Introduction
Short peptides are forming an increasingly
significant portion of the biopharmaceutical market (1–4).
Humanin (HN) was first identified by the functional screening of
genes that protect neuronal cells from apoptotic death caused by a
familial Alzheimer’s disease (AD)-linked mutant amyloid precursor
protein (5). A gene coding for HN
was identified in a cDNA library derived from the occipital lobe of
a brain from a patient with AD (5). HN is a 24-amino acid residue peptide
(MAPRGFSCLLLLTSEIDLPVKRRA), whose sequence has characteristics of
signal peptides (6). Unexpectedly,
the endogenous HN produced by the cells transfected with its
cDNA-containing plasmid has not only been identified to be
partitioned into the cell membrane, but also secreted into the
culture media, indicating that the mechanism via which HN acts is
that of an extracellular ligand, although its intracellular
function cannot be excluded (5,6). The
secreted and synthetic HN peptides have shown neuroprotective
activities against various toxic stresses (5–8).
Synthetic HN was demonstrated to protect neurons from cell death
caused by familial AD-linked mutant proteins in neuronal cell lines
(5,7,8) and
by amyloid-β peptides in mouse cortical primary neurons (5,8–10).
Alanine scanning analysis has revealed several
inactive analogs, including S7A-HN and C8A-HN (10). It has been demonstrated that this
cysteine at amino acid position eight is in the free SH form and
hence is not involved in a disulfide bond (11). The binding of zinc ions to HN
through C8 has been indicated to be important in neuroprotection
(11). An artificial HN dimer was
created by interchain disulfide bond formation, which resulted in
reduced activity, meaning that disulfide-linked dimer formation was
not the mechanism behind the action of HN and that the artificial
dimer may present the functional structure, although with reduced
affinity (8). We have previously
shown that the strong tendency of S7A-HN to irreversibly change its
conformation and aggregate under physiological conditions may be,
at least in part, responsible for the observed loss of its
neuroprotective activity (12,13).
Such aggregation would obscure the results of the biological
activity of C8A-HN and possibly other HN analogs, and hence, in the
present study, the structure of this analog in comparison with HN
and other HN analogs was characterized.
Materials and methods
Peptide analog preparation
HN, S7A-HN and S14G-HN were obtained from Peptide
Institute, Inc. (Osaka, Japan) and C8A-HN was purchased from KNC
Laboratories Co., Ltd. (Kobe, Japan). The purity was confirmed to
be at least 95% by the vendor using an amino acid and liquid
chromatography-mass spectrometry analysis. These peptides were
dissolved at 1 mg/ml in cold water and kept cold until use.
Phosphate-buffered saline (PBS; 1.1×) was prepared from 10× PBS and
also kept cold until use. The peptide solution in cold water was
diluted with the cold 1.1× PBS to a final concentration of 0.1
mg/ml and 1× PBS. The diluted solution was immediately delivered to
the circular dichroism (CD) cell and placed in the Peltier cell
holder set at 5ºC. CD spectral or time-course measurements were
then acquired as described in the results section.
CD spectroscopy measurements
CD measurements were recorded on a Jasco J-715
spectropolarimeter (Jasco Inc., Easton, MD, USA) at the indicated
temperatures using a 0.1-cm cell throughout with the following
parameters: A 10-nm/min scan rate, five accumulations, a 0.1-nm
data pitch and a 4-sec time constant. The sample temperature was
controlled with a Peltier cell holder (Jasco Inc.) and a PTC-348WI
temperature controller (Jasco Inc.). The CD spectra were converted
to the mean residue ellipticity following subtraction of the PBS
spectrum using the peptide concentration (0.1 mg/ml), the mean
residue weight (112 g/mol for HN, S7A-HN and C8A-HN and 111 g/mol
for S14G-HN) and the pathlength of the cell (0.1 cm).
