Introduction
Congenital deafness is one of the most common human
genetic birth defects, occurring in 1 or 2 of every 1,000 births.
Connexins (Cxs) are membrane proteins that form intercellular
channels known as gap junctions (GJs). Six Cx subunits form a
connexon (hemichannel). Two hemichannels in adjacent cells align to
form a complete GJ allowing the exchange of ions and molecules with
a molecular weight <1,000 Da (1). Genetic linkage and mouse genomic
studies have demonstrated that normal functions of Cx26 are
essential for hearing, although the mechanisms underlying deafness
caused by Cx mutations remain unclear. A number of subtypes of Cxs
are reported to be expressed in the mammalian inner ear, with Cx26
being one of the most predominant (2). Previous studies found expression of
Cx26 in the stria vascularis, spiral ligament, spiral limbus and
supporting cells of the human cochlea (3) Among >100 deafness genes identified
thus far, mutations in the gap junction protein Cx26, coded the by
GJB2 gene, account for more than half of hereditary
non-syndromic deafness in humans (4,5).
Loss of Cx26 is hypothesized to prevent recycling of
K+ following sound stimulation, with elevated
K+ in the extracellular perilymph inhibiting uptake of
the neurotransmitter glutamate, which ultimately results in cell
death. Generating Cx26 mutant mouse models has been crucial in
understanding deafness mechanisms. Complete knockout of Cx26 in
mice results in neonatal lethality, preventing examination of its
function in the adult inner ear (6). Cohen-Salmon et al(7) performed targeted ablation of Cx26 in
the epithelial gap junction network in the cochlea using
otogelin-driven Cre expression. In this previous study, deafness in
the mutant mice was reported to be the result of cell death in the
organ of Corti beginning at postnatal day 14 (P14), soon after the
onset of hearing, which is ~P13 in mice. The initial site of cell
death is found near the inner hair cells, consistent with the
K+ accumulation hypothesis. Other conditional Cx26
knockout mouse models have been developed. Kudo et
al(8) established a mouse
model expressing the dominant-negative Cx26 mutant R75W in the
inner ear. Wang et al(9)
generated three different lines of conditional mouse models. Cx26
mutant mice from the Kudo et al and Wang et al
studies developed histological abnormalities prior to P14, which is
in contrast to the initial report from Cohen-Salmon et
al(7).
Since different phenotypes are reported for
different conditional Cx26 knockout mouse models, the pathological
mechanisms underlying deafness caused by Cx26 mutations remain
unclear. Pathological changes in the organ of Corti observed at the
ultrastructural level in Cx26 mutant mice are particularly lacking.
The aim of the current study was to examine and document
ultrastructural pathological changes of cochlear cells in
previously generated Cx26 conditional knockout (cCx26ko) mice
(9).
Materials and methods
cCx26ko mice
The cCx26ko mice were provided by Xi Lin at Emory
University, Atlanta, Georgia. Data presented previously
demonstrated that the hearing of cCx26ko mice is severely impaired
(9). Detailed descriptions of the
hearing of cCx26 mutant mice and light microscopy of the morphology
of their cochlea have been published (9,10).
The following experimental groups of cCx26ko mice were observed in
the current study (two animals/time point): P8, P10, P18, P30, P60,
P90, P120, P180 and one cCx26ko mouse aged 360 days. The control
groups were two littermate-controlled wild-type mice at P10, P18,
P30 and P360. The study protocol was approved by the Institutional
Animal Care and Use Committee of Emory Univerity, Atlanta, GA, USA
(protocol no. 255-2009).
Immunostaining
Cochlear tissue was dissected using microdissecting
tools under a stereomicroscope and fixed in 4% paraformaldehyde in
PBS (pH 7.4) overnight at 4°C. Tissues were embedded in 10% gelatin
dissolved in water for <2 h at room temperature, cut into small
blocks (<3-mm cubes) and dehydrated by submerging in 2.3 M
sucrose solution overnight at 4°C in an Eppendorf tube fixed on an
orbital rotor. Cochlear cryosections of 8 μm were prepared (model
CM1900; Leica Microsystems, Bannockburn, IL, USA). Antibodies
against pillar cell marker P75 (11) (1:200 dilution) and the supporting
cell marker prox1 (12) (1:800
dilution) were obtained from Chemicon (Temecula, CA, USA). Hair
cell markers myosin 6 and phalloidin were labeled with antibodies
from Proteus Bioscience (Ramona, CA, USA) and Sigma-Aldrich (St.
Louis, MO, USA). The secondary antibody used was donkey anti-mouse
conjugated to rhodamine (1:200 dilution, Jackson ImmunoResearch
Lab. Inc., West Grove, PA, USA) or goat anti-rabbit IgG conjugated
to Alexa Fluor 488 (Jackson ImmunoResearch Lab. Inc., West Grove,
PA, USA) 1:500 dilution).
Transmission electron microscopy
The organ of Corti was dissected under a dissecting
microscope and transferred to a rinse solution (0.18 M sucrose in
0.1 PBS, 3 washes). Tissues were immersed in 1% osmium tetroxide
for 2 h. Specimens were dehydrated in increasing alcohol
concentrations (50–100%) and embedded in Epon618. Viewing was by
contrast phase microscopy, where the sample was implanted in the
encasement with the apex of the cochlea upwards and the cochlear
axis parallel with the incisal surface. Solidification was achieved
by drying in an oven overnight. Embedded samples were placed to the
central axis under the anatomical microscope. Semithin sections (1
μm) were prepared with an ultramicrotome (Reichert-Jung, Munich,
Germany). Samples were dried at 70–80°C, stained with toluidine
blue (1%) and observed for cochlear morphology. Ultrathin sections
(50–60 nm) were prepared with an ultramicrotome (Bromma 2088; LKB
Produkter, Ontario, Canada). Samples were stained with
uracyl-acetate and lead-citrate and images were captured with a
Philips CM-120 transmission electron microscope (Philips,
Amsterdam, Holland).
