown CLU in 786-O cells in subsequent
experiments.
CLU knockdown inhibits the proliferation
of RCC cells
To investigate whether CLU regulates the
proliferation of RCC cells, 786-O cells were infected with either
an si-CLU lentivirus or a control lentivirus, and cell
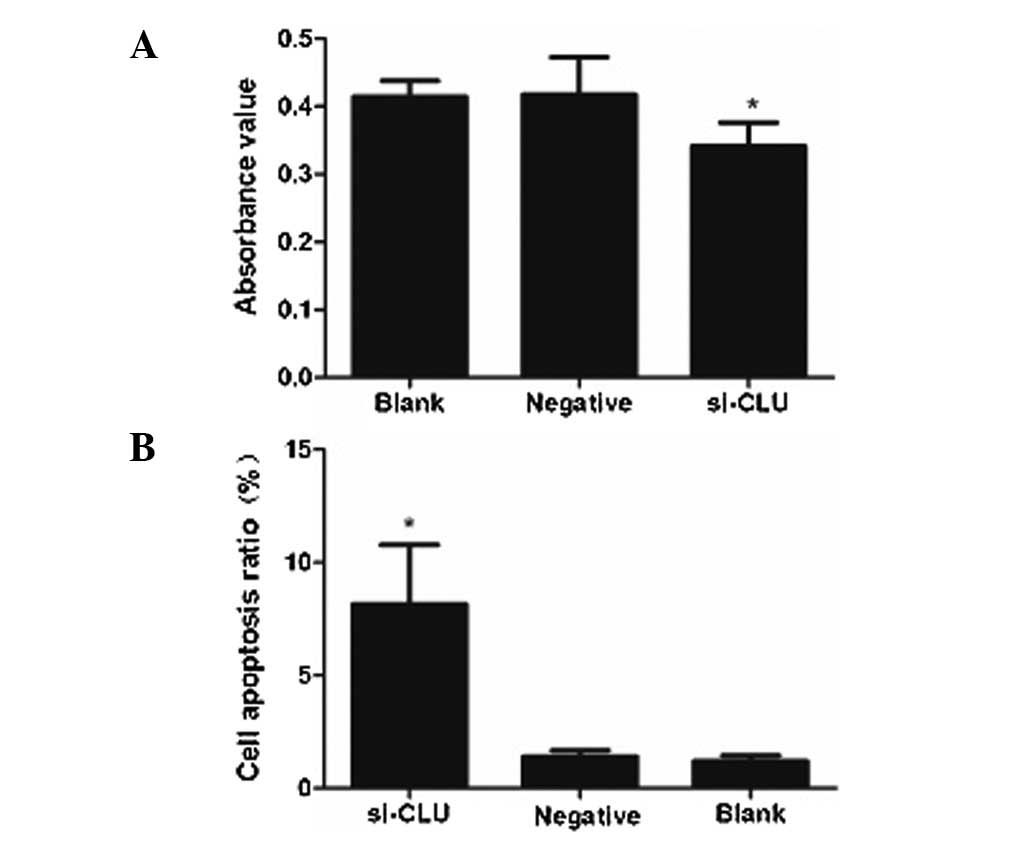
proliferation was evaluated with a WST-1 assay. The results
revealed that CLU knockdown reduced the proliferation of 786-O
cells over the 72 h period (Fig.
2A).
The effect of CLU knockdown on apoptosis
in RCC cells
Flow cytometry analysis revealed that the apoptotic
ratio was 6.30±3.17% in 786-O cells infected by the si-CLU
lentivirus, which was significantly lower than that in cells
infected by the negative control lentivirus (1.20±0.40%) or
uninfected cells (1.01±0.37%; Fig.
2B). These results suggest that CLU plays an anti-apoptotic
role to promote the proliferation of RCC cells.
