Introduction
Phosphoinositide 3-kinase-γ (PI3Kγ) and PI3Kδ, which
belong to the class I PI3K family, are essential for inflammatory
cell signaling (1). PI3Kδ and
PI3Kγ are primarily limited to hematopoietic and endothelial cells
(2,3). Activated by downstream receptor
tyrosine kinases and G-protein coupled receptors, respectively
(4), PI3Kδ and PI3Kγ lead to the
formation of phosphatidylinositol-(3,4,5)-triphosphate (PIP3) and the
phosphorylation of Akt. The interaction of phospho-Akt with PIP3 at
the cell membrane stimulates the phosphorylation of downstream
targets that regulate several inflammatory and immune functions,
including the recruitment of activated T cells, macrophages and
neutrophils (2). Studies using
genetic deletion and benchmark compounds have demonstrated that the
most effective anti-inflammatory PI3K inhibitor inhibits PI3Kδ and
PI3Kγ by markedly affecting anti-inflammatory markers in T cell and
monocyte-driven environments, with concurrent antiproliferative
effects (5). Therefore, studies
concerning PI3Kγ and PI3Kδ have generated interest in the
development of PI3Kδ/γ inhibitors as therapeutic agents for the
treatment of autoimmune and inflammatory diseases.
Regardless of numerous studies, the mechanisms of
PI3Kδ/γ in inflammatory diseases, such as hepatitis, remain to be
elucidated. Pharmacological inhibition of PI3Kγ has been
demonstrated to exert a therapeutic effect on hepatic injury in
mice (6); however, the effect of
PI3Kδ/γ in this process is unclear. ConA-induced acute hepatitis,
which is characterized by hepatic necrosis and inflammatory cell
infiltration, is commonly used as an experimental animal model for
human liver disease (7). In the
present study, we hypothesized that the pharmacological
inactivation of PI3Kδ/γ may interfere with the pathology of
ConA-induced hepatitis and thereby impact the functional recovery
of the injured liver.
TG100-115, a dual PI3Kγ and δ-selective inhibitor
was used to investigate its potential effect on ConA-induced acute
hepatitis in mice. Notably, this study determined that the
pharmacological inhibition of PI3Kδ/γ by TG100-115 did not prevent
liver damage following ConA challenge. Furthermore, TG100-115
aggravated liver damage by elevating transaminase activity and IL-2
levels in sera, and by promoting hepatic necrosis and hepatocyte
apoptosis. This suggested that the inhibition of PI3Kγ and PI3Kδ
may be involved in the pathogenesis of acute hepatitis induced by
ConA. To the best of our knowledge, these results may be the first
to warn against the development of PI3Kδ/γ inhibitors as
therapeutic agents for the prevention and treatment of hepatitis in
humans.
Materials and methods
Reagents
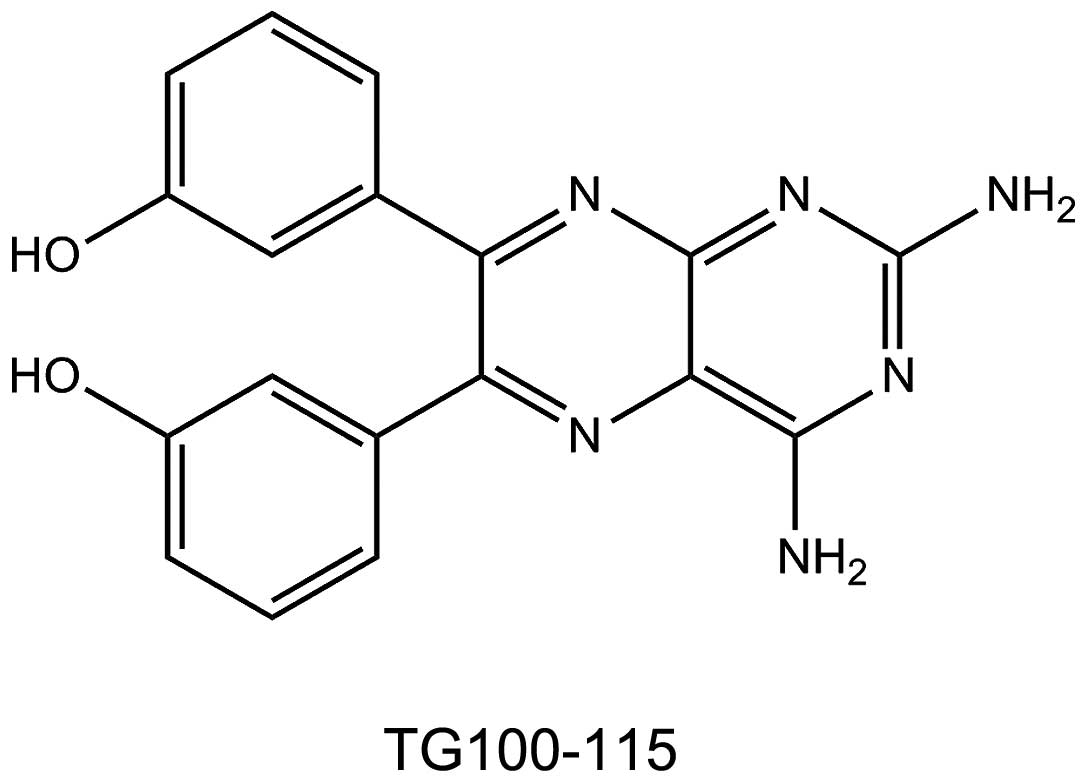
TG100-115 was synthesized (purity, >99.5%) in our
laboratory (Sichuan University) according to previous studies
(8,9). The purity and structural
identification of TG100-115 were confirmed by high performance
liquid chromatography, mass spectrometry and nuclear magnetic
resonance (Bruker Avance 400 NMR system; Bruker, Billerica, MA,
USA) (Fig. 1). ConA (type IV) was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Female BALB/C mice (age, 6–8 weeks; weight, 20–24 g)
were obtained from the Western China Experimental Animal Center
(Sichuan, China) and maintained in a temperature and humidity
controlled environment in a 12-h light-dark cycle, and were allowed
free access to water and food. All mice received human care and all
experimental procedures in the study were handled according to the
guidelines for the humane care of laboratory animals established by
Sichuan University (Chengdu, Sichuan, China), and the study
protocol was approved by the local ethics committee (Chengdu,
Sichuan, China).