Results
Hair cell and supporting cell markers
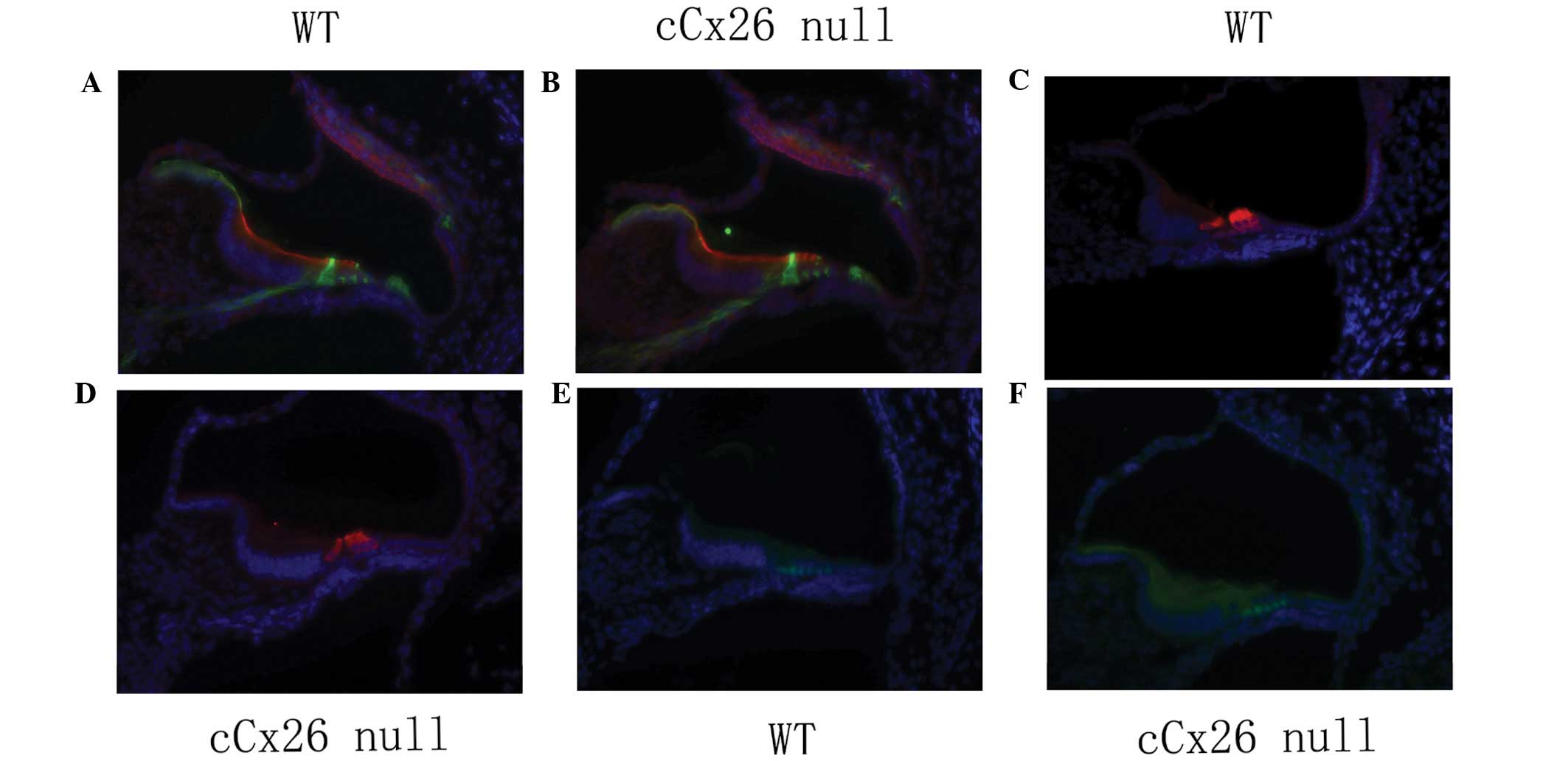
show no marked changes
Cochlear frozen sections at P3 showed that the organ
of Corti and Nuel’s space were not open (Fig. 1A and B)and pillar cells in
cochlear-supporting cells had no marked changes between wild-type
and mutant mice. (Fig. 1A and B).
Prox1 staining of cochlear sertoli cells also showed no marked
changes (Fig. 1E and F). The
cochlear hair cell marker myosin 6 was not different between
cCx26ko and wild-type mice (Fig. 1C
and D).
Tunnel of the organ of Corti and Nuel’s
space does not develop prior to hearing onset in cCx26ko mice
The opening of the tunnel of Corti between the inner
pillar cells and outer pillar cells was observed at P8, and Nuel’s
space formed at P10 in wild-type mice (Fig. 2A). These are important hallmarks in
cochlear development. However, the tunnel of Corti was not formed
at P8 and Nuel’s space was only partly formed at this postnatal
stage in cCx26ko mice (Fig. 2B).
As shown in Fig. 2C, the tunnel
and Nuel’s space had not yet formed at P10 in cCx26ko mice. The
space at the tunnel of Corti and Nuel’s space was occupied by the
processes of Deiter’s cells. At P18, the tunnel of Corti and Nuel’s
space remained immature in cCx26ko mice. The space was filled with
neighboring enlarged supporting cells (Fig. 2D). The changes in microtubules were
examined in this region of the organ of Corti, which is significant
in the opening of the tunnel and the Nuel’s space. The inner and
outer pillar cells showed abundant microtubules in wild-type mice
(Fig. 3A). In cCx26ko mice, the
numbers of microtubules in the inner and outer pillar cells were
reduced following P10 (Fig. 3B)
and were even lower at P18 (Fig.
3C). At P30, the microtubules of the inner pillar cells had
almost disappeared (Fig. 3D).
Thus, the abnormal development of microtubules in pillar cells may
be an underlying factor in the inability to generate the opening of
the intercellular space between the outer and inner pillar
cells.
