Introduction
Ovarian cancer is the most malignant type of
gynecological cancer. There are 225,000 new cases worldwide every
year, and 140,000 females succumb to this disease each year
(1). Most patients are diagnosed
in advanced stages, since the anatomical position of the ovary is
deep within the abdomen. Treatment of ovarian cancer includes
cytoreductive surgery and chemotherapy based on a combination of
paclitaxel and cisplatin. More than 80% of patients respond to
initial therapy, although some patients with advanced stages
develop recrudescent ovarian cancer that is resistant to
platinum-based chemotherapies (2,3).
Therefore, it is important to develop more effective therapies for
treating ovarian cancer.
Acetyl-CoA carboxylase (ACC) is a rate-limiting,
biotin-dependent enzyme involved in fatty acid synthesis. ACC
catalyzes the carboxylation of cytosolic acetyl-CoA to malonyl-CoA.
This is the first step in the fatty acid synthesis pathway that is
controlled by a balance between active and less active forms of
ACC. Malonyl-CoA is a dual-functioning compound that donates
two-carbon units to fatty acid synthesis and controls fatty acid
oxidation in the cytoplasm by inhibiting acylcarnitine transfer to
mitochondria. Adjustment of malonyl-CoA levels ensures that these
two processes do not occur simultaneously. Eukaryotic ACC contains
biotin carboxylase (BC), biotin carboxyl carrier protein (BCCP) and
carboxyltransferase (CT) (4–6).
ACC has two isoforms, ACCA (ACC-α) and ACCB (ACC-β),
which are encoded by different genes. In mammals, ACCA is enriched
in the liver, adipose tissue and mammary glands, since they are
abundant in fatty acids. ACCA is located at the 17q21 locus, while
ACCB is located at the 12q24 locus. ACCB is expressed in skeletal
muscles and the heart. ACCA catalyzes fatty acid synthesis, while
ACCB controls fatty acid oxidation progression (7,8). ACC
activity is tightly regulated by reversible phosphorylation,
modulation of gene expression, and metabolite-mediated allosteric
mechanisms. Several critical ACCA phosphorylation sites have been
identified (Ser79, Ser1200 and
Ser1215) and they are phosphorylated by adenosine
monophosphate-activated protein kinase (AMPK) (9,10).
Notably, ACCA is overexpressed in many types of human cancer, such
as liver and breast tumors in which lipogenesis is highly activated
(10–12). To date, there have been no studies
on ACCA expression in ovarian cancer tissues. However, an
increasing body of evidence has shown that fatty acid synthesis is
required for cancer cell proliferation and survival (13). When ACCA expression is
downregulated by small interfering RNA (siRNA), prostate, breast
and colon cancer cell proliferation and fatty acid synthesis are
immediately inhibited, and apoptosis is induced (13–15).
These findings suggest that the regulation of ACCA plays an
important role in cell growth and apoptosis, and that ACCA may be a
potential target in cancer therapy.
5-Tetradecyloxy-2-furoic acid (TOFA) is a
cell-permeable small molecule that is an allosteric inhibitor of
ACCA. TOFA reduces the availability of endogenous fatty acids
required for the generation of phosphatidylcholine, the major
phospholipid of cell membranes (16–19).
TOFA has been reported to be cytotoxic and to induce apoptosis in
the lung cancer cell line NCI-460, the colon cancer cell lines
HCT-8 and HCT-15, and the prostate cancer cell line LNCaP. However,
the effect of TOFA on ovarian cancer cells has yet to be elucidated
(15,20). Therefore, the aim of the present
study was to investigate the effects of TOFA on the proliferation
and cell cycle progression of the ovarian cancer cell lines COC1
and COC1/DDP.
Materials and methods
Cell culture
The human ovarian cancer cell lines COC1 and
COC1/DDP were purchased from the China Center for Type Culture
Collection (CCTCC; Wuhan, China) and were cultured in RPMI-1640
medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal calf
serum (FCS; Hyclone) and penicillin/streptomycin (1:100, Hyclone)
in a 5% CO2 air-humidified atmosphere at 37°C.
Cis-platinum (CDDP, 0.5 μg/ml dissolved in RPMI-1640; Qilu, Inc.,
Shandong, China) was added to the culture medium of COC1/DDP
cells.