Experimental protocol
TG100-115 was dissolved in vehicle (10%
hydroxypropyl-β-cycloamylose) and a 1.5 mg/ml solution of ConA was
produced in sterile saline. For the investigation of the
dose-response relationship for TG100-115, mice were treated with
TG100-115 [10 or 50 mg/kg; intraperitoneally (ip)], followed by
ConA (15 mg/kg) or solvent [intravenously (iv)] after 0.5 h
(10). Mice treated with ConA (15
mg/kg, iv) were sacrificed 2, 4, 8 or 20 h following treatment.
Sera were collected 20 h following ConA challenge to determine the
levels of serum alanine aminotransaminase (ALT), asparate
aminotransaminase (AST) and albumin (ALB), and livers were
harvested for hematoxylin and eosin (H&E) staining. The levels
of tumor necrosis factor (TNF)-α and interleukin (IL)-2 in the sera
were analyzed at 2 and 4 h with commercial enzyme-linked
immunosorbent assay (ELISA) kits (Mouse TNF-α ELISA and Rouse IL-2
ELISA kits, respectively; Dakewe Biotech, Co., Ltd., Nanshan,
Shenzen, China) according to the manufacturer’s instructions. The
apoptosis of hepatocytes at 2 and 4 h was determined by Hoechst
staining. In a survival test, mice were treated with a lethal dose
of ConA (30 mg/kg, iv), followed by TG100-115 (10 mg/kg, ip) or
vehicle (11) after 0.5 h, and
then monitored for survival every 2 h.
Evaluation of liver enzymes
Liver injury was determined by the measurement of
serum ALT, AST and ALB levels by the Chemical Lab in the National
Chengdu Center for Safety Evaluation of Drugs (Chengdu, Sichuan,
China).
H&E and Hoechst staining
Formalin-fixed liver samples were embedded in
paraffin, and 4-μm sections were stained with H&E or Hoechst
staining (Beyotime Biotech, Jiangsu, China) according to the
manufacturer’s instructions (6).
The stained liver sections were examined using light microscopy
followed by fluorescence microscopy. Histological scores of
centrilobular necrosis for individual mice (n=10) 20 h following
ConA injection are presented. Histological slides were evaluated by
a board of certified veterinary pathologists without the knowledge
of the treatment groups. Histopathological changes were graded and
recorded on a severity scale of 0 to 5 (0, no lesion; 1, minimal;
2, mild; 3, moderate; 4, marked; 5, severe) (11).
Detection of cytokines by ELISA
Mice treated with TG100-115 (10 mg/kg, ip) 0.5 h
following ConA challenge were sacrificed 2 or 4 h following ConA
injection. Serum levels of TNF-α and IL-2 were analyzed by ELISA
using commercially available kits (Dakewe Biotech, Co., Ltd.)
according to the manufacturer’s instructions. TNF-α and IL-2 levels
were measured 2 and 4 h following ConA challenge, respectively
(12,13).
Assessment of the cytotoxicity of
TG100-115 in vitro
The direct effect of TG100-115 on hepatocytes was
determined by a colorimetric method using methyl thiazolyl
tetrazolium (MTT) dye. Human liver cell line, L-02 (HL-7702), was
used to assess the cytotoxicity of TG100-115. TG100-115 was added
to cells in a doubling dilution manner at a series of doses (100,
50, 25, 12.5, 6.25, 3.13, 1.56 and 0.75 μM) for a 24 h coculture.
The percentage of viable cells was determined by the conversion of
MTT to its formazan derivative in each well. This was conducted by
comparing the optical density at 570 nm (OD570) of the wells with
that of the drug-free control based on the following equation:
(A570 of wells that contained the drug/A570 of the drug-free wells)
× 100%. IC50 (the concentration of 50% maximal
inhibition of cell growth) was determined by interpolation of the
dose-response curves.