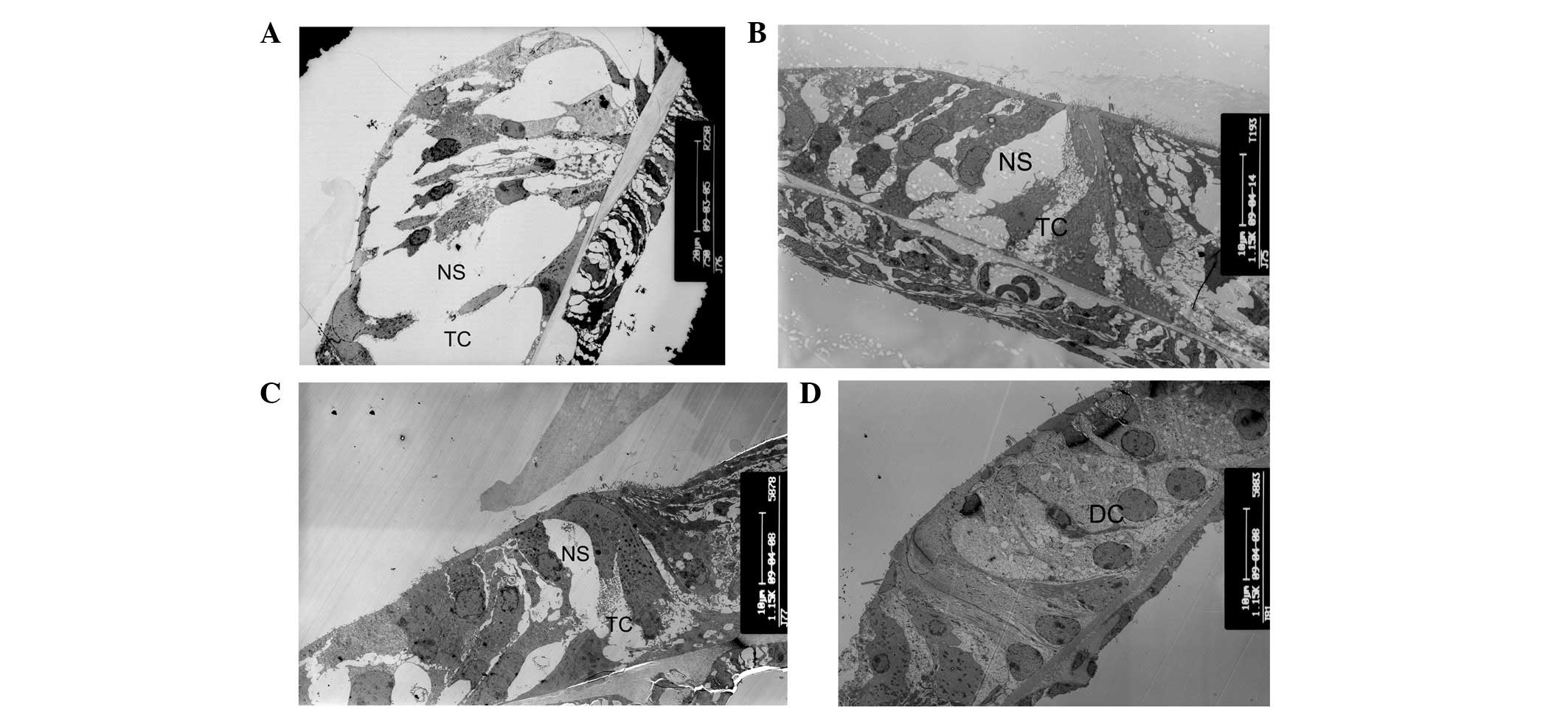 | Figure 2Alterations of tunnel of Corti and
Nuel’s space. (A) Tunnel of Corti and Nuel’s space at postnatal day
10 (P10) in wild-type mice (magnification, ×750; apical turn). (B)
The tunnel of Corti was not formed at P8 and Nuel’s space was
partly formed in cCx26ko mice (magnification, ×1,150; apical turn).
(C) The tunnel and Nuel’s space were not formed at P10 in cCx26ko
mice (magnification, ×1,150; middle turn). (D) The tunnel of Corti
and Nuel’s space at P18 in cCx26ko mice were filled with
neighboring enlarged supporting cells (magnification, ×1,150;
middle turn). TC, Corti tunnel; NS, Nuel’s space; DC, Deiter’s
cell; cCx26ko, conditional connexin 26 knockout. |
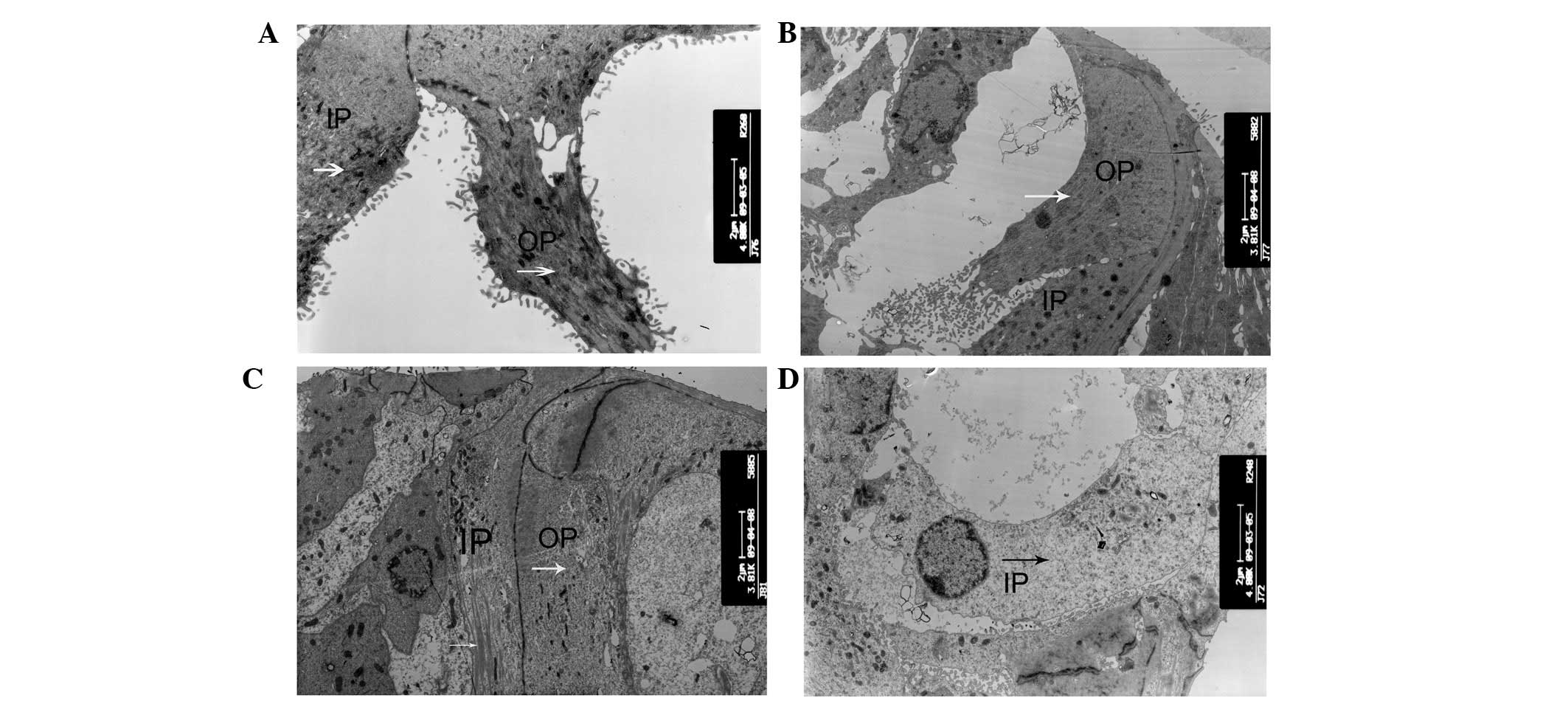 | Figure 3Alterations of microtubules. (A) IPCs
and OPCs showed abundant microtubules in P10 wild-type mice
(magnification, ×4,800; apical turn). (B and C) Numbers of
microtubules in IPCs and OPCs following P10 in cCx26ko mice
(magnification, ×3,800; middle turn). (D) Microtubules of IPCs were
almost absent in P30 cCx26 mice (magnification, ×4,800; middle
turn). IPC, inner pillar cells; OPC, outer pillar cells; arrows,
microtubules; cCx26ko, conditional connexin 26 knockout. |
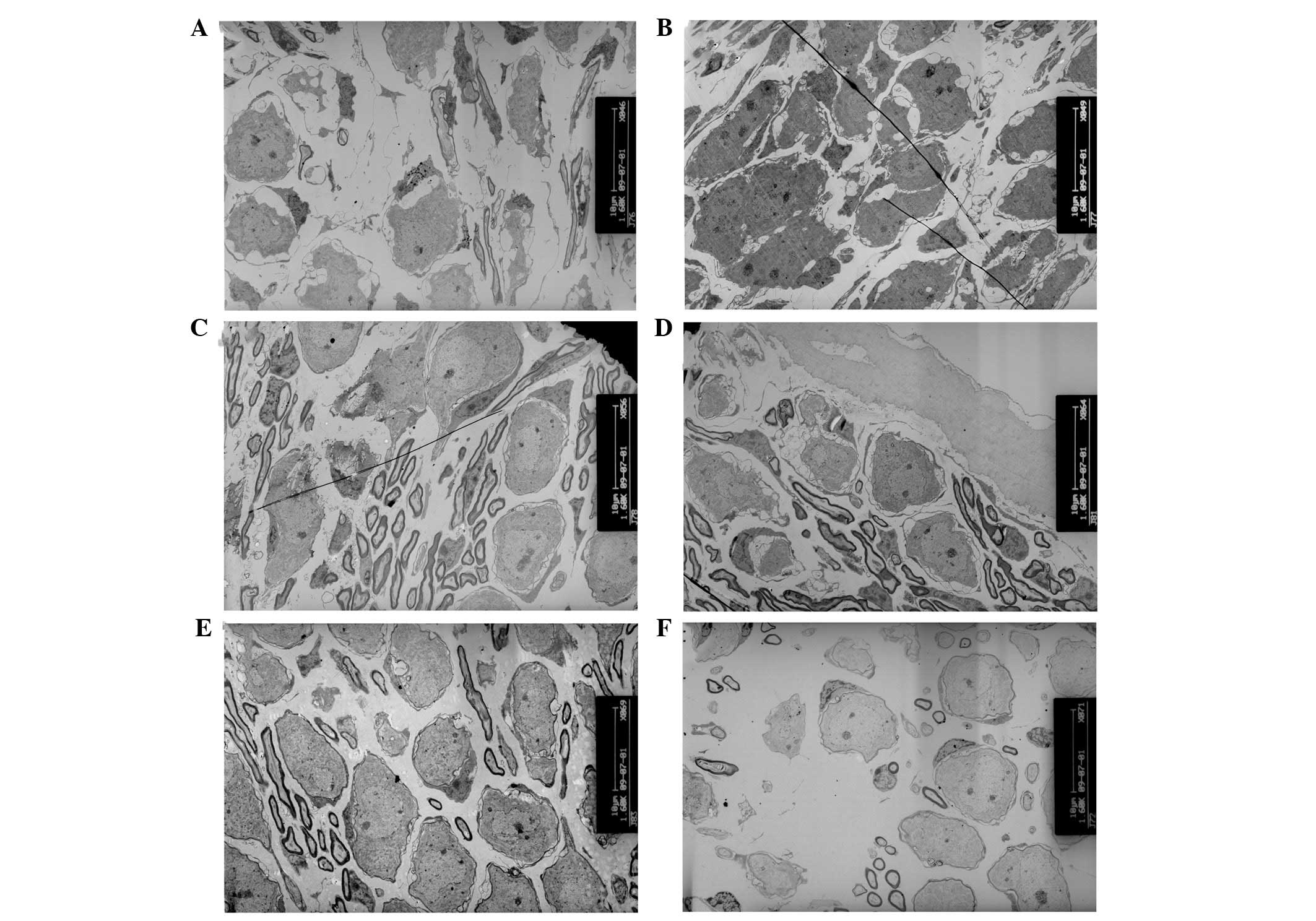
Alterations of the cellular
ultrastructure in the organ of Corti
The degeneration process of hair cells and
supporting cells was systematically examined at various
developmental stages of cCx26ko mice. At P10, only a small number