Cell proliferation assay
TOFA (Sigma, St. Louis, MO, USA) was dissolved in
dimethylsulphoxide (DMSO) to make a 50 mg/ml stock solution. The
cells (1×104 cells/well) were seeded in 96-well plates
and incubated with TOFA at various concentrations (0, 1, 5, 10, 20
and 50 μg/ml). Cell proliferation was assessed 24, 48 and 72 h
following TOFA treatment by measuring the reduction of the
tetrazolium salt WST-8 to formazan using the Cell Counting kit-8
(CCK-8) purchased from Dojindo (Kumamoto, Japan) according to the
manufacturer’s instructions. At each time point, 10 μl of the CCK-8
solution was added to each well and cultured at 37°C for 2 h. The
supernatant from each plate was collected and the absorbance was
measured at 450 nm. The inhibition rate of cell proliferation and
the 50% inhibitory concentration (IC50; calculated by
nonlinear regression) was determined according to the following
equation: Inhibition rate (%) = [(Ac−Ae)/(Ac−Ab)] × 100%, where Ae,
absorbance of the culture media in experimental wells; Ac,
absorbance of the culture medium in control wells and Ab,
absorbance of the culture medium in blank wells (21).
Cell cycle analysis
COC1 and COC1/DDP cells were seeded in 6-well plates
at a density of 2×105 cells/well and treated with or
without TOFA (5 or 10 μg/ml) for 24, 48 and 72 h. For cell cycle
analysis, the cells were centrifuged at 466 × g for 5 min and
washed twice with cold phosphate-buffered saline (PBS). The cells
were fixed with 70% ice-cold ethanol and stored at −20°C. The cells
were washed with PBS 24 h later, treated with RNase A and incubated
at 37°C for 30 min according to the manufacturer’s protocol.
Propidium iodide (PI) was added (400 μl of a 20-μg/ml solution),
the cells were incubated in the dark for 30 min and the absorbance
was measured at 488 nm.
Apoptosis assay
Ovarian cancer cells COC1 and COC1/DDP
(2×105 cells/well) were treated with TOFA (0, 1, 5, 10,
20 and 50 μg/ml) for 24, 48 and 72 h. Subsequently, an apoptosis
detection kit (Becton-Dickinson, San Diego, CA, USA) was used
according to the manufacturer’s instructions. The cells were
collected by centrifugation at 466 × g for 5 min, washed twice with
cold PBS and suspended in 100 μl binding buffer. Annexin V-FITC (5
μl) and PI (5 μl) were added to the culture media of cells and the
cells were incubated for 15 min at room temperature in the dark.
Binding buffer (400 μl) was added and fluorescent intensities were
determined by flow cytometry at 488 nm (FACSCalibur;
Becton-Dickinson).
Western blot analysis
The cells were washed with cold PBS and harvested by
scraping into 50 μl cell lysis solution (Merck, Darmstadt,
Germany). The cell lysates were incubated on ice for 5 min. After
centrifugation at 15,000 × g for 5 min at 4°C, supernatants were
collected and protein concentration was determined using the
Bradford assay (Roche, Indianapolis, IN, USA). The proteins were
subjected to electrophoresis on SDS-polyacrylamide gels and then
transferred to nitrocellulose membranes. The membranes were blocked
in 5% (w/v) bovine serum albumin (BSA) containing 0.1% (v/v)
Tween-20 (TBST). The membranes were then probed with primary
antibodies in TBST containing 5% BSA overnight at 4°C.
Subsequently, the membranes were incubated with horseradish
peroxidase (HRP)-linked secondary antibodies at room temperature
for 1 h and washed with TBST thrice. The signals were detected by a
chemiluminescence phototope-HRP kit according to the manufacturer’s
instructions (Millipore, Boston, MA, USA).
COC1/DDP tumor xenografts in mice
Female athymic BALB/c nude mice, 4–5 weeks old,
weighing 17–20 g were purchased from the Chinese Academy of
Sciences (Beijing, China). The study was approved by the ethics
committee of School of Medicine, Shanghai Jiao Tong University
Shanghai, China. COC1/DDP cells were collected by centrifugation at
300 × g for 5 min and suspended in RPMI-1640 medium at a density of
2×106 cells/100 μl. The cells were subcutaneously
injected into both right and left flanks of each mouse. Twenty days
later, mice were randomly allocated into one of the 2 groups (n=5
mice/group): i) mice treated with 50 μl DMSO (control group) or ii)
mice treated with TOFA (50 mg/kg). The drugs were injected
intraperitoneally daily for two weeks. Tumor volumes were recorded
every two days by measuring dimensions (length and width) with
Vernier calipers. The mice were sacrificed four weeks after the
final treatment. Tumor weights were measured by a scale. The
formula used to determine tumor sizes was A × B2 ×
0.5236 (A, length; B, width; all measured in mm) (22). For histopathological examination,
tumors and multiple organs were fixed in 10% neutral-buffered
formalin and wax embedded. The tissues were cut into 4-mm sections
and stained with hematoxylin and eosin (H&E).
Statistical analysis
SPSS 18.0 software was used for statistical
analysis. Statistical analyses between the control and treatment
groups were performed using t-tests. P≤0.05 was considered to
indicate a statistically significant difference.