Statistical analysis
Student’s t-test was used to determine the
significance of differences between vehicle controls and
experimental groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
PI3Kδ/γ inhibition aggravates acute liver
injury of mice induced by ConA
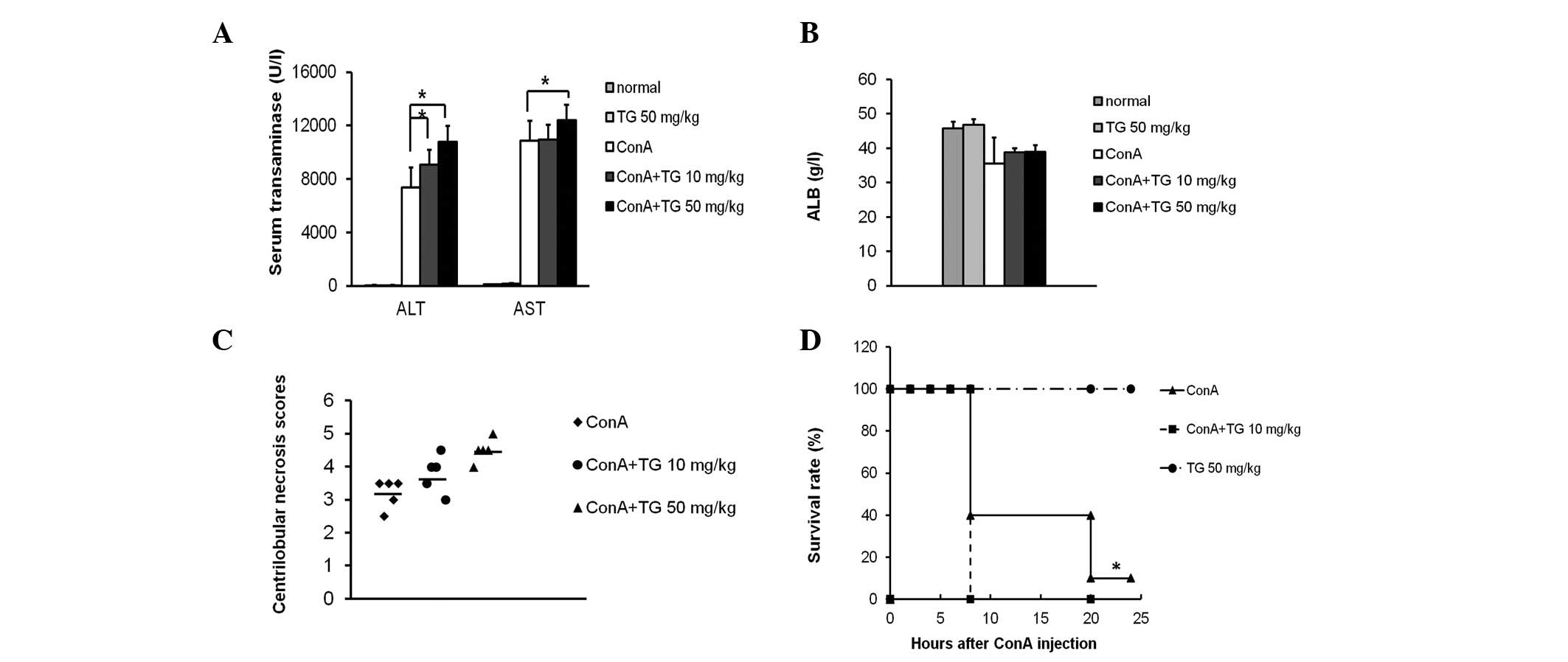
TG100-115 (10 and 50 mg/kg) significantly increased
the serum ALT and AST levels in ConA-induced mice (Fig. 2A). ALB (synthesized in the liver),
ALT and AST are indicators of liver function. The average level of
ALB in normal mice is 47 g/l, while that in ConA-treated mice
decreased to 35 g/l, indicating that ConA induced hypohepatia, in
accordance with the change in the transaminase levels. However,
there was no significant difference in the ALB levels between the
control and TG100-115 groups (Fig.