of vacuoles in the inner hair cells were observed (Fig. 4A) and the cell shape was intact. By
contrast, at the same developmental stage, the majority of the
outer hair cells appeared cuboid with an enlarged cytoplasm and a
remaining intercellular gap (Fig. 5A
and B). This change was first observed in the Claudius cells at
the middle turn. At P8, the Claudius cells of Cx26 knockout mice
appeared normal (Fig. 6A). In the
Claudius cells at P10, the mitochondria of wild-type mice were
dense and robust (Fig. 6B), while
the mitochondria of cCx26ko mice appeared to be enlarged (Fig. 6C). Notably, the Hensen cells of
cCx26ko mice had more cytoplasm compared with wild-type mice
(Fig. 7A and B), indicating an
abnormal intracellular metabolism. Following P18, the number of
lysosomes increased and mitochondria of the inner hair cells became
swollen in the cells of cCx26ko mice (Fig. 4B). The Claudius cells began
degenerating, in particular, the cytoplasm became scarce and
scattered (Fig. 6D). The dense
cuticular plate of the outer hair cells was thinner and the
intercellular space had almost disappeared (Fig. 5C). Thinning of dense cuticular
plates in inner hair cells occurred at P30 (Fig. 4C and D). At this stage, the outer
hair cells became cuboid with unclear cell boundaries (Fig. 5D). The cytoplasm of Hensen cells
became scarce (Fig. 7C). In
addition, fewer mitochondria were observed in Claudius cells in
knockout mice compared with wild-type mice (Fig. 6E).