CLU knockdown inhibits the migration of
RCC cells
To investigate whether CLU regulates the migration
of RCC cells, an important behavior involved in RCC metastasis,
786-O cells were infected with either an si-CLU lentivirus or a
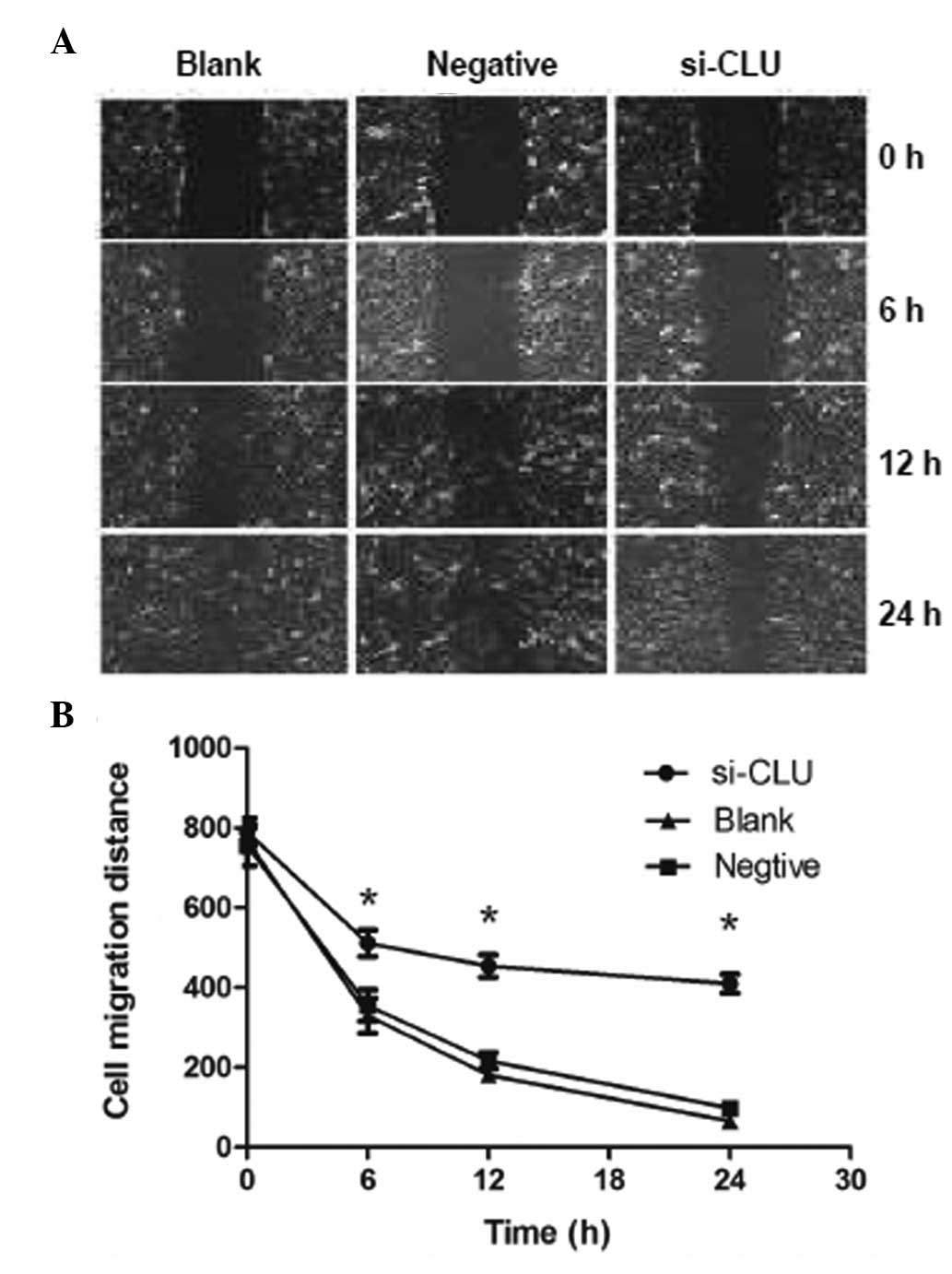
control lentivirus, and cell migration was evaluated using a wound
healing assay. We observed that CLU knockdown reduced the migration
of 786-O cells at 12 and 24 h after scratches were created
(Fig. 3), suggesting that CLU
promotes the migration and invasion of RCC cells.