2B), indicating that TG100-115 does not effect the ALB levels
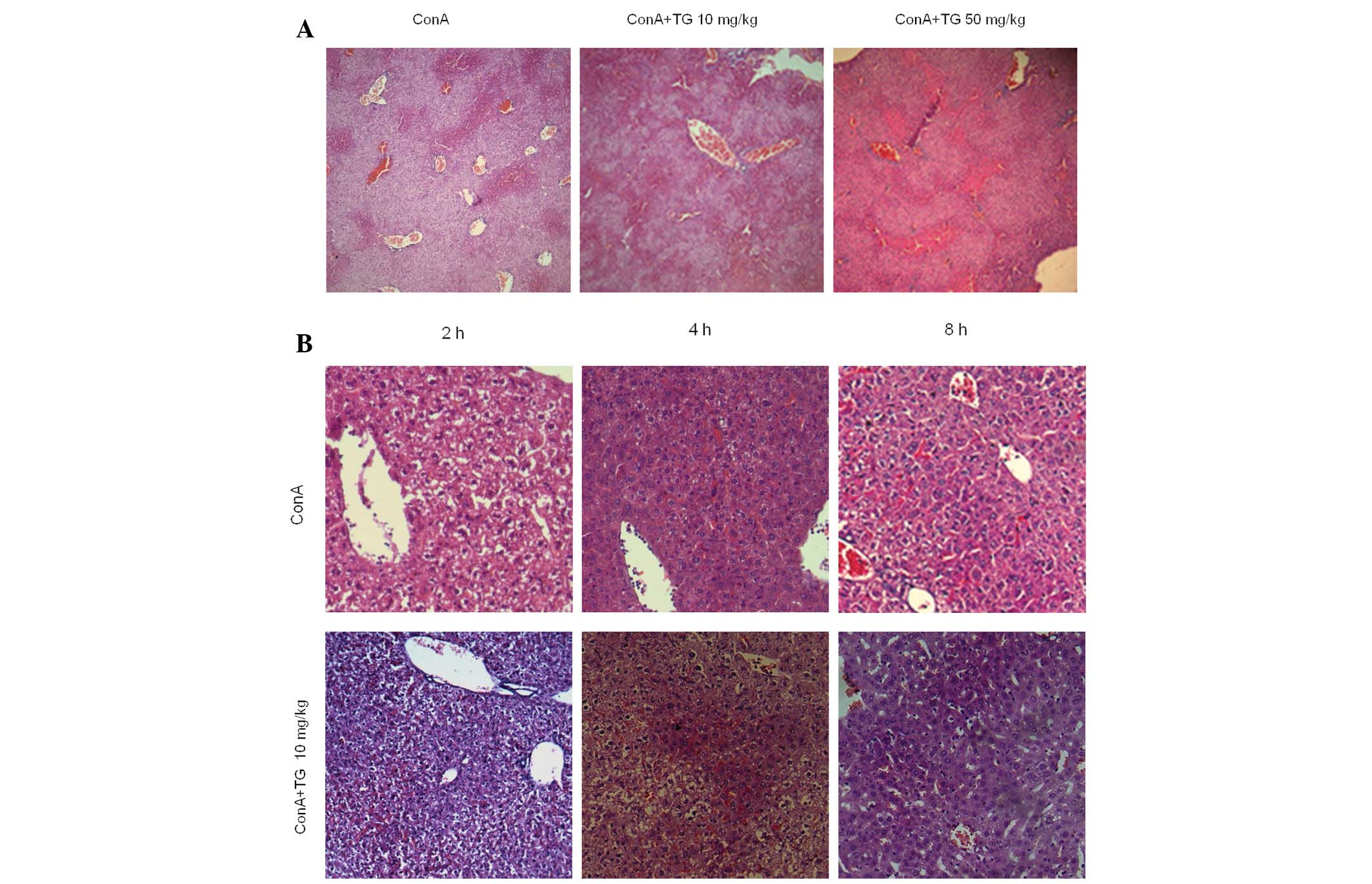
in acute hepatitis. The livers from ConA-treated mice with
hepatitis exhibited inflammatory cell infiltration, dilatation and
congestion of the blood vessels, as well as widespread
hepatocellular necrosis in the liver lobules. In addition, the
inflammatory cell infiltration and the necrotic regions were also
markedly increased by TG100-115 (50 mg/kg) treatment (Fig. 3A). Histological scores of
centrilobular necrosis for individual mice demonstrated that ConA
induced a lower score (3.29±0.49) compared with that for TG100-115
treatment (10 or 50 mg/kg), which significantly increased the
scores to 4.09±0.51 and 4.50±0.71, respectively (Fig. 2C). As severe liver damage may be
fatal in mice, increased liver damage is correlated with an
increased mortality rate. Following a lethal dose of 30 mg/kg ConA,
90% of the ConA-treated mice succumbed within 24 h. By contrast,
mice pretreated with TG100-115 (15 mg/kg) demonstrated an increased
mortality rate with 100% of mice no longer surviving within 8 h.
These results indicated that TG100-115 exerted a detrimental effect
on ConA-induced hepatitis in mice (Fig. 2D).
Histological analysis of the effect of
TG100-115 on liver inflammatory infiltration and hepatocyte
apoptosis
Liver tissues of TG100-115-treated and untreated
mice were harvested for H&E staining 2 and 4 h following ConA
challenge (13). An increased
number of inflammatory cells in the central veins and the sinusoids
of the livers from mice with ConA-induced hepatitis were observed.