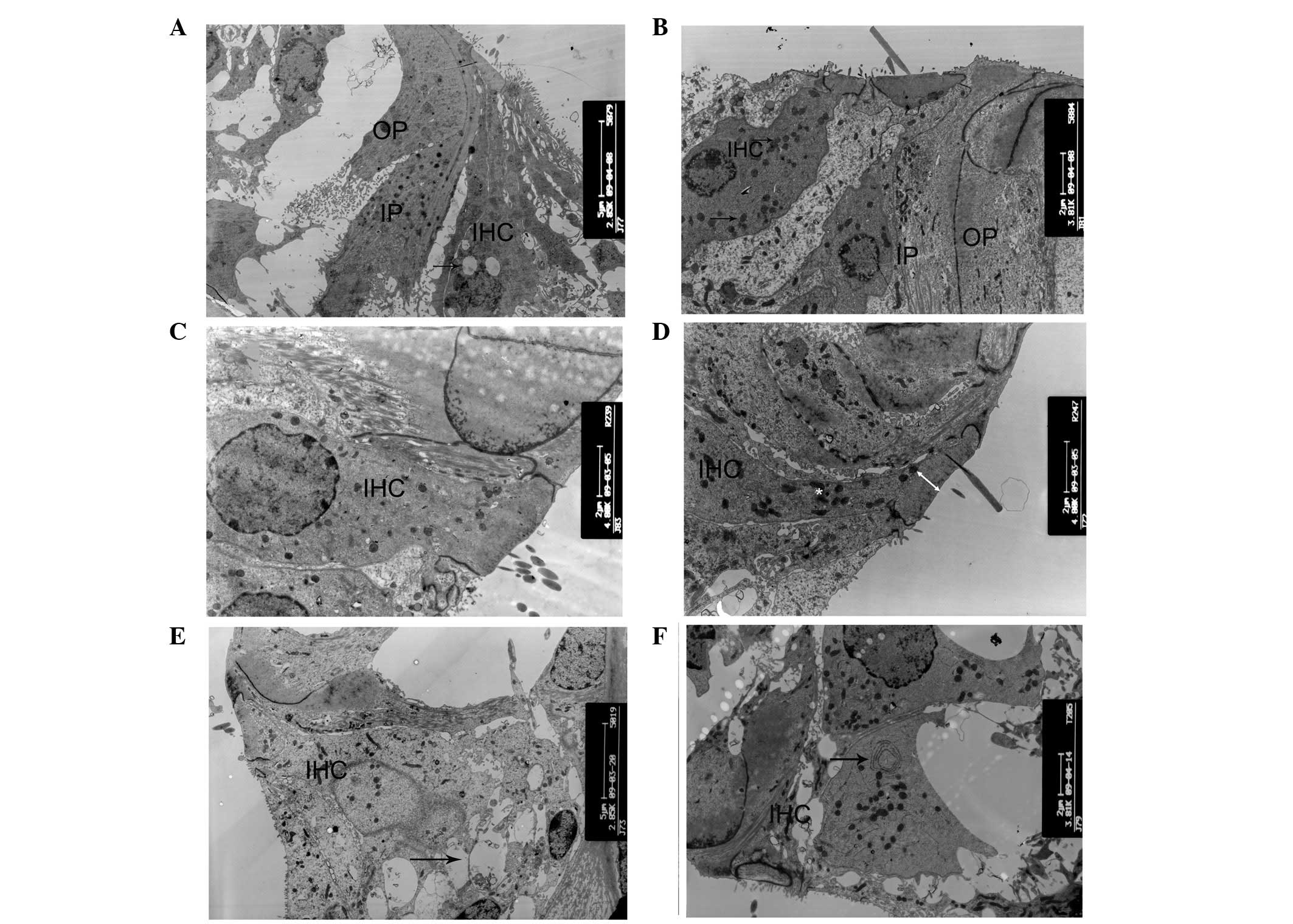 | Figure 4Ultrastructural changes of inner
cells. (A) Small number of vacuoles in inner hair cells and intact
cell shape in P10 cCx26ko mice (magnification, ×2,850; middle
turn). Arrow, vacuoles. (B) Increased number of lysosomes and
swollen mitochondria in inner hair cells in P18 cCx26ko mice
(magnification, ×3,800; middle turn). Arrows, mitochondria. (C and
D) Thinning of dense cuticular plate in inner hair cells at P30 in
cCx26ko mice compared with P30 wild-type mice (magnification,
×4,800; middle turn). Arrows, dense cuticular plate. (E and F)
Deformed inner hair cells of the basal turn with (E) empty spaces,
arrows, from degenerated cells in P60 cCx26ko mice (magnification,
×2,800; basal turn). (F) Arrows, mitochondria with myelin in inner
hair cells of P90 cCx26ko mice (magnification, ×2,800, apical
turn). IPC, inner pillar cells; OPC, outer pillar cells; IHC, inner
hair cells; cCx26ko, conditional connexin 26 knockout. |
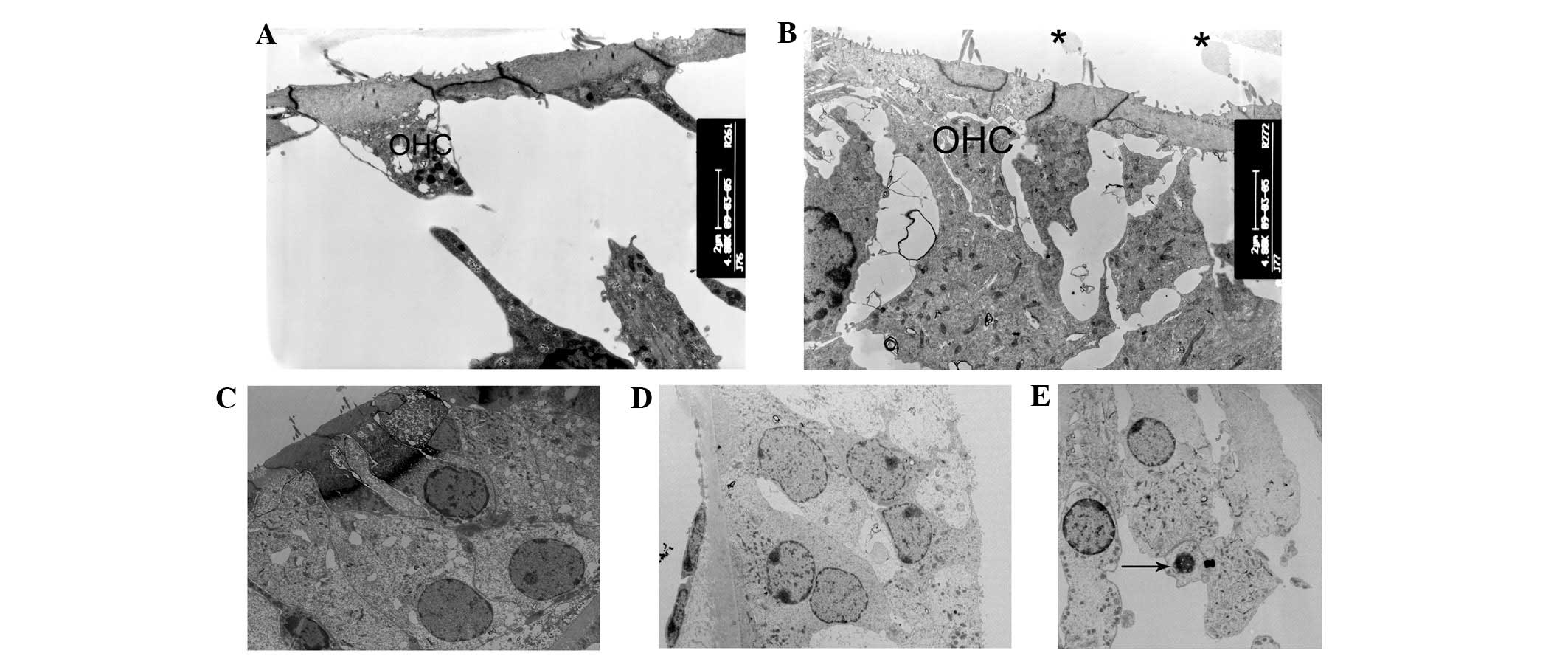 | Figure 5Ultrastructural changes of the outer
hair cells regions. (A and B) At postnatal day 10 (P10), the
majority of outer hair cells appeared cuboid with enlarged
cytoplasm in cCx26ko mice compared with wild-type mice
(magnification, ×4,800; middle turn). (C and D) Following P18,
outer hair cells became cuboid with unclear cell boundaries and
little intercellular space (magnification, ×1,680; middle turn).