Results and Discussion
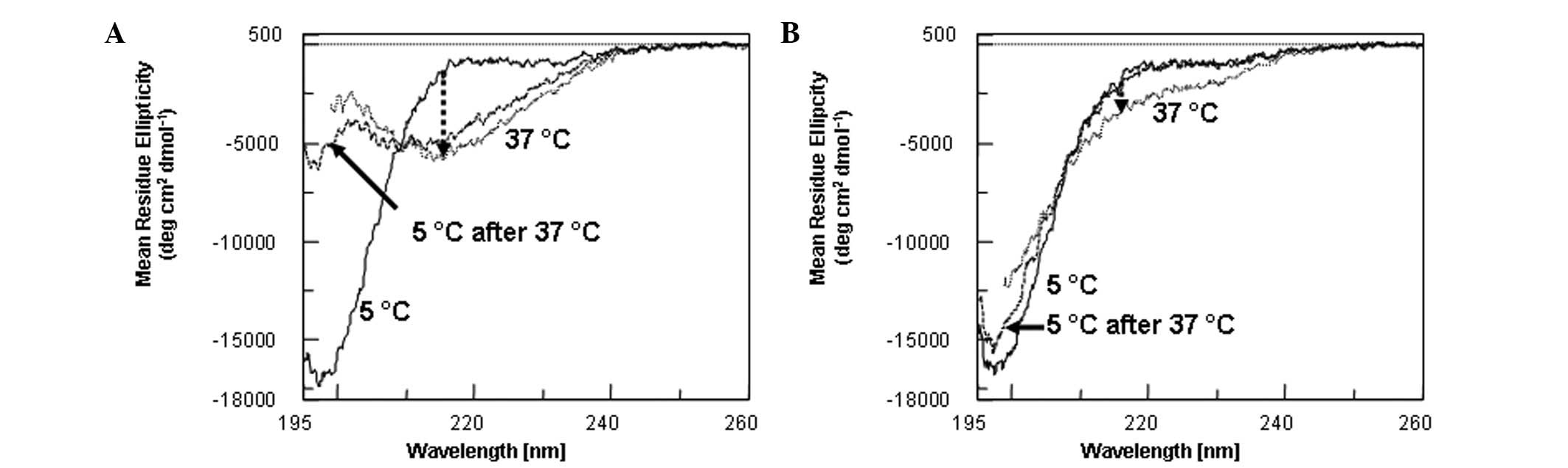
Far-ultraviolet (UV) CD spectra of HN were
determined as a function of temperature, i.e., at 5, 37 and then
5ºC again following the 37ºC measurements (Fig. 1A). The spectrum at 5ºC (solid
curve) was essentially identical to the previous data and was
characterized as primarily disordered (1). The cell temperature was then raised
to 37ºC and the sample was kept at 37ºC for 15 min. The spectrum at
37ºC was entirely different from the 5ºC spectrum, with a valley
around 217 nm (dotted curve). The change at 217 nm is indicated by
an arrow in Fig. 1A. The spectrum
is characteristic of the β-sheet structure that may be observed in
antibody structures (14), most
likely due to the aggregation of the peptide, leading to
intermolecular β-sheet formation. We have previously shown using a
sedimentation velocity technique that the HN and HN analog
extensively aggregate into different sizes in PBS at 25ºC (13). Following the 37ºC measurement, the
sample temperature was cooled to 5ºC and then maintained at 5ºC for
15 min. The cooled sample (Fig. 1,
dashed curve) demonstrated no recovery of the original spectrum,
largely retaining the shape of the 37ºC spectrum. Such a spectrum
is consistent with the irreversible nature of structural change at
37ºC due to aggregation. A similar result was obtained for S7A-HN
(data not shown): Namely, the disordered structure at 5ºC was
converted to the spectrum characteristic of the interpeptide
β-sheet at 37ºC and was not recovered upon cooling. This analog
also revealed extensive aggregation, as determined by the
sedimentation velocity technique (13).
By contrast, the results for S14G-HN and C8A-HN were
qualitatively different from the aforementioned two samples. The
far-UV CD spectra of S14G-HN were determined at 5ºC (solid curve),
37ºC (dotted curve) and 5ºC subsequent to heating (dashed curve)
(Fig. 1B). It is evident that
these three spectra were not much different from each other. The
spectrum at 5ºC was similar to the 5ºC spectrum for HN (Fig. 1A), i.e., that of a disordered
structure. There were only small changes upon heating to 37ºC (as
indicated by a small arrow at 217 nm). The spectrum at 37ºC showed
the structure to have remained largely disordered, without the
appearance of a 217 nm valley, and was almost restored upon
cooling; namely, the small changes induced by heating to 37ºC were
largely reversible. A similar conclusion may be made for C8A-HN
(data not shown). In the case of C8A-HN, the original 5ºC spectrum
was completely restored upon cooling.