Furthermore, TG100-115 treatment increased the apoptosis of
hepatocytes in the portal area compared with that of the control
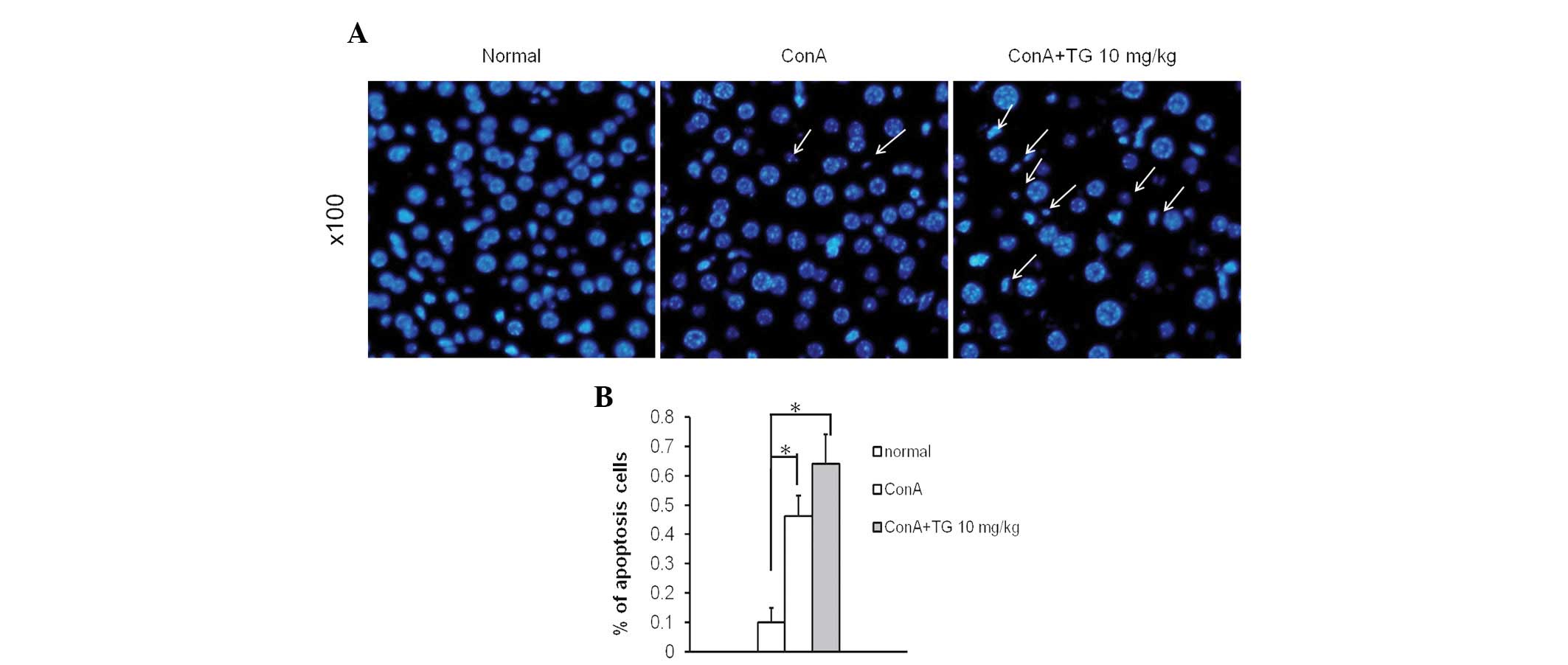
(Fig. 3B). Hoechst staining was
used to further investigate the apoptosis of hepatocytes. The liver
tissue sections from ConA and TG100-115-treated mice demonstrated
greater quantities of condensed chromatin and nuclear fragmentation
(which are characteristics of cells undergoing apoptosis) than
those with ConA treatment alone (Fig.
4). These results suggested that interference of PI3Kδ/γ
signaling by TG100-115 modified the inflammatory cell recruitment
that is responsible for the development of ConA-induced hepatic
injury. Thus, TG100-115 may aggravate hepatocyte apoptosis induced
by ConA in vivo.