Results
TOFA suppresses ovarian cancer cell
proliferation
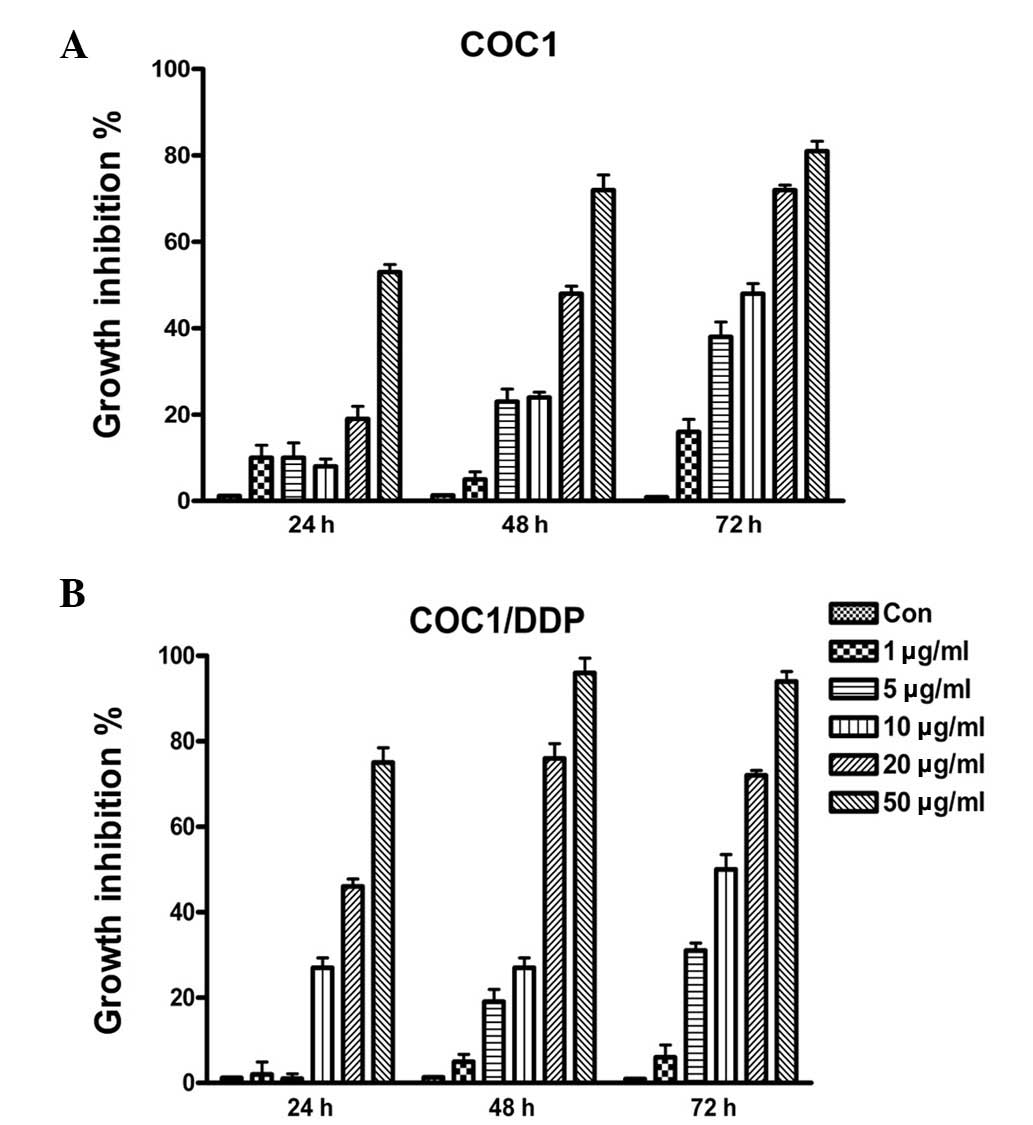
To investigate the effect of TOFA on ovarian cancer
cells, COC1 and COC1/DDP cell lines were treated with various
concentrations of TOFA (1–50 μg/ml) for 24–72 h, and cell
proliferation was assessed. The various concentrations of TOFA used
were found to inhibit COC1 and COC1/DDP cell growth in a
concentration- and time-dependent manner (Fig. 1). TOFA was also shown to be highly
cytotoxic to ovarian cancer cells. The IC50 values of
COC1 and COC1/DDP cells for TOFA were ~26.1 and ~11.2 μg/ml,
respectively, after 48 h of treatment. These data indicate that
fatty acid synthesis plays an important role in the proliferation
of the ovarian cancer cell lines COC1 and COC1/DDP.