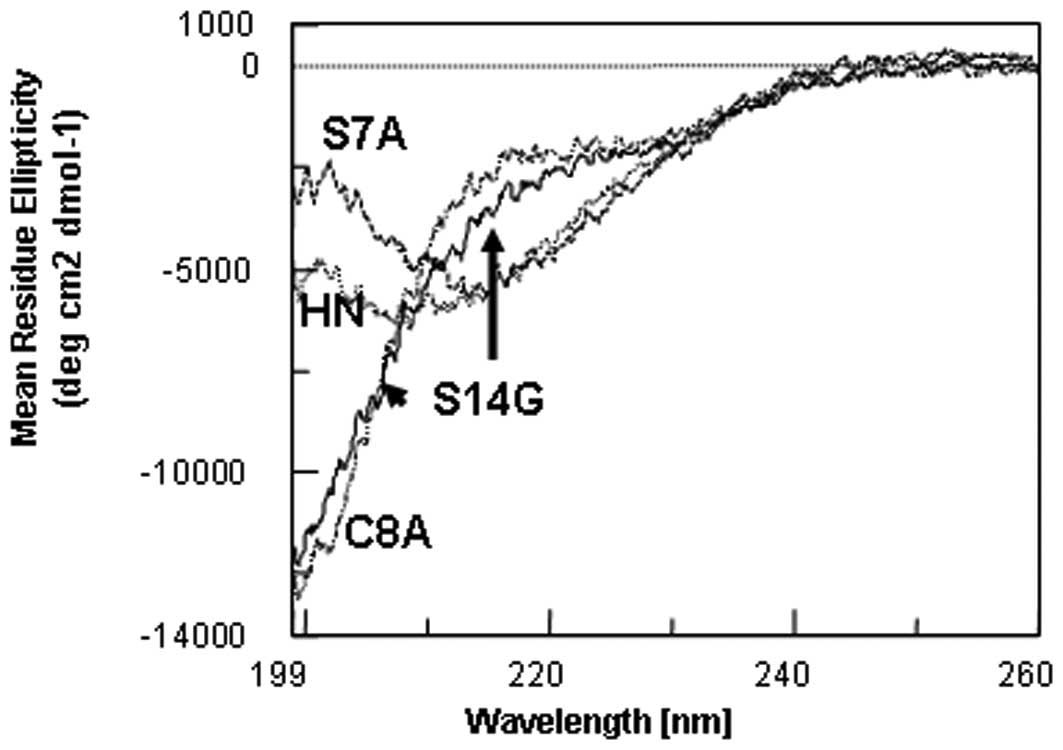
The differences in reversibility between these
peptides appear to be ascribed to the structure at 37ºC. The far-UV
CD spectra at 37ºC were compared among these four peptides
(Fig. 2). The spectra of HN
(dotted curve, also shown by the arrow) and S7A-HN (dashed curve)
have a valley, although at different wavelengths, indicating that
they undergo a transition into β-sheet aggregates. It appears that
the analogs have marginally different secondary structures.
Conversely, the spectra of S14G-HN (solid curve) and C8A-HN (double
dashed curve) are similarly disordered, indicating that these HN
analogs have an identical secondary structure at 37ºC. This
indicates that S14G-HN and C8A-HN have less or no tendency to form
β-sheet aggregates upon heating, although a sedimentation analysis
of C8A-HN has not yet been performed. This explains why the
structural changes are reversible upon heating, i.e., the lack of
aggregation. Such aggregation for HN and S7A-HN occurs only at
37ºC, as all these HN forms are similar and largely disordered at
5ºC (data not shown).