CLU knockdown leads to differential gene
expression in RCC cells
To identify gene regulation networks that contribute
to the various biological behaviors of 786-O cells upon CLU
knockdown, we performed microarray analysis to compare the gene
expression profiling in 786-O cells infected by si-CLU lentivirus
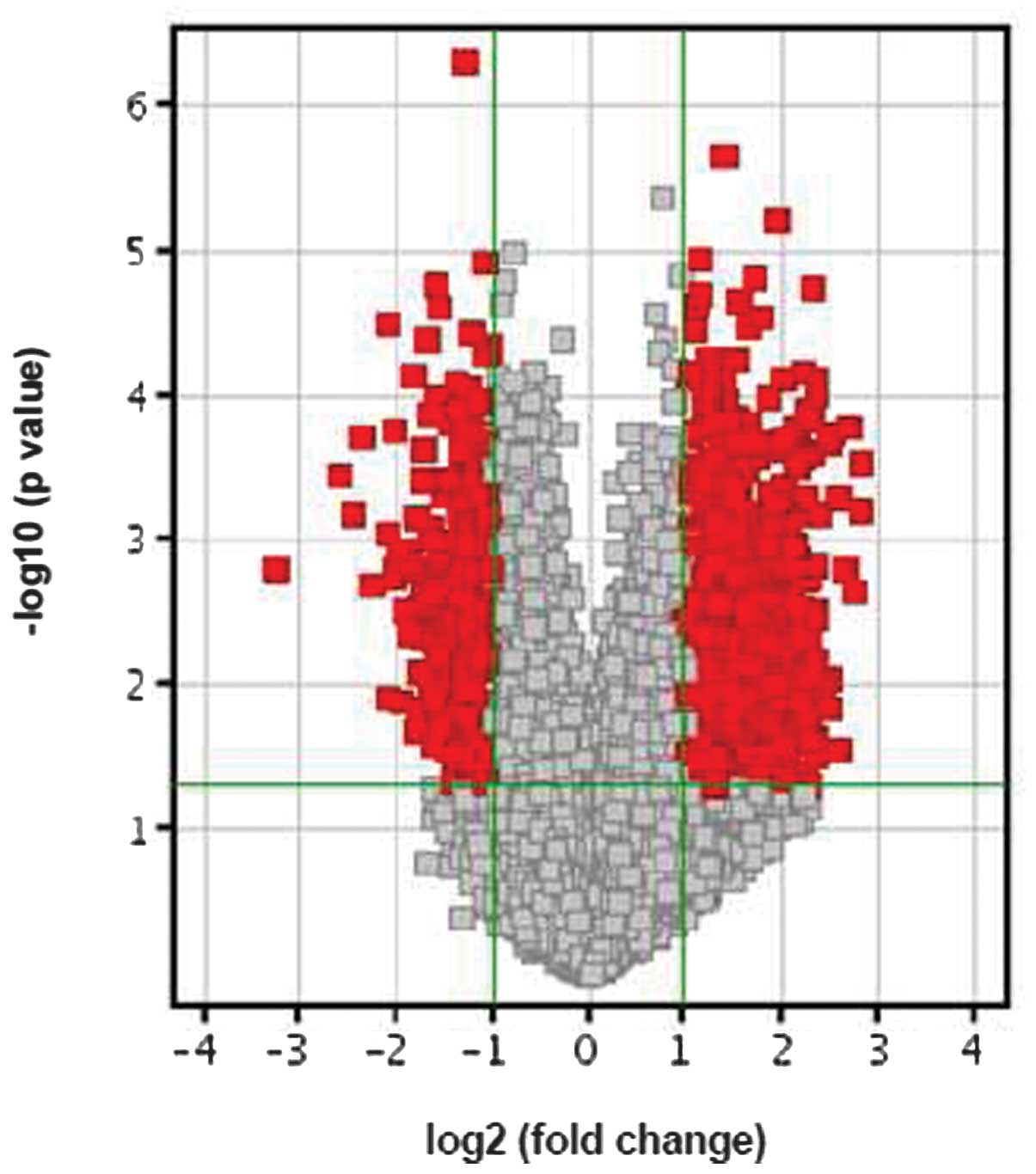
vs. cells infected by a negative control lentivirus. Notably, 588
genes showed significant changes in expression between the 786-O
cells infected with an si-CLU lentivirus and the control cells
(P<0.01), with 356 genes upregulated and 232 downregulated
(Fig. 4). These differentially
expressed genes were distributed in almost all chromosomes, with
the exception of the Y chromosome, but were enriched in chromosome
1 (9.97%), chromosome 2 (7.58%), chromosome 6 (5.92%), chromosome
11 (8.1%), chromosome 14 (5.4%) and chromosome 19 (5.92%).