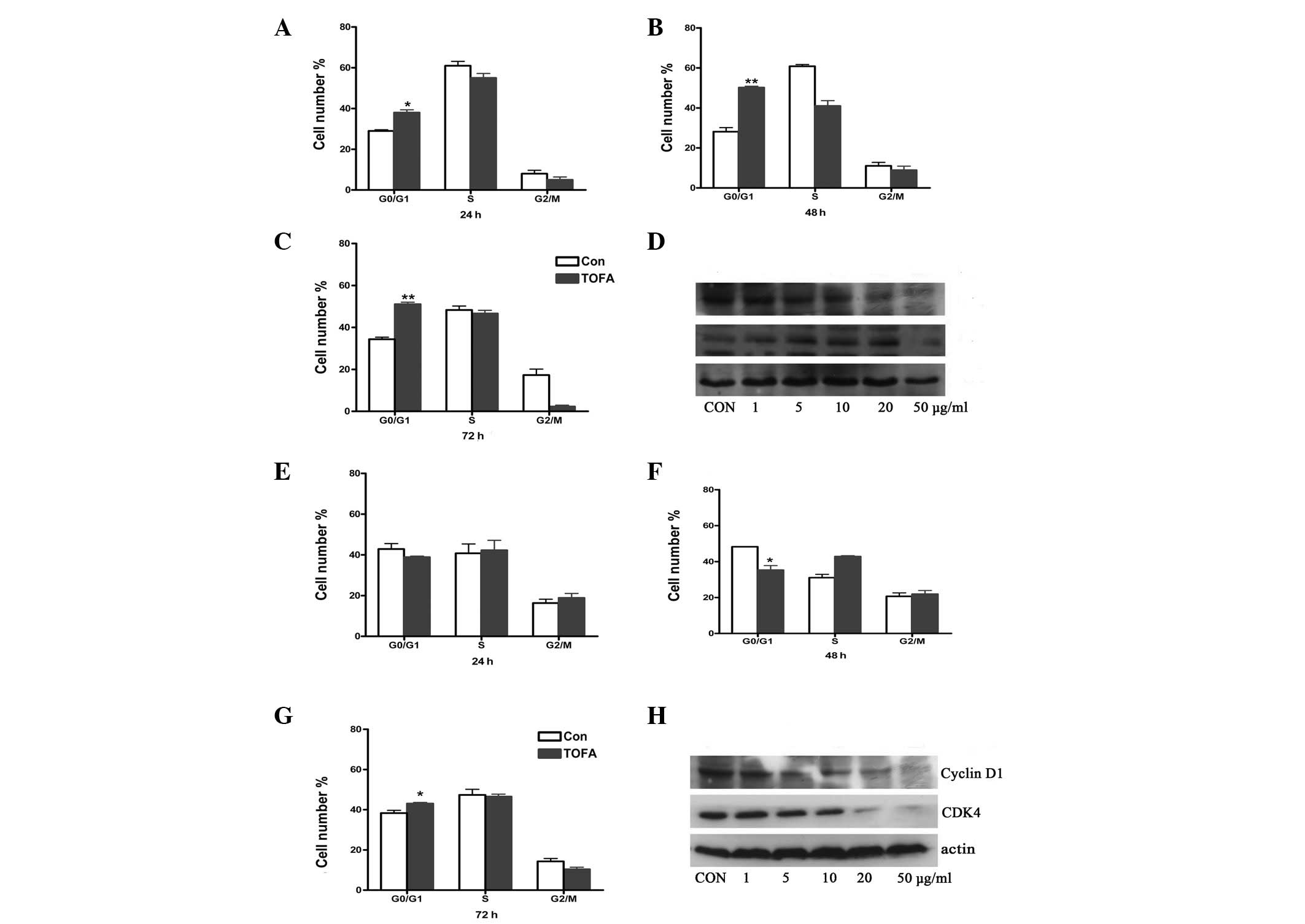
TOFA induces cell cycle arrest
To investigate whether TOFA induced cell cycle
arrest in ovarian cancer cells, COC1 and COC1/DDP cells were
cultured with TOFA for 24, 48 and 72 h, followed by cell counting
using FACScan analysis. COC1 cells treated with TOFA were arrested
in the G0/G1 phase. Particularly, treatment with 5 μg/ml TOFA for
48 and 72 h increased the percentage of COC1 cells in this phase to
50.2 and 51.1%, compared with the percentage of the untreated
control groups, 28.1 and 34.4% (P<0.01), respectively (Fig. 2B and C). The treatment of COC1/DDP
cells with TOFA (10 μg/ml) for 72 h increased the percentage of
cells in the G0/G1 phase from 38.3 (control) to 43.0% (P<0.05;
Fig. 2G). These results indicate
that cell cycle progression from the G1 to the S phase is
associated with fatty acid synthesis. To investigate the mechanism
underlying cell cycle arrest, the protein levels of cyclin D1 and
cyclin-dependent kinase (CDK) 4, two proteins that regulate
progression from the G1 to the S phase, were examined using western
blot analysis (Fig. 2D and H).