The time-course of heat-induced structural changes
and their reversibility were examined as follows. Previously, we
have used a wavelength at 201 nm to follow the time-course, as the
change in CD intensity is greatest at this wavelength (12). However, it does not correspond to a
change in specific secondary structures, e.g., an α-helix or
β-sheet. As observed in Fig. 1,
the signal at 217 nm significantly changed upon heating and
corresponded to an induction of a β-sheet structure for HN and
S7A-HN (see arrow for the changes in the CD signal). However, this
wavelength was not informative for S14G-HN and C8A-HN, as observed
in Fig. 1B (smaller arrow). The
changes at 217 nm were too small to follow the structural changes
for S14G-HN and C8A-HN. The time-course of signal changes at 201 nm
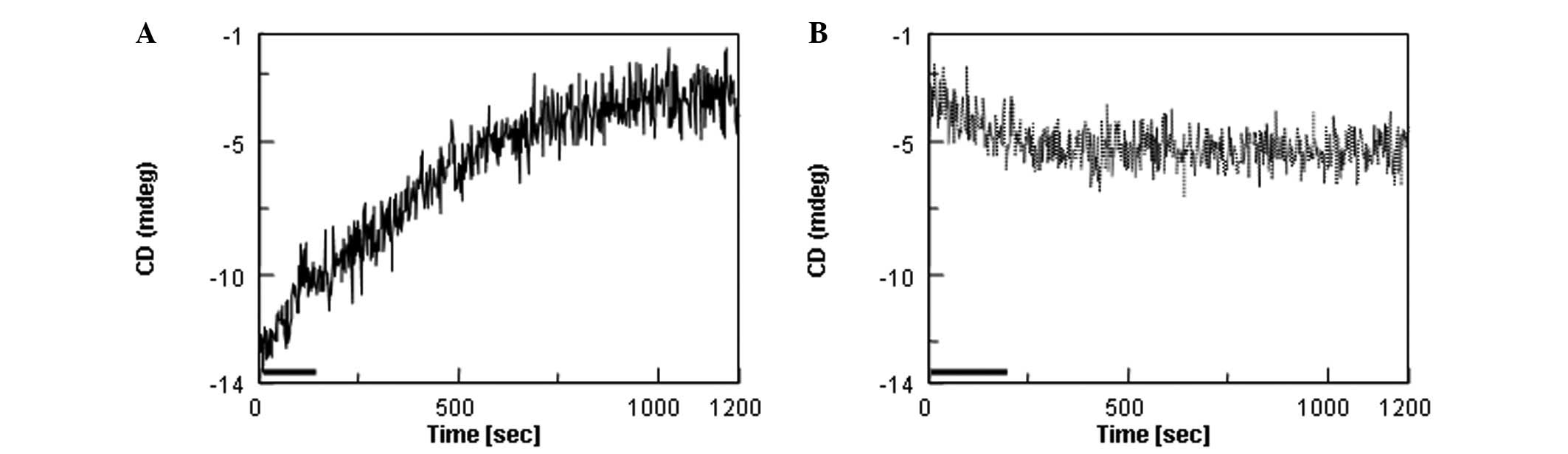
upon heating was determined for HN (Fig. 3). In Fig. 3A, the sample temperature was raised
from 5 to 37ºC at time zero. It took ~120 sec to bring the
temperature to 37ºC, as indicated by the black bar. There were slow
increases in the CD intensity, consistent with the spectral changes
observed in Fig. 1. The signals
appeared to reach a plateau around 1,000 sec. Following completion
of a 20-min time-course measurement, the sample temperature was
immediately dropped to 5ºC and then the CD signal at 201 nm was
followed in Fig. 3B. The signal at
time zero was close to the signal at 1,200 sec (Fig. 3A), meaning that the structure at
37ºC remained intact immediately after changing the temperature to
5ºC. There were only small decreases in the 201-nm intensity with a
5ºC incubation, the final value at 1,200 sec being far from the
value of −13 mdeg for the starting structure (prior to heating to
37ºC). The results were consistent with the spectral changes in
Fig. 1A. The changes in CD
intensity upon the temperature shift are summarized in Table I. It is evident that the structural
changes caused by heating at 37ºC are largely irreversible for
HN.
 | Table ICD signal change due to a temperature
shift. |
Table I
CD signal change due to a temperature
shift.