(E) At P60, nuclei of outer hair cells showed pycknosis and cells
were severely degenerated (magnification, ×3,800; basal turn).
Arrows, degenerated cells; OHC, outer hair cells; cCx26ko,
conditional connexin 26 knockout. |
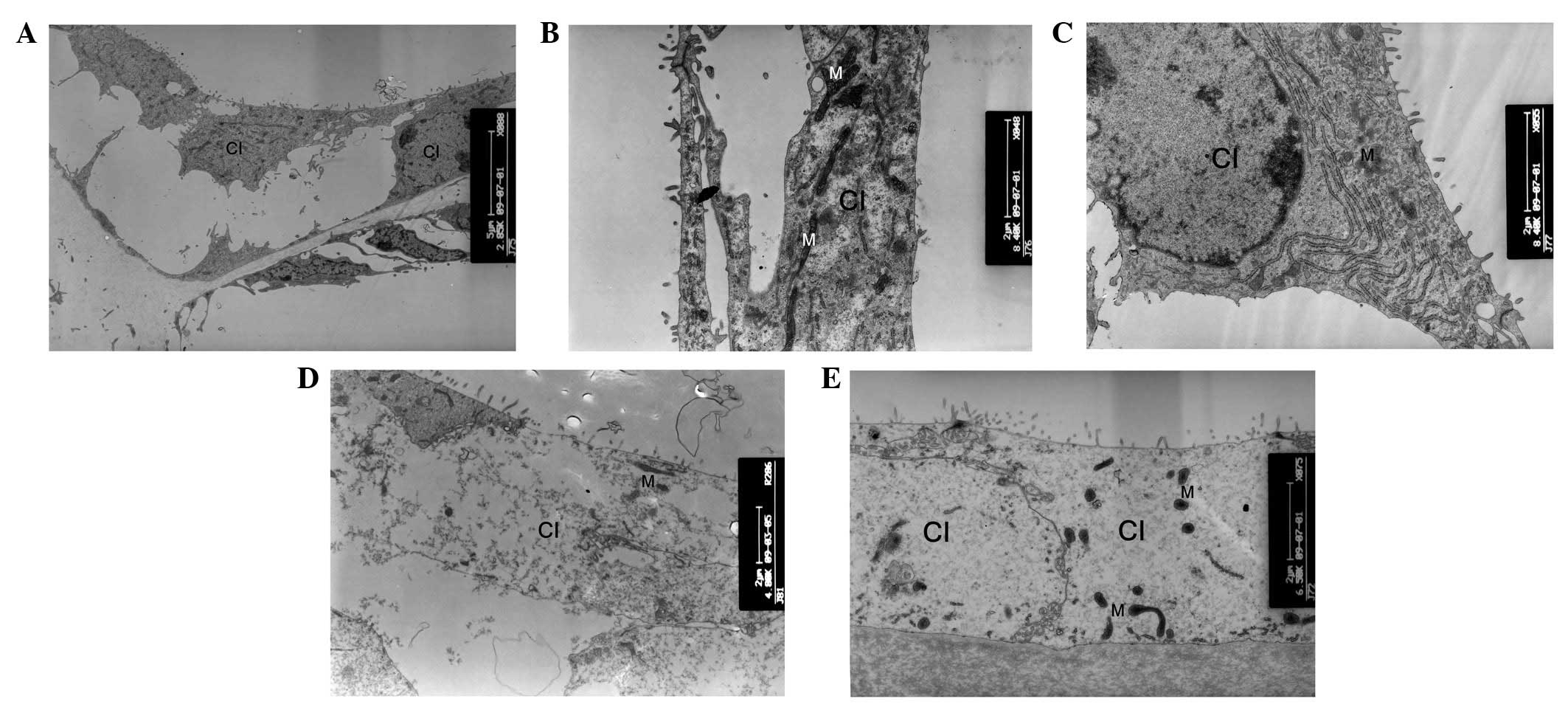 | Figure 6Ultrastructural changes of Claudius
cells. (A) At postnatal day 8 (P8), the Claudius cells of cCx26ko
mice appeared normal (magnification, ×2,850; apical turn). At P10,
(B) mitochondria of wild-type mice were dense and robust
(magnification, ×8,400; apical turn) and (C) mitochondria of
cCx26ko mice were enlarged (magnification, ×8,400; middle turn).
Following P18, (D) Claudius cells degenerated and (E) cytoplasm
became scarce and scattered (magnification, ×4,800; middle turn).