We performed GO and KEGG pathway analyses to
classify the differentially expressed genes in the 786-O cells
after CLU knockdown. The results demonstrated that 17 pathways were
upregulated and 12 were downregulated (Table I).
 | Table IBiological pathways of differentially
expressed genes in clusterin (CLU) knockdown 786-O cells. |
Table I
Biological pathways of differentially
expressed genes in clusterin (CLU) knockdown 786-O cells.
| Pathways | Genes |
|---|
| Upregulated |
| Viral
myocarditis | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G, ITGB2, MYH13 |
| Hematopoietic cell
lineage | CD7, GP1BA, GP9,
HLA-DRB1, HLA-DRB1, HLA-DRB3, IL5RA, IL9R, ITGAM, TFRC |
| Graft-versus-host
disease | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G |
| Basal cell
carcinoma | DVL1, PTCH2, WNT1,
WNT2, WNT3, WNT3A |
| Type 1 diabetes
mellitus | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G |
| Leishmaniasis | FCGR2C, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, ITGAM, ITGB2, NCF1 |
| Staphylococcus
aureus infection | FCGR2C, FPRL2,
HLA-DPB1, HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, ITGAM, ITGB2 |
| Hedgehog
signaling | BMP8B, PRKACG, PTCH2,
WNT1, WNT2, WNT3, WNT3A |
| Autoimmune thyroid
disease | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G, IFNA4 |
| Allograft
rejection | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G |
| Phagosome | CLEC4M, COMP, FCGR2C,
HLA-A, HLA-DPB1, HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G,
ITGAM, ITGB2, NCF1, TFRC, TUBB2B |
| Antigen processing
and presentation | HLA-A, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HLA-G, HSPA6, KIR2DL4 |
| Intestinal immune
network for Ig A | CCL25, HLA-DPB1,
HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, MADCAM1 |
| ECM-receptor
interaction | COL11A2, COMP, GP1BA,
GP9, ITGB4, LAMA5, LAMC3, SDC3 |
| Cytokine-receptor
interaction | BLR1, CCL1, CCL25,
CCL4L2, CCL4L2, CLC, EDA, IFNA4, IL17B, IL5RA, IL9R, PF4, TNFRSF25,
TNFRSF6B, TNFSF14, XCL1 |
| Toxoplasmosis | AKT1, BIRC4,
HLA-DPB1, HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3, HSPA6, LAMA5,
LAMC3, PLA2G2F |
| HTLV-I
infection | AKT1, ATM, BIRC4,
DVL1, HLA-A, HLA-DPB1, HLA-DQB1, HLA-DRB1, HLA-DRB1, HLA-DRB3,
HLA-G, ITGB2, PRKACG, WNT1, WNT2, WNT3, WNT3A |
| Downregulated |
| Focal adhesion | AKT3, ARHGAP5, EGFR,
FN1, ITGA5, ITGB3, PARVA, PDGFC, PDPK1, PIK3CD, PRKCA, RAP1B,
VEGF |
| Carbohydrate
digestion and absorption | AKT3, ATP1A1, ATP1A1,
HK2, PIK3CD |
| Small cell lung
cancer | AKT3, CDK6, FN1,
PIAS2, PIK3CD, RB1, RXRA |
| Glioma | AKT3, CDK6, EGFR,
MDM2, PIK3CD, PRKCA, RB1 |
| Prostate cancer | AKT3, CREB3L2, EGFR,
MDM2, PDGFC, PDPK1, PIK3CD, RB1 |
| mTOR signaling
pathway | AKT3, DDIT4, PDPK1,
PIK3CD, RICTOR, RPS6KA2, VEGF |
| MAPK signaling
pathway | AKT3, ATF2, CASP3,
DUSP5, EGFR, FGF5, IL1R1, MAP3K7IP2, MAP3K8, MAPK14, MAPKAPK2, NF1,
PRKACB, PRKCA, RAP1B, RAPGEF2, RPS6KA2 |