| Sample | Change at 201 nm,
mdeg | Recovery, % (n/total
n) |
|---|
|
|---|
| 5→37ºC | 37→5ºC |
|---|
| HN | 10 | 2 | 20 (2/10) |
| S7A-HN | 8 | 2 | 25 (2/8) |
| S14G-HN | 2 | 2 | 100 (2/2) |
| C8A-HN | 2 | 2 | 100 (2/2) |
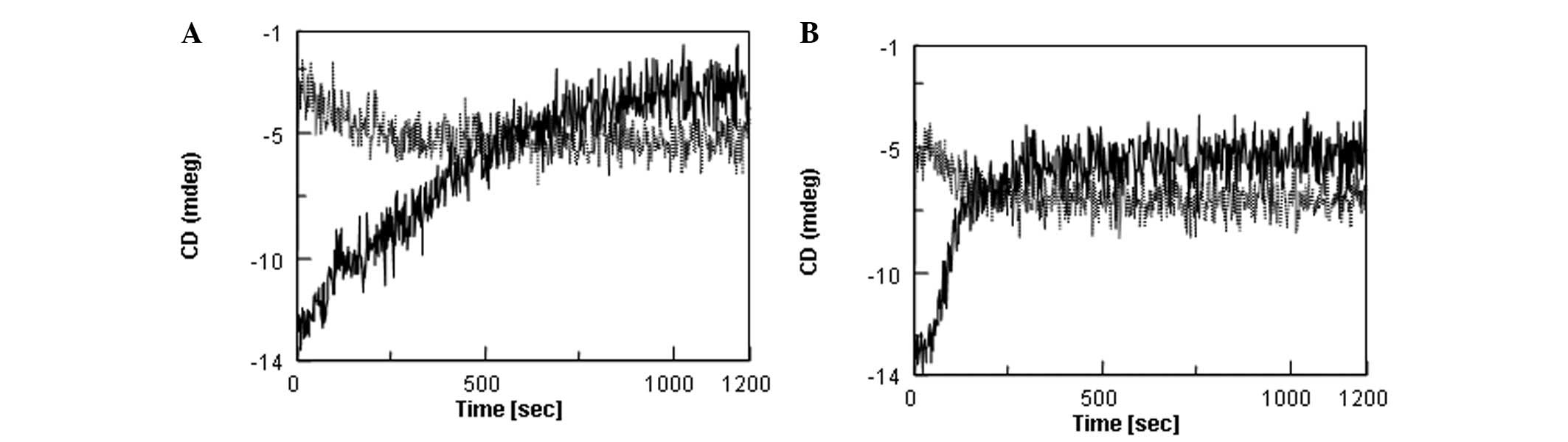
The same results were replotted in Fig. 4A for HN; changes due to a
temperature increase (solid curve, 5→37ºC) and subsequent decrease
(dotted curve, 37→5ºC) are plotted on the same panel. Fig. 4B shows the results for S7A-HN. The
signal change upon heating was marginally smaller for S7A-HN than
HN. However, these peptides were similar in reversibility. The
signal changes induced by heating at 37ºC were irreversible upon
cooling. Additionally, it is evident that these two peptides were
marginally different with regard to the time-course. The 201-nm
signal change appeared to occur faster for S7A-HN than for HN (when
comparing Fig. 4A and B). If, in
fact, this signal change was due to aggregation, the result
indicates that S7A-HN aggregates faster upon incubation at
37ºC.
The changes at 201 nm upon heating and cooling for
S14G-HN and C8A-HN were also determined as a function of incubation
time (data not shown). For S14G-HN and C8A-HN, the 201 nm signal
changed marginally upon heating to 37ºC and was fully recovered
upon cooling. For S14G-HN and C8A-HN, the signal changes were much
smaller than HN and S7A-HN (Table
I), ~20–25% those for HN and S7A-HN. As demonstrated by the
changes upon cooling (Table I),
the recovery of the CD signal was ~100%, indicating an apparent
full reversibility. Such reversibility is consistent with the
theory that S14G-HN and C8A-HN do not aggregate upon heating at
37ºC under the experimental conditions. The time-course of signal
change upon heating appears to be identical for S14G-HN and C8A-HN
(data not shown). Thus, it may be concluded that there is no
difference between S14G-HN and C8A-HN in terms of the stability at
37ºC.
The present study also analyzed how the
temperature-dependent structural changes and aggregation affect the
biological activity of the HN and HN analogs. We have previously
suggested the possibility that the instability of S7A-HN at 37ºC is
at least one of the factors responsible for the observed lack of
biological activity (12); namely,
that it may aggregate in a physiological solution prior to reaching
its target site during in vitro or in vivo assays.
Thus, those peptides that have a greater tendency to aggregate at
37ºC may lose activity. In the present study, the inactive C8A-HN
behaved differently from the inactive S7A-HN and was similar to the
potent S14G-HN analog, meaning that the stability at 37ºC did not
correlate with the loss of neuroprotective activity for C8A-HN. One
possibility is that C8A-HN in fact lost its ability to bind to the
target site to which the active HN and S14G-HN bind. However, it
should be noted that the S14G-HN that revealed no apparent
aggregation tendency in the present study does aggregate under
harsher conditions, e.g., prolonged incubation, higher temperature,
higher peptide concentration and higher ionic strength (13,15–17).