TG100-115 modulates ConA-induced cytokine
production
TNF-α and IL-2, key inflammatory cytokines mainly
secreted by neutrophils and T cells, are essential in the
development of ConA-induced acute hepatitis (14). To determine the effect of TG100-115
in modulating the production of these key inflammatory cytokines,
we analyzed the TNF-α and IL-2 protein levels in serum of mice with
ConA-induced hepatitis and observed their changes following
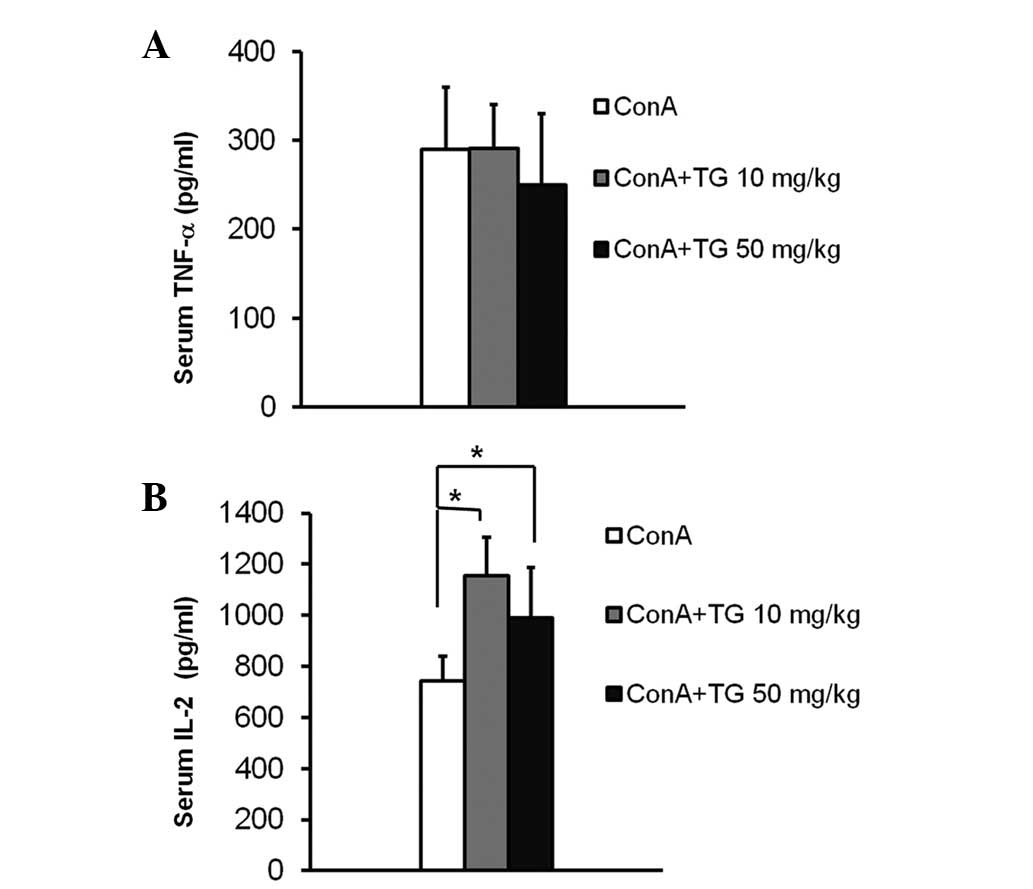
TG100-115 treatment. TNF-α levels in the serum were elevated to 300
pg/ml 2 h following ConA injection, the peak time point according
to previous studies (12), and
marginally changed following TG100-115 treatment (Fig. 5A). In a previous study, the peak
serum level of IL-2 was detected at 4 h following ConA injection
(13). TG100-115 treatment
resulted in an increase in IL-2 serum levels compared with those of
ConA-treated mice 4 h following ConA injection (Fig. 5B). This increase suggested that the
inhibition of PI3Kδ/γ by TG100-115 may modulate ConA-induced T cell
activation in vivo.
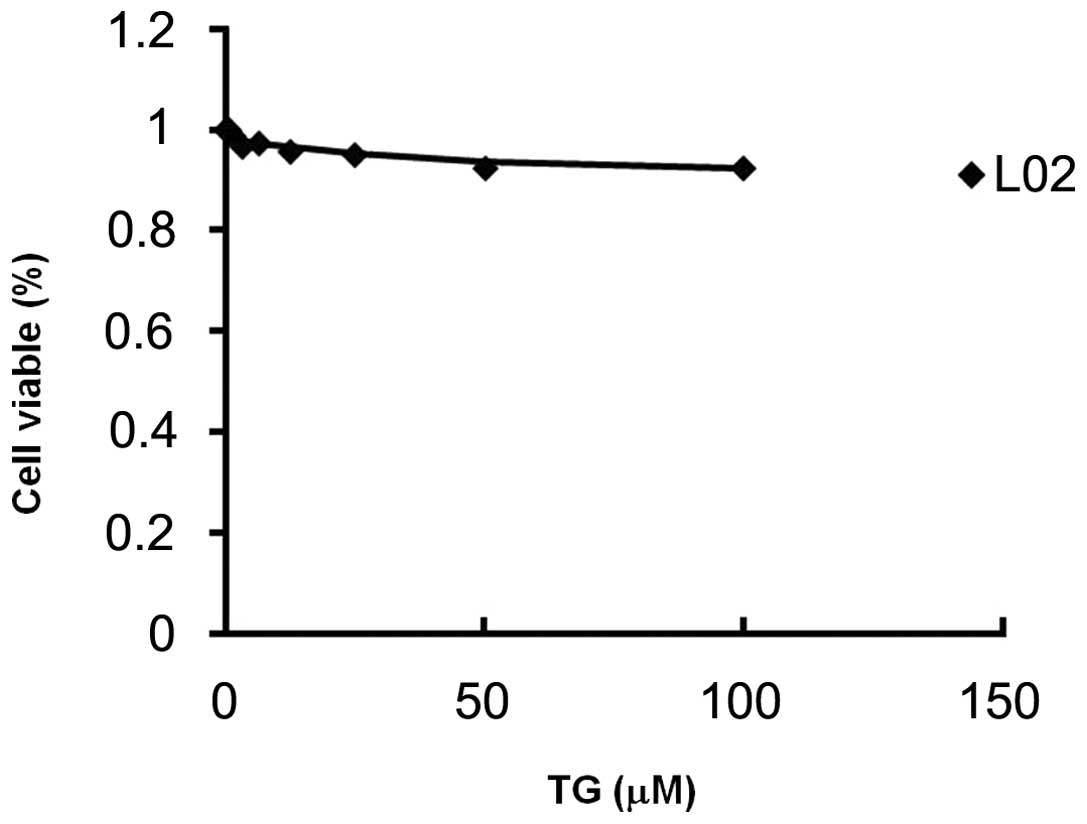
Toxicity of TG100-115
Following exposure to various concentrations of
TG100-115 for 24 h, the percentage of viable L-02 cells was
assessed by MTT assay. Results revealed that TG100-115 (at a
concentration of 100 μM) had no effect on the percentage of viable
L-02 cells (Fig. 6). The
IC50 value of TG100-115 on L-02 cells was >100 μM,
indicating that TG100-115 did not significantly reduce the
viability of hepatic cells in vitro (Table I).