| Melanoma | AKT3, CDK6, EGFR,
FGF5, MDM2, MITF, PDGFC, PIK3CD, RB1 |
| Non-small cell lung
cancer | AKT3, CDK6, EGFR,
PDPK1, PIK3CD, PRKCA, RB1, RXRA |
| Chronic myeloid
leukemia | AKT3, CBLB, CDK6,
MDM2, PIK3CD, RB1, RUNX1 |
| Aldosterone
regulated sodium reabsorption | ATP1A1, ATP1A1,
PDPK1, PIK3CD, PRKCA |
| Pathways in
cancer | AKT3, AXIN2, CASP3,
CBLB, CCDC6, CDK6, CUL2, EGFR, EGLN1, FGF5, FN1, GLI2, KITLG, MDM2,
MITF, PIAS2, PIK3CD, PRKCA, RB1, RUNX1, RXRA, VEGF, WNT5A |
Discussion
CLU is overexpressed in a variety of tumors and
promotes tumorigenesis. Furthermore, 3-CLU has been proposed as a
prognostic marker for RCC (10). A
recent study reported that an antisense oligodeoxynucleotide
targeting clusterin exhibited antitumor activity in an RCC model
(11). These studies suggest that
CLU plays an oncogenic role in RCC. Consistent with this theory, in
the present study, we employed a loss-of-function approach to
knockdown CLU in the 786-O cell line and observed that CLU
knockdown induces apoptosis and inhibits the proliferation and
migration of 786-O cells. These results provide further evidence
for the oncogenic role of CLU in RCC. However, the detailed
molecular mechanisms by which CLU promotes RCC development remain
largely unknown.
Cancer development is known to be a multi-step
process involving sequential changes in a variety of genes and
cellular pathways (12).
Microarray techniques have been applied widely in cancer research
due to their advantages in revealing the dynamics of gene
expression and gene regulation networks from a global perspective,
which contributes to our understanding of cancer initiation,
progression and metastasis (9,13,14).
In this study, we used a NimbleGen microarray to
screen the differentially expressed genes in 786-O cells after CLU
knockdown vs. the parental 786-O cells. We revealed that 356 genes
were upregulated and 232 were downregulated. Although these
differentially expressed genes were distributed in almost all
chromosomes, with the exception of the Y chromosome, they were
relatively enriched in chromosomes 1, 2, 6, 11 and 14, consistent
with previous molecular genetics studies on RCC. For example,
Beroukhim et al(15)
identified 7 regions of deletion (1p, 3p, 4q, 6q, 8p, 9p and 14q)
in hereditary and sporadic clear-cell RCC. Moreover, Monzon et
al(16) identified deletions
in chromosomes 1, 5, 6, 9, 13 and 14 in renal cancer patients.
Furthermore, we classified the differentially
expressed genes into different biological pathways in order to
characterize their functional role in RCC. As expected,
approximately half of the downregulated pathways are
cancer-related, including the PI3K/Akt, MAPK and VEGF pathways,
known to promote cancer cell proliferation, survival and tumor
angiogenesis and metastasis. After CLU knockdown, the
downregulation of these pathways may have contributed to the
observed inhibition of 786-O cell proliferation and migration.