Thus, it is possible that C8A-HN aggregates under certain bioassay
conditions, resulting in activity loss. In other words, this analog
may be active depending on the bioassay system that is capable of
reducing exposure to harsh conditions and thereby minimize
aggregation. In fact, the preliminary bioassay results of C8A-HN,
based on phosphorylation of early response kinase stimulated by the
HN peptides through their receptor, indicated C8A-HN to be active
(Kita and Niikura, unpublished data).
In the present study, an attempt was made to relate
the effects of these mutations on HN aggregation to the physical
properties of the side chain of each mutated amino acid. Since HN
is disordered, it is more likely that these amino acid side chains
are solvent-exposed. The solvent-exposed sequence may be analyzed
by a computer program to predict the aggregation or fibrillation
tendency. In the present study, the Waltz program was used
(18). Using the highest
sensitivity for the propensity of aggregation, the following
sequences were shown to be prone to aggregation: S7-I16 for HN,
F6-I16 for S7A-HN, G5-I16 for C8A-HN and S7-G14 for S14G-HN. The
observed aggregation of HN, S7A-HN and S14G-HN appears to agree
with the prediction that a greater tendency for aggregation
correlates with the longer predicted sequence. However, the
observed deviation for C8A-HN (the longest sequence) requires
further explanation.
As expected, all these sequences contain L9–L12,
indicating that the hydrophobicity is also involved in the observed
aggregation. Side chain hydrophobicity has been determined by a
pioneering study by Nozaki and Tanford (19). The free energy of the transfer of
the glycine side chain from ethanol (i.e., core environments of the
folded protein structure or aggregated structure) to water was
assumed to be zero; in other words, the glycine side chain has no
preference for water over hydrophobic environments. A serine side
chain is more stable in water than a glycine side chain by a
transfer free energy of −300 cal/mol, meaning that a S14G mutation
should render S14G-HN less polar or more hydrophobic, as this
mutation loses favorable interaction free energy of the serine side
chain with water. This disagrees with the lower observed
aggregation tendency of S14G-HN or the Waltz prediction. This may
be explained by the unique structural nature of glycine, as pointed
out by Richardson and Richardson (20); glycine is well known to form a
local structure around glycine making it more mobile, facilitating
backbone motion and favoring the disordered structure. While the
S14G mutation renders S14G-HN less polar, it may stabilize the
disordered structure and prevent conversion into a more folded
β-sheet, and thereby into an aggregated structure. This may be why
the Waltz prediction demonstrated termination at residue G14 in the
present study. With regard to S7A-HN, the alanine side chain is
non-polar by 500 cal/mol, i.e., its side chain interaction with
water is highly unfavorable. Combining the polar nature of the
serine side chain and the non-polar nature of the alanine side
chain, the S7A mutation renders S7A-HN highly hydrophobic,
consistent with a one amino acid excess over the parental HN in the
Waltz prediction. No free energy of transfer data is available for
the cysteine side chain. Using the hydropathy scale of Kyte and
Doolittle (21) shows, however,
that a cysteine side chain is more hydrophobic than an alanine side
chain, meaning that the C8A mutation reduces the hydrophobicity,
consistent with its weaker aggregation, although inconsistent with
the Waltz prediction.
A number of short peptides behave similarly to HN,
i.e., they are largely disordered in aqueous solutions and have the
tendency to aggregate, often leading to structures termed β-amyloid
fibrils (22–26). Peptides are clinically significant
biopharmaceuticals (1–4) and their development requires stable
formulations, physically and chemically, for long-term storage.
Considering the irreversible nature of aggregation, as observed in
the present study for HN, there is a requirement to design a
formulation of short peptides in order to minimize aggregation.
In conclusion, less active HN and inactive S7A-HN
demonstrated larger and irreversible structural changes upon
heating than the more active S14G-HN analog. Notably, the C8A-HN
analog, which has been previously reported to be inactive, revealed
a weak aggregation tendency similar to S14G-HN, indicating that the
activity of C8A-HN may be compromised due to factors other than
instability under physiological conditions.
References
|
1
|
Fields K, Falla TJ, Rodan K and Bush L:
Bioactive peptides: signaling the future. J Cosmet Dermatol.