 | Table IIC50 of TG100-115 on L0-2
cells. |
Table I
IC50 of TG100-115 on L0-2
cells.
| Cell line | IC50
(μM) |
|---|
| L0-2 | >100 |
Discussion
Studies using genetic deletion and benchmark
compounds have demonstrated that the most effective
anti-inflammatory PI3K inhibitor inhibits PI3Kδ and PI3Kγ (5) by affecting anti-inflammatory markers
in T cell- and monocyte-driven environments, with concurrent
antiproliferative effects. This raises the possibility that
pharmacological inactivation of PI3Kδ and PI3Kγ may be used for the
treatment of hepatitis. However, regardless of its demonstrated
preventive effect, also confirmed by us (data not shown), TG100-115
failed to reduce, and even aggravated, the progression of hepatic
injury. This aggravation occurred in a time- and dose-dependent
manner, as indicated by the elevated transaminase activity in the
serum and the increased hepatic necrosis. In the survival test,
TG100-115 resulted in increased mortality compared with that of
mice challenged with ConA alone.
PI3Kδ/γ is important in the development of T
lymphocytes and other related cellular functions (15). The dual PI3Kδ/γ knockout mouse has
been demonstrated to be viable but to exhibit serious defects in
thymocyte survival and T cell development (16,17).
Dual targeting of PI3Kδ/γ has resulted in enhanced inhibition of
overall T cell activation and LPS-induced TNF-α production
(1). TG100-115, a dual PI3Kδ/γ
inhibitor, has been identified to reduce the severity of asthma in
a murine model by reducing pulmonary eosinophilia and the
concomitant interleukin-13 levels (18). T lymphocyte activation is the
predominant pathology in human and ConA-induced hepatitis (19). In the present study, it was
demonstrated that TG100-115 treatment did not block ConA-induced T
cell accumulation in the liver. Contrary to this, T cell hepatic
infiltration appeared to increase following TG100-115 treatment in
ConA-hepatitis mice. This was in accordance with the aggregated
IL-2 secretion in the serum of TG100-115 treated mice in
vivo. IL-2, IFN-γ and TNF-α are cytokines secreted by Th1 T
cells, which enhance the Th1 immune response. Therefore, it was
hypothesized that aggravated progression of hepatic injury by
TG100-115 in ConA-induced hepatitis may be due to the stimulation
of T cells or a T cell subtype. Mice immunized with ConA-activated
autologous T cells demonstrated dampened regulatory T cell (Treg)
function, which may have contributed to an enhanced Th1 response
in vivo(20). It has been
determined that Tregs are important in immune system homeostasis
and that the Th1–Th2 balance was partially impaired in
p110γKO/δD910A mice compared with the wild
type (15). In the present study,
the pharmaceutical inhibition of PI3Kδ/γ by TG100-115 (as a result
of a loss of Th1–Th2 balance) and the increased levels of IL-2 in
the serum may have resulted in a dominant Th1 immune response with
TG100-115 treatment following ConA injection. Therefore, it may be
beneficial to identify the precise mechanism of PI3Kδ/γ in T
lymphocyte differentiation, and the balance of Th1 and Th2 during
ConA-induced hepatitis development.
Numerous clinical anti-inflammatory agents function
through different targets, yet all inhibit immune cell function
while leaving nonimmune cells relatively unaffected. An MTT
analysis on the liver cell line L-02 was performed to determine
whether the PI3Kδ/γ inhibitors exhibited this property. The
IC50 value of TG100-115 on L-02 cells was >100 μM,
which indicated that TG100-115 did not demonstrate growth
inhibition in L-02 cells in vitro. However, mice treated
with TG100-115 resulted in earlier and markedly greater hepatocyte
apoptosis than that of mice receiving ConA alone. This indicated
that TG100-115 aggravated ConA-induced hepatitis via an
immune-mediated mechanism, irrespective of TG100-115 alone.
In this study, it was demonstrated that the
pharmacological inhibition of PI3Kδ/γ did not prevent ConA-induced
acute hepatic injury in vivo. This indicates that PI3Kγ and
PI3Kδ may be involved in the process of acute hepatitis induced by
ConA. These results may suggest against the development of
inhibitors of PI3Kδ/γ as therapeutic agents for the treatment of
human hepatitis.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 8100093).