TOFA treatment of COC1 and COC1/DDP cells decreased cyclin D1
protein levels in a dose-dependent manner. However, CDK4 protein
levels were increased in TOFA-treated COC1 cells (1–20 μg/ml),
followed by a subsequent decrease when 50 μg/ml TOFA was used
(Fig. 2D).
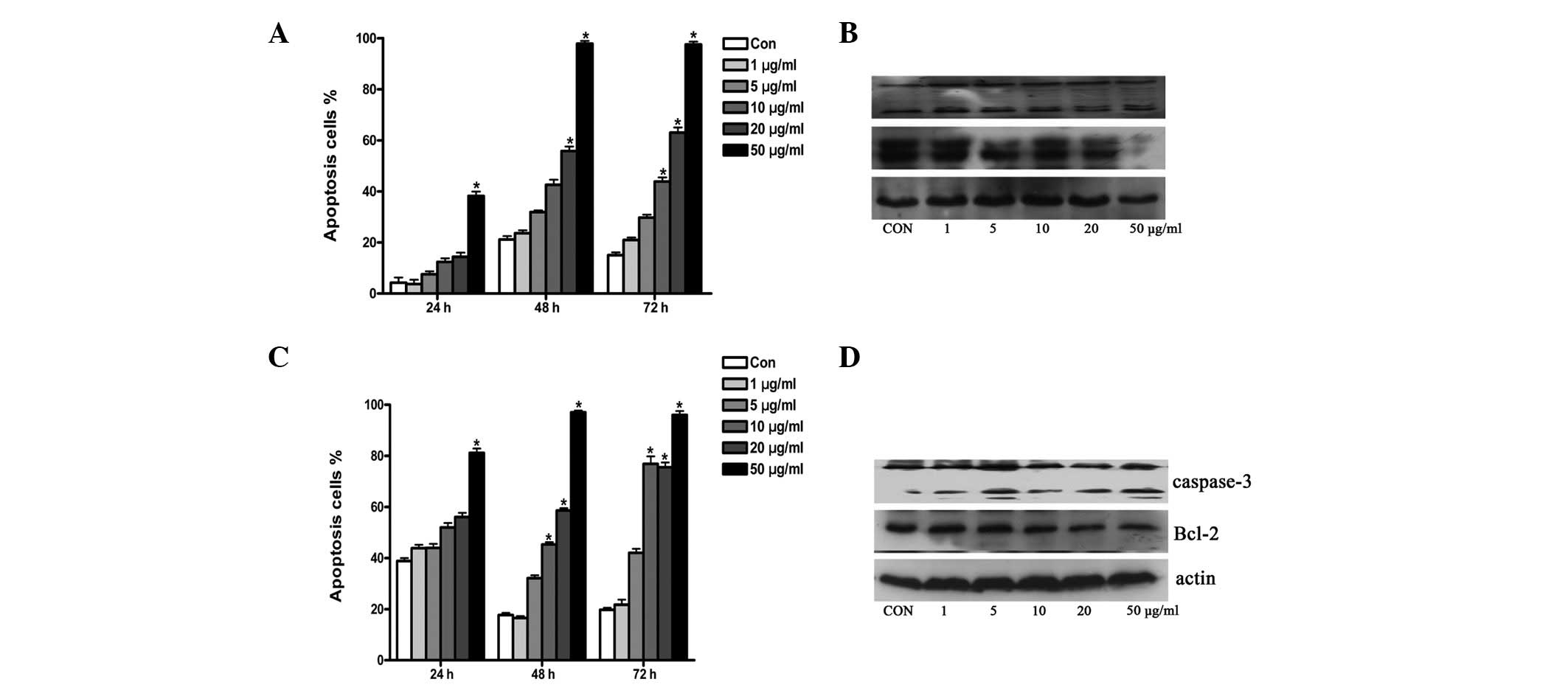
TOFA induces apoptosis
TOFA induces apoptosis in human lung and colon
cancer cells by inhibiting fatty acid synthesis and inducing
poly(ADP-ribose) polymerase (PARP) cleavage and DNA fragmentation
(15). To investigate the
underlying mechanism of the cytotoxic effect of TOFA on ovarian
cancer cells, the ovarian cancer cell lines COC1 and COC1/DDP were
treated with TOFA as previously described. Apoptosis was detected
by Annexin V staining and FACScan analysis. Western blot analysis
was also used to investigate the expression of the
apoptosis-related proteins caspase-3 and Bcl-2. Low concentrations
of TOFA were found to induce apoptosis in COC1 and COC1/DDP cells.