Notably, we identified that a number of pathways involved in
immunity and infection were upregulated. Future studies exploring
the correlation between RCC development and immunological function
may shed new light on the functional role of CLU in
tumorigenesis.
In conclusion, this study presents evidence that CLU
acts as an oncogene in RCC by promoting cancer cell proliferation
and migration and inhibiting cancer cell apoptosis. We identified
differentially expressed genes after CLU knockdown and classified
them according to their related biological pathways. Our findings
provide a platform for further characterization of the individual
genes implicated in RCC development, which may provide new insights
into the oncogenic role of CLU.
Acknowledgements
This work was supported by the grants from the
Science and Technology Fund of Guangdong Province (no.
2009B030801053) and the Science and Technology Fund of Guangzhou
City (no. 2009Z1-E381-02).
References
|
1
|
Patel C, Ahmed A and Ellsworth P: Renal
cell carcinoma: a reappraisal. Urol Nurs. 32:182–190.
2012.PubMed/NCBI
|
|
2
|
Cohen HT and McGovern FJ: Renal cell
carcinoma. N Engl J Med. 353:2477–2490. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosenberg ME and Silkensen J: Clusterin:
physiologic and pathophysiologic considerations. Int J Biochem Cell
Biol. 27:633–645. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shannan B, Seifert M, Leskov K, et al:
Challenge and promise: roles for clusterin in pathogenesis,
progression and therapy of cancer. Cell Death Differ. 13:12–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Busam KJ, Kucukgol D, Eastlake-Wade S, et
al: Clusterin expression in primary and metastatic melanoma. J
Cutan Pathol. 33:619–623. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bi J, Guo AL, Lai YR, et al:
Overexpression of clusterin correlates with tumor progression,
metastasis in gastric cancer: a study on tissue microarrays.
Neoplasma. 57:191–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miyake H, Gleave ME, Arakawa S, et al:
Introducing the clusterin gene into human renal cell carcinoma
cells enhances their metastatic potential. J Urol. 167:2203–2208.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei L, Xue T, Wang J, et al: Roles of
clusterin in progression, chemoresistance and metastasis of human
ovarian cancer. Int J Cancer. 125:791–806. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai J and Hu S: Transcriptome network
analysis reveals potential candidate genes for squamous lung
cancer. Int J Mol Med. 29:95–101. 2012.PubMed/NCBI
|
|
10
|
Sakai I, Miyake H, Takenaka A and Fujisawa
M: Expression of potential molecular markers in renal cell
carcinoma: impact on clinicopathological outcomes in patients
undergoing radical nephrectomy. BJU Int. 104:942–946. 2009.
View Article : Google Scholar
|
|
11
|
Kususda Y, Miyake H, Gleave ME and
Fujisawa M: Clusterin inhibition using OGX-011 synergistically
enhances antitumour activity of sorafenib in a human renal cell
carcinoma model. Br J Cancer. 106:1945–1952. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaikhibrahim Z, Lindstrot A, Langer B,
Buettner R and Wernert N: Comprehensive gene expression microarray
analysis of Ets-1 blockade in PC3 prostate cancer cells and
correlations with prostate cancer tissues: Insights into genes
involved in the metastatic cascade. Int J Mol Med. 27:811–819.
2011.
|
|
14
|
Tabuchi Y, Wada S, Furusawa Y, Ohtsuka K
and Kondo T: Gene networks related to the cell death elicited by
hyperthermia in human oral squamous cell carcinoma HSC-3 cells. Int
J Mol Med. 29:380–386. 2012.PubMed/NCBI
|
|
15
|
Beroukhim R, Brunet JP, Di Napoli A, et
al: Patterns of gene expression and copy-number alterations in
von-hippel lindau disease-associated and sporadic clear cell
carcinoma of the kidney. Cancer Res. 69:4674–4681. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Monzon FA, Alvarez K, Gatalica Z, et al:
Detection of chromosomal aberrations in renal tumors: a comparative
study of conventional cytogenetics and virtual karyotyping with
single-nucleotide polymorphism microarrays. Arch Pathol Lab Med.
133:1917–1922. 2009.
|