References
|
1
|
Williams O, Houseman BT, Kunkel EJ, et al:
Discovery of dual inhibitors of the immune cell PI3Ks p110delta and
p110gamma: a prototype for new anti-inflammatory drugs. Chem Biol.
17:123–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deane JA and Fruman DA: Phosphoinositide
3-kinase: diverse roles in immune cell activation. Annu Rev
Immunol. 22:563–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puri KD, Doggett TA, Douangpanya J, et al:
Mechanisms and implications of phosphoinositide 3-kinase delta in
promoting neutrophil trafficking into inflamed tissue. Blood.
103:3448–3456. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katso R, Okkenhaug K, Ahmadi K, White S,
Timms J and Waterfield MD: Cellular function of phosphoinositide
3-kinases: implications for development, homeostasis, and cancer.
Annu Rev Cell Dev Biol. 17:615–675. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marone R, Cmiljanovic V, Giese B and
Wymann MP: Targeting phosphoinositide 3-kinase: moving towards
therapy. Biochim Biophys Acta. 1784:159–185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang ZL, Wu XH, Song LF, et al:
Phosphoinositide 3-kinase gamma inhibitor ameliorates concanavalin
A-induced hepatic injury in mice. Biochem Biophys Res Commun.
386:569–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tiegs G, Hentschel J and Wendel A: A T
cell-dependent experimental liver injury in mice inducible by
concanavalin A. J Clin Invest. 90:196–203. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Palanki MS, Dneprovskaia E, Doukas J, et
al: Discovery of 3,3′-(2,4-diaminopteridine-6,7-diyl) diphenol as
an isozyme-selective inhibitor of PI3K for the treatment of
ischemia reperfusion injury associated with myocardial infarction.
J Med Chem. 50:4279–4294. 2007.
|
|
9
|
Ichihara M, Suzuki H, Mohr B and Ohta K:
Different disk structures in the hexagonal columnar mesophases of
2,3-dicyano-6,7,10,11-tetraalkoxy-1,4-diazatriphenylenes and
2,3-dicyano-6,7,10,11-tetraalkoxytriphenylenes. Liq Cryst.
34:401–410. 2007. View Article : Google Scholar
|
|
10
|
Wolf AM, Wolf D, Rumpold H, et al: The
kinase inhibitor imatinib mesylate inhibits TNF-α production in
vitro and prevents TNF-dependent acute hepatic inflammation. Proc
Natl Sci USA. 102:13622–13627. 2005.
|
|
11
|
Bozza M, Bliss JL, Maylor R, et al:
Interleukin-11 reduces T-cell-dependent experimental liver injury
in mice. Hepatology. 30:1441–1447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fougerat A, Gayral S, Gourdy P, et al:
Genetic and pharmacological targeting of phosphoinositide
3-kinase-gamma reduces atherosclerosis and favors plaque stability
by modulating inflammatory processes. Circulation. 117:1310–1317.
2008. View Article : Google Scholar
|
|
13
|
Casini A, Ricci OE, Paoletti F and
Surrenti C: Immune mechanisms for hepatic fibrogenesis.
T-lymphocyte-mediated stimulation of fibroblast collagen production
in chronic active hepatitis. Liver. 5:134–141. 1985. View Article : Google Scholar
|
|
14
|
Camps M, Rückle T, Ji H, et al: Blockade
of PI3Kgamma suppresses joint inflammation and damage in mouse
models of rheumatoid arthritis. Nat Med. 11:936–943.
2005.PubMed/NCBI
|
|
15
|
Ji H, Rintelen F, Waltzinger C, et al:
Inactivation of PI3Kgamma and PI3Kdelta distorts T-cell development
and causes multiple organ inflammation. Blood. 110:2940–2947. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Swat W, Montgrain V, Doggett TA, et al:
Essential role of PI3Kdelta and PI3Kgamma in thymocyte survival.
Blood. 107:2415–2422. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Webb LM, Vigorito E, Wymann MP, Hirsch E
and Turner M: Cutting edge: T cell development requires the
combined activities of the p110gamma and p110delta catalytic
isoforms of phosphatidylinositol 3-kinase. J Immunol.
175:2783–2787. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doukas J, Eide L, Stebbins K, et al:
Aerosolized phosphoinositide 3-kinase gamma/delta inhibitor
TG100-115 [3-[2, 4-diamino-6-(3-hydroxyphenyl) pteridin-7-yl]
phenol] as a therapeutic candidate for asthma and chronic
obstructive pulmonary disease. J Pharmacol Exp Ther. 328:758–765.
2009.PubMed/NCBI
|
|
19
|
Dienes HP, Hütteroth T, Hess G and Meuer
SC: Immunoelectron microscopic observations on the inflammatory
infiltrates and HLA antigens in hepatitis B and non-A, non-B.
Hepatology. 7:1317–1325. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao Q, Wang L, Du F, et al: Downregulation
of CD4+ CD25+ regulatory T cells may underlie enhanced Th1 immunity
caused by immunization with activated autologous T cells. Cell Res.
17:627–637. 2007.
|