The ratio of apoptosis to cell proliferation was 12.4, 42.6 and
43.9% in COC1 cells treated with 10 μg/ml TOFA for 24, 48 and 72 h,
respectively (Fig. 3A). The ratio
of apoptosis to cell proliferation was 32.2 and 42.1% for COC1/DDP
cells treated with 5 μg/ml TOFA for 48 and 72 h, respectively
(Fig. 3C). Caspase-3 was cleaved
and activated while Bcl-2 expression decreased following treatment
with TOFA of both COC1 and COC1/DDP cells (Fig. 3B and D).
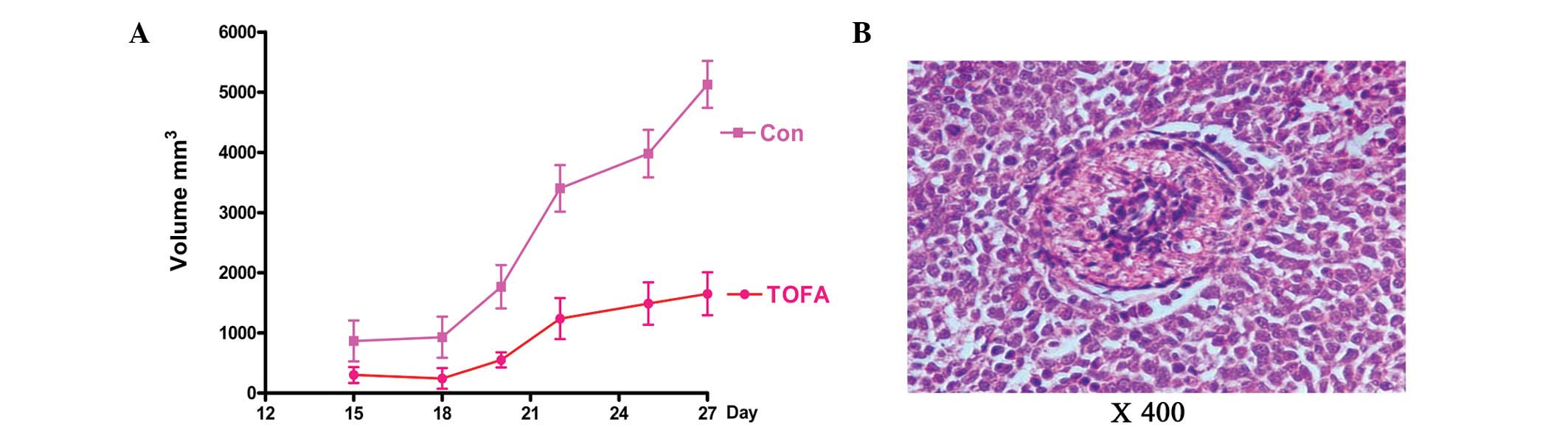
TOFA inhibits growth of ovarian cancer
xenografts in mice
To investigate whether TOFA suppresses tumor growth
in vivo, the effects of TOFA were investigated using a human
ovarian cancer mouse xenograft model. COC1/DDP cells were injected
into female nude mice, and tumor sizes were measured following TOFA
treatment. The tumor growth rate was significantly inhibited by
TOFA compared with the DMSO-treated control mice (1649±356.3 vs.
5128±390.4 mm3; Fig.
4A). To test for TOFA toxicity, the effect of TOFA was examined
on multiple mouse organs with H&E staining (Fig. 4B). No toxicity was observed in the
heart, liver, spleen, lung, kidney and intestinal tissues (data not
shown).
Discussion
Several studies have suggested that fatty acid
synthesis is important in the cellular proliferation of many types
of cancer. Long-chain fatty acids are the building blocks of
biomembranes, and ~95% of saturated and monosaturated fatty acids
constitute nutrients for cancer cell growth. Thus, fatty acid
synthesis plays an important role in carcinogenesis (23). ACCA is a key enzyme in fatty acid
synthesis. ACCA RNAi reduces the pool of palmitic acid, and ACCA
gene expression is upregulated in many cancer tissues. High ACCA
expression has been associated with poor prognoses in patients
(13–15). Therefore, fatty acid synthesis may
be a potential target for cancer therapy.
TOFA is a specific ACCA inhibitor and it increases
fatty acid oxidation and ketogenesis by exerting allosteric
inhibition on ACCA (17,18). According to Wang et
al(15), TOFA was shown to be
cytotoxic to human lung and colon cancer cells via inhibition of
the fatty acid synthesis pathway. The present study showed that
TOFA exerts a highly cytotoxic effect on ovarian cancer COC1 and
COC1/DDP cell proliferation in a dose- and time-dependent manner.
TOFA was also shown to arrest cancer cells in the G1 phase as well
as to induce apoptosis. These results are in agreement with similar
findings in the lung cancer cell line NCI-H460 and the colon cancer
cell lines HCT-8 and HCT-15.