Fewer mitochondria were observed in Claudius cells at P30 in
cCx26ko mice (magnification, ×6,500; middle turn). Cl, Claudius
cells; M, mitochondria; cCx26ko, conditional connexin 26
knockout. |
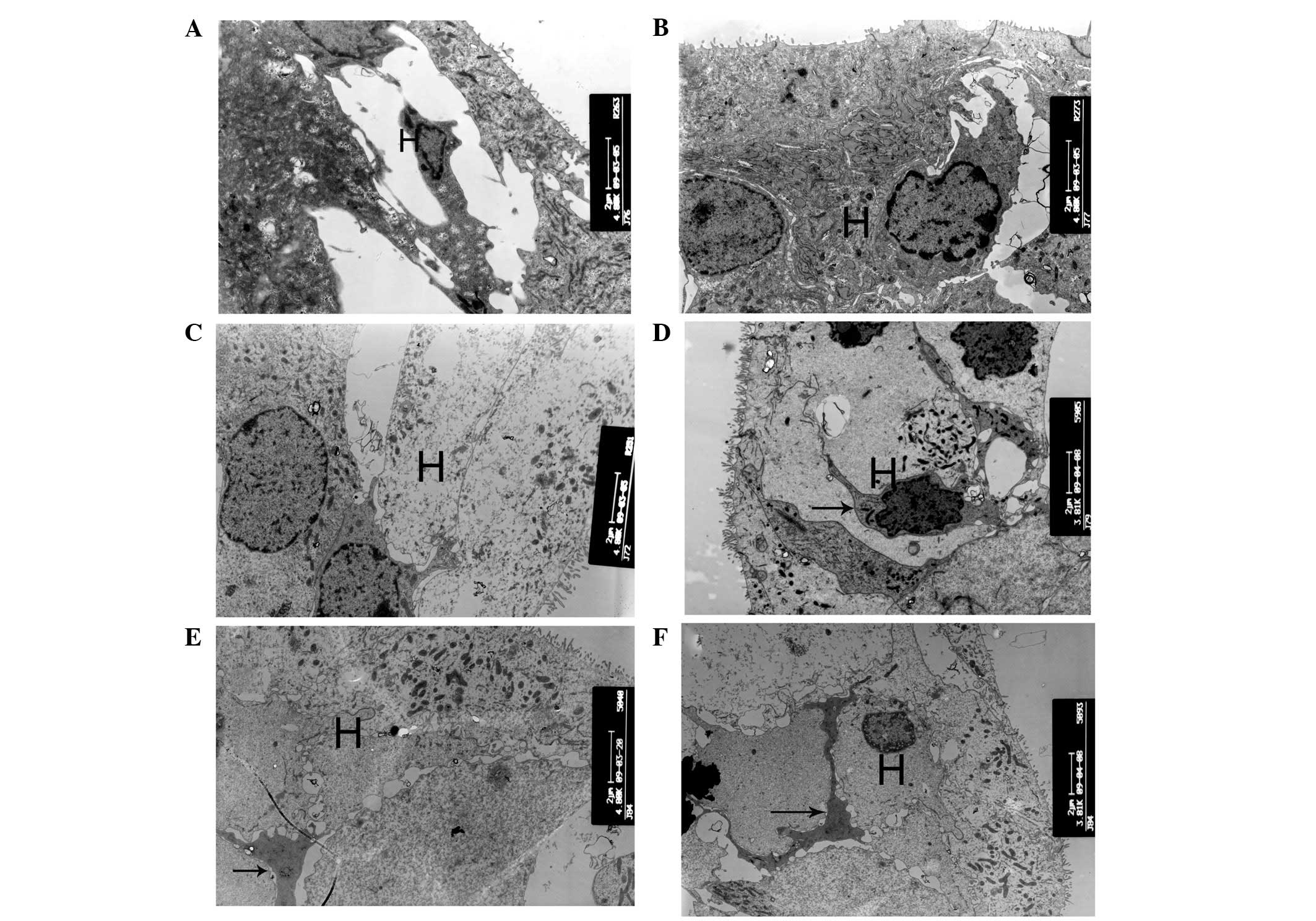 | Figure 7Ultrastructural changes of Hensen cell
regions. (A and B) At postnatal day 10 (P10), the Hensen cells of
cCx26ko mice had more cytoplasm than P10 wild-type mice
(magnification, ×4,800; middle turn). (C) At P30, cytoplasm of
Hensen cells was scarce (magnification, ×4,800; middle turn). (D)
At P90, cells around the outer hair cell region showed necrosis
(magnification, ×3,800; apical turn). Arrows, necrosis. (E and F)
Following P180, microglia-like cells appeared in the Hensen cell
region at the apical turn (magnification, ×4,800 and ×3,800,
respectively; apical turn). Arrows, microglia-like cell. H, Hensen
cells; cCx26ko, conditional connexin 26 knockout. |
At P60, the inner hair cells of the basal turn
became deformed in cCx26ko mice, with large empty spaces from
degenerated cells (Fig. 4E). The
nuclei of the outer hair cells showed pyknosis and the cells were
severely degenerated (Fig. 5E). At
P90, the cells around the outer hair cell region showed signs of
necrosis (Fig. 7D) and the
majority of other cells in the sensory epithelium at the basal turn
had died. At the apical turn, mitochondria with myelin fibers in
the inner hair cells were observed (Fig. 4F), indicating damage to the
mitochondrial membrane. Following P180, microglia-like cells
appeared in the Hensen cell region at the apical turn (Fig. 7E and F), which was the only region
in the cochlea where surviving cells were found. The histological
features of the apical organ of Corti were anachromasis and
enlarged cytoplasm, with pleomorphism with pseudopodia.
These particular cells were first reported in the
rat organ of Corti following aminoglycoside ototoxicity (13). Studies showed that these types of
cells contain numerous microfilaments and microtubules (14). The cells appear in the Hensen cell
region in older stages and were found to be active supportive cells
participating in repair and cleanup following damage.
Ultrastructural changes of the spiral
ganglion
Between P10 and P18, no noticeable differences were
observed in the numbers of spiral ganglion neurons (Fig. 8A–D). However, at P30, the numbers
of spiral ganglion neurons in cCx26ko mice were distinctively lower
compared with Cx26 wild-type mice (Fig. 8E and F). The thickness of the
myelin sheath was also found to be decreased in cCx26ko mice.
Discussion
A decrease in Cx26 protein expression affects the
development of structures in the organ of Corti (9), but does not affect the early
development of hair cells and supporting cells. No differences were
found in markers of hair cells and supporting cells between cCx26ko
mice and wild-type mice. The hair cell marker myosin 6, the
supporting cell marker prox1 and the pillar cell marker p75 were
unchanged. Although the tunnel of Corti was not open in cCx26ko
mice, this anomaly was not caused by changes in the three marker
proteins. Shim et al(15)
found that a lack of Sprouty2 leads to ectopic tunnel and tunnel
development abnormalities, thus the organ of Corti developmental
abnormalities in cCx26ko mice may be associated with the Sprouty2
gene. This hypothesis requires further investigation.
Epithelial gap junctions formed by Cx26 are known to
be necessary for normal cochlear functions (16). However, the mechanisms of deafness
caused by Cx26 mutations remain unknown. A landmark morphological
development immediately prior to the onset of hearing is the
opening of the tunnel of Corti and formation of Nuel’s space. The
current results are consistent with a study by Inoshita et
al(17) that observed a
different cCx26ko mouse model. Cx26 may directly or indirectly
regulate the genes necessary for differentiation of supporting
cells at the transcriptional or translational level. Abnormal
release of ATP or cell-signaling molecules through Cx26 gap
junction hemichannels may also cause the abnormal structure of the
pillar cells.
The present observations for the organ of Corti
showed that microglia-like cells around the Hensen cells were
involved in the cell degeneration process. Following hair cell
degeneration, the surrounding supporting cells migrate to fill the
spaces left to maintain the integrity of the epithelial cells.
Microglia-like cells appeared in later stages of development while
the hair cells were undergoing apoptosis. Degeneration is
hypothesized to be first initiated by phagocytosis to clear debris
or metabolic waste generated by apoptotic cells. The morphological
characteristics of irregular protrusions may fill spaces following
hair cell damage and form scar-like tissue (14).
GJs are widely hypothesized to connect supporting
cells in the organ of Corti, primarily to provide ionic pathways
for rapid removal of K+ around the base of hair cells.