To investigate whether TOFA suppresses tumor
proliferation in vivo, we examined the effect of TOFA using
a human ovarian cancer mouse xenograft model (24). The results showed that the tumor
growth rate was significantly inhibited by TOFA when compared with
the control mice, and no toxicity was observed in the histological
samples of several important organs.
The present study showed that ovarian cancer cell
growth was effectively inhibited in vitro when TOFA was used
at concentrations of 1–50 μg/ml (Fig.
1). The number of cells in the cell cycle phases was counted
using FACScan analysis. When the cells were treated with 10 μg/ml
TOFA for 72 h, the percentage of cells arrested in the G0/G1 phase
was higher compared with that of the control cells (Fig. 2). Therefore, we hypothesized that
TOFA suppresses ovarian cancer cell proliferation by arresting
cells in the G0/G1 phase. Cyclin D1 is an integral regulator of the
G1 phase and is overexpressed in several types of cancer, such as
ovarian, lung and breast cancer (25). Cyclin 1 may be closely associated
with invasive ovarian cancer, although no impact on clinical
outcomes has been observed. CDK proteins, a family of
serine/threonine kinases, tightly regulate the cell cycle at
multiple points. Activation of the CDK protein by binding to its
cyclin partner controls cell cycle progression. Overexpression of
CDK4 is often associated with increased cyclin D1 expression
(25). Growth stimulators induce
cyclin D1 expression and the formation of a cyclin D1-CDK4 complex,
leading to G1 to S phase transition. Cyclin D1/CDK4 complex plays
an important role in maintaining normal cell division (26). Ray et al(27) confirmed that the BRCA1/ACC complex
regulates the cell cycle by phosphorylating the Ser1263
residue of ACC (27). In the
present study, cyclin D1 expression was shown to be decreased by
exposure to TOFA and the cells were found to be arrested in the G1
phase. Therefore, the inhibition or regulation of ACCA activity
constitutes a potential mechanism that could suppress cancer cell
proliferation.
According to a previous study (24), TOFA was shown to have no cytotoxic
effect on the ovarian cancer cell line SKOV-3, after a 24-h
treatment. Moreover, TOFA treatment did not lead to AMPK
phosphorylation (24). However,
according to the results of the present study, TOFA induced
apoptosis in the ovarian cancer cell lines COC1 and COC1/DDP, as
measured by FACScan analysis. The cells were exposed to TOFA (10
μg/ml) for 72 h, and the percentage of apoptotic cells was higher
compared with that of the untreated cells. Moreover, we observed
that caspase-3 was cleaved and activated, and that Bcl-2 protein
levels were slightly decreased after TOFA treatment of both COC1
and COC1/DDP cells. Consequently, the apoptosis pathway is
suggested to constitute one of the mechanisms underlying TOFA
action in inducing cell death.
To the best of our knowledge, this is the first
study indicating that the ACCA inhibitor TOFA is a suppressor of
ovarian cancer cell proliferation in vitro and in
vivo. TOFA was shown to arrest the cells in the G0/G1 phase as
well as to induce apoptosis. Therefore, the inhibition of ACCA
activity is suggested to be a potential target of future cancer
therapies.
Acknowledgements
This work was supported by grants from the Shanghai
Health Bureau Key Disciplines and Specialties Foundation, Shanghai
Education Commission Key Disciplines Foundation, Key Discipline
Project of Ren Ji Hospital, Shanghai Jiao Tong University School of
Medicine, the National Natural Science Foundation of China (no.
81072137), and the Funding Scheme for Training Young Teachers in
Colleges and Universities in Shanghai (no. ZZjdyx12066).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Eltabbakh GH and Awtrey CS: Current
treatment for ovarian cancer. Expert Opin Pharmacother. 2:109–124.
2001. View Article : Google Scholar
|
|
3
|
Herzog TJ: The current treatment of
recurrent ovarian cancer. Curr Oncol Rep. 8:448–454. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wakil SJ, Stoops JK and Joshi VC: Fatty
acid synthesis and its regulation. Annu Rev Biochem. 52:537–579.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barber MC, Price NT and Travers MT:
Structure and regulation of acetyl-CoA carboxylase genes of
metazoa. Biochim Biophys Acta. 1733:1–28. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Griffin MJ and Sul HS: Insulin regulation
of fatty acid synthase gene transcription: roles of USF and
SREBP-1c. IUBMB Life. 56:595–600. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hardie DG and Pan DA: Regulation of fatty
acid synthesis and oxidation by the AMP-activated protein kinase.
Biochem Soc Trans. 30(Pt 6): 1064–1070. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abu-Elheiga L, Brinkley WR, Zhong L,
Chirala SS, Woldegiorgis G and Wakil SJ: The subcellular
localization of acetyl-CoA carboxylase. Proc Natl Acad Sci USA.
97:1444–1449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen ZP, McConell GK, Michell BJ, Snow RJ,
Canny BJ and Kemp BE: AMPK signaling in contracting human skeletal
muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am
J Physiol Endocrinol Metab. 279:E1202–E1206. 2000.PubMed/NCBI
|
|
10
|
Milgraum LZ, Witters LA, Pasternack GR and
Kuhajda FP: Enzymes of the fatty acid synthesis pathway are highly
expressed in in situ breast carcinoma. Clin Cancer Res.
3:2115–2120. 1997.PubMed/NCBI
|
|
11
|
Witters LA, Widmer J, King AN, Fassihi K
and Kuhajda F: Identification of human acetyl-CoA carboxylase
isozymes in tissue and in breast cancer cells. Int J Biochem.
26:589–594. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yahagi N, Shimano H, Hasegawa K, Ohashi K,
Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y,
Iizuka Y, Ohashi K, Nagai R, Ishibashi S, Kadowaki T, Makuuchi M,
Ohnishi S, Osuga J and Yamada N: Co-ordinate activation of
lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer.
41:1316–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chajès V, Cambot M, Moreau K, Lenoir GM
and Joulin V: Acetyl-CoA carboxylase alpha is essential to breast
cancer cell survival. Cancer Res. 66:5287–5294. 2006.PubMed/NCBI
|
|
14
|
Brusselmans K, De Schrijver E, Verhoeven G
and Swinnen JV: RNA interference-mediated silencing of the
acetyl-CoA-carboxylase-alpha gene induces growth inhibition and
apoptosis of prostate cancer cells. Cancer Res. 65:6719–6725. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang C, Xu C, Sun M, Luo D, Liao DF and
Cao D: Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human
cancer cell apoptosis. Biochem Biophys Res Commun. 385:302–306.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McCune SA and Harris RA: Mechanism
responsible for 5-(tetradecyloxy)-2-furoic acid inhibition of
hepatic lipogenesis. J Biol Chem. 254:10095–10101. 1979.PubMed/NCBI
|
|
17
|
Tong L and Harwood HJ Jr: Acetyl-coenzyme
A carboxylases: versatile targets for drug discovery. J Cell
Biochem. 99:1476–1488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harwood HJ Jr: Treating the metabolic
syndrome: acetyl-CoA carboxylase inhibition. Expert Opin Ther
Targets. 9:267–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Halvorson DL and McCune SA: Inhibition of
fatty acid synthesis in isolated adipocytes by
5-(tetradecyloxy)-2-furoic acid. Lipids. 19:851–856. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guseva NV, Rokhlin OW, Glover RA and Cohen
MB: TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid
synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and
kills prostate cancer cells independent of p53 status. Cancer Biol
Ther. 12:80–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oda T, Hayano T, Miyaso H, Takahashi N and
Yamashita T: Hsp90 regulates the Fanconi anemia DNA damage response
pathway. Blood. 109:5016–5026. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhong Q, Wen YJ, Yang HS, Luo H, Fu AF,
Yang F, Chen LJ, Chen X, Qi XR, Lin HG, Wan Y, Chen XC, Wei YQ and
Zhao X: Efficient inhibition of cisplatin-resistant human ovarian
cancer growth and prolonged survival by gene transferred vesicular
stomatitis virus matrix protein in nude mice. Ann Oncol.
19:1584–1591. 2008. View Article : Google Scholar
|
|
23
|
Kuhajda FP: Fatty-acid synthase and human
cancer: new perspectives on its role in tumor biology. Nutrition.
16:202–208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou W, Han WF, Landree LE, Thupari JN,
Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, El Meskini
R, Ronnett GV, Townsend CA and Kuhajda FP: Fatty acid synthase
inhibition activates AMP-activated protein kinase in SKOV3 human
ovarian cancer cells. Cancer Res. 67:2964–2971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
D’Andrilli G, Kumar C, Scambia G and
Giordano A: Cell cycle genes in ovarian cancer: steps toward
earlier diagnosis and novel therapies. Clin Cancer Res.
10:8132–8141. 2004.PubMed/NCBI
|
|
26
|
Aggarwal P, Vaites LP, Kim JK, Mellert H,
Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB and
Diehl JA: Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression
and triggers neoplastic growth via activation of the PRMT5
methyltransferase. Cancer Cell. 18:329–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ray H, Suau F, Vincent A and Dalla Venezia
N: Cell cycle regulation of the BRCA1/acetyl-CoA-carboxylase
complex. Biochem Biophys Res Commun. 378:615–619. 2009. View Article : Google Scholar : PubMed/NCBI
|