However, the precise function of GJs in the cochlea remains
unknown. A working hypothesis explaining hair cell apoptosis is the
K+ recycling theory (18). A previous study showed that
K+ recirculation in the cochlea may be affected by the
level and properties of GJ intercellular communication (19). Stimulation of hair cells by sound
generates an increase in extracellular K+, which
transports into outer hair cells and then into Deiters’ cells and
adjacent supporting cells by the K-Cl co-transporter Kcc4, as well
as extracellular signals, including ATP-induced IP3 production and
Ca2+ release from the ER compartment. Ca2+
may activate the Cl− channels of supporting cells,
allowing Cl− to move to the extracellular side, favoring
K+ transport to the endolymph. The absence of Cx26
blocks GJ-mediated K+ recycling, which is driven by
transduction events around the inner hair cells. This model
predicts that cellular pathological changes in the organ of Corti
of cCx26ko mice should begin at a site close to the inner hair
cells. However, the pathological changes were observed to occur
initially around the outer hair cells. Specifically, Claudius cells
were the first to degenerate. Wang et al(9) reported first observing cell
degeneration in Claudius cells at ~P8. In the current study,
swollen mitochondria were initially observed in Claudius cells at
~P10 in the middle turn cochlea. High metabolism in cochlear cells
may produce reactive oxygen species (ROS), regulated by an
antioxidative enzyme system. ROS are negative regulators of GJs,
reducing intercellular coupling. Ablation of Cx26 in the cochlea
may increase the accumulation of the ROS in the cochlea, resulting
in mitochondrial swelling and an increase in lysosome numbers.
Swollen mitochondria were observed in numerous cells types in the
organ of Corti, implying that abnormal energy metabolism may be
correlated with a decrease in ATP synthesis. In addition,
mitochondria are involved in cellular apoptosis (20), accelerating the progress of
degeneration and necrosis. Apoptosis of hair cells is hypothesized
to cause metabolic stress and energy deficiency.
The current results support the hypothesis that a
number of pathological changes in the organ of Corti of cCx26 mice
occur prior to the formation of the endocochlear potential and
prior to the onset of hearing, when endocochlear potential is low
and K+ recycling is lacking. The timing of these changes
indicates that the basis for deafness in Cx26 mutant mice is not a
lack of K+ recycling.
Acknowledgements
This study was supported by grants from the Major
State Basic Research Development Program of China (973 Program; no.
2011CB504506), the National Natural Science Foundation of China
(no. 81230019), the Program for Changjiang Scholars and Innovative
Research Team in Universities (no. IRT1010), the Medical Guiding
Fund of the Science and Technology Commission of Shanghai
Municipality (no. 10411962100), the Program of Outstanding Shanghai
Academic Leaders (no. 11XD1401300) and the Research Fund for the
Doctoral Program of Higher Education of China (RFDP, no.
20120071110077). The study was supported also by grants to Yunfeng
Wang from the National Nature Science Foundation of China (no.
81100721) and to Xi Lin from the National Institute on Deafness and
other Communication Disorders (nos. NIDCD 4R33DC010476 and RO1
DC006483). Xi Lin and Huawei Li also received grant support from
the National Science Foundation of China (no. 30728029).
References
|
1
|
Nicholson SM and Bruzzone R: Gap
junctions: getting the message through. Curr Biol. 7:R340–R344.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoang Dinh E, Ahmad S, Chang Q, et al:
Diverse deafness mechanisms of connexin mutations revealed by
studies using in vitro approaches and mouse models. Brain Res.
1277:52–69. 2009.PubMed/NCBI
|
|
3
|
Kelsell DP, Dunlop J, Stevens HP, et al:
Connexin 26 mutations in hereditary non-syndromic sensorineural
deafness. Nature. 387:80–83. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Denoyelle F, Weil D, Maw MA, et al:
Prelingual deafness: high prevalence of a 30delG mutation in the
connexin 26 gene. Hum Mol Genet. 6:2173–2177. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rabionet R, Gasparini P and Estivill X:
Molecular genetics of hearing impairment due to mutations in gap
junction gene encoding beta connexin. Hum Mutat. 16:190–202. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gabriel HD, Jung D, Bützler C, et al:
Transplacental uptake of glucose is decreased in embryonic lethal
connexin26-deficient mice. J Cell Biol. 140:1453–1461. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cohen-Salmon M, Ott T, Michel V, et al:
Targeted ablation of connexin26 in the inner ear epithelial gap
junction network causes hearing impairment and cell death. Curr
Biol. 12:1106–1111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kudo T, Kure S, Ikeda K, et al: Transgenic
expression of a dominant-negative connexin26 causes degeneration of
the organ of Corti and non-syndromic deafness. Hum Mol Genet.
12:995–1004. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Chang Q, Tang W, et al: Targeted
connexin26 ablation arrests postnatal development of the organ of
Corti. Biochem Biophys Res Commun. 385:33–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohyama T and Groves AK: Generation of
Pax2-Cre mice by modification of a Pax2 bacterial artificial
chromosome. Genesis. 38:195–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gestwa G, Wiechers B, Zimmermann U, et al:
Differential expression of trkB. T1 and trkBT2, truncated trkC, and
p75(NGFR) in the cochlea prior to hearing function. J Comp Neurol.
414:33–49. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jones C, Roper VC, Foucher I, et al:
Ciliary proteins link basal body polarization to planar cell
polarity regulation. Nat Genet. 40:69–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z and Li H: Microglia-like cells in
rat organ of Corti following aminoglycoside ototoxicity.
Neuroreport. 11:1389–1393. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Wang Z, Wei W, et al: Research on
the origin and ultrastructure microglia-like cell in SD rat Corti’s
organ after neomycin ototoxicity. Zhonghua Er Bi Yan Hou Tou Jing
Wai Ke Za Zhi. 40:618–619. 2005.PubMed/NCBI
|
|
15
|
Shim K, Minowada G, Coling DE, et al:
Sprouty2, a mouse deafness gene, regulates cell fate decisions in
the auditory sensory epithelium by antagonizing FGF signaling. Dev
Cell. 8:553–564. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Tang W, Ahmad S, et al: Gap
junction-mediated intercellular biochemical coupling in cochlear
supporting cells is required for normal cochlear functions. Proc
Natl Acad Sci USA. 102:15201–15206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inoshita A, Iizuka T, Okamura HO, et al:
Postnatal development of the organ of Corti in dominant-negative
Gjb2 transgenic mice. Neuroscience. 156:1039–1047. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schulte BA and Adams JC: Distribution of
immunoreactive Na+, K+-ATPase in gerbil
cochlea. J Histochem Cytochem. 37:127–134. 1989. View Article : Google Scholar
|
|
19
|
Martinez AD, Acuña R, Figueora V, et al:
Gap-junction channels dysfunction in deafness and hearing loss.
Antioxid Redox Signal. 11:309–319. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ott M, Gogvadze V, Orrenius S, et al:
Mitochondria, oxidative stress and cell death. Apoptosis.
12:913–922. 2007. View Article : Google Scholar : PubMed/NCBI
|