8:8–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aneiros A and Garateix A: Bioactive
peptides from marine sources: pharmacological properties and
isolation procedures. J Chromatogr B Analyt Technol Biomed Life
Sci. 803:41–53. 2004.PubMed/NCBI
|
|
3
|
Mason JM: Design and development of
peptides and peptide mimetics as antagonists for therapeutic
intervention. Future Med Chem. 2:1813–1822. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Otvos L Jr: Synthesis of a multivalent,
multiepitope vaccine construct. Methods Mol Biol. 494:263–273.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hashimoto Y, Niikura T, Tajima H, et al: A
rescue factor abolishing neuronal cell death by a wide spectrum of
familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci
USA. 98:6336–6341. 2001.PubMed/NCBI
|
|
6
|
Yamagishi Y, Hashimoto Y, Niikura T and
Nishimoto I: Identification of essential amino acids in Humanin, a
neuroprotective factor against Alzheimer’s disease-relevant
insults. Peptides. 24:585–595. 2003.PubMed/NCBI
|
|
7
|
Hashimoto Y, Kurita M and Matsuoka M:
Identification of soluble WSX-1 not as a dominant-negative but as
an alternative functional subunit of a receptor for an
anti-Alzheimer’s disease rescue factor Humanin. Biochem Biophys Res
Commun. 389:95–99. 2009.
|
|
8
|
Hashimoto Y, Niikura T, Ito Y, et al:
Detailed characterization of neuroprotection by a rescue factor
humanin against various Alzheimer’s disease-relevant insults. J
Neurosci. 21:9235–9245. 2001.PubMed/NCBI
|
|
9
|
Ikonen M, Liu B, Hashimoto Y, et al:
Interaction between the Alzheimer’s survival peptide humanin and
insulin-like growth factor-binding protein 3 regulates cell
survival and apoptosis. Proc Natl Acad Sci USA. 100:13042–13047.
2003.
|
|
10
|
Terashita K, Hashimoto Y, Niikura T, et
al: Two serine residues distinctly regulate the rescue function of
Humanin, an inhibiting factor of Alzheimer’s disease-related
neurotoxicity: functional potentiation by isomerization and
dimerization. J Neurochem. 85:1521–1538. 2003.PubMed/NCBI
|
|
11
|
Armas A, Sonois V, Mothes E, Mazarguil H
and Faller P: Zinc(II) binds to the neuroprotective peptide
humanin. J Inorg Biochem. 100:1672–1678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arakawa T, Niikura T and Kita Y: The
biological activity of Humanin analogs correlates with structure
stabilities in solution. Int J Biol Macromol. 49:93–97. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arisaka F, Arakawa T, Niikura T and Kita
Y: Active form of neuroprotective Humanin, HN, and inactive analog,
S7A-HN, are monomeric and disordered in aqueous phosphate solution
at pH 6.0; No correlation of solution structure with activity.
Protein Pept Lett. 16:132–137. 2009. View Article : Google Scholar
|
|
14
|
Thies MJ, Talamo F, Mayer M, et al:
Folding and oxidation of the antibody domain C(H)3. J Mol Biol.
319:1267–1277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arakawa T, Niikura T, Tajima H and Kita Y:
The secondary structure analysis of a potent Ser14Gly analog of
antiAlzheimer peptide, Humanin, by circular dichroism. J Pept Sci.
12:639–642. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arakawa T, Kita Y and Niikura T: A rescue
factor for Alzheimer’s diseases: discovery, activity, structure,
and mechanism. Curr Med Chem. 15:2086–2098. 2008.
|
|
17
|
Pistolesi S, Rossini L, Ferro E, Basosi R,
Trabalzini L and Pogni R: Humanin structural versatility and
interaction with model cerebral cortex membranes. Biochemistry.
48:5026–5033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maurer-Stroh S, Debulpaep M, Kuemmerer N,
et al: Exploring the sequence determinants of amyloid structure
using position-specific scoring matrices. Nature Methods.
7:237–242. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nozaki Y and Tanford C: The solubility of
amino acids and two glycine peptides in aqueous ethanol and dioxane
solutions. Establishment of a hydrophobicity scale. J Biol Chem.
246:2211–2217. 1971.PubMed/NCBI
|
|
20
|
Richardson JS and Richardson DC:
Principles and patterns of protein conformation. Prediction of
Protein Structure and the Principles of Protein Conformation.
Fasman G: Plenum Press; New York: pp. 43–75. 1989
|
|
21
|
Kyte J and Doolittle RF: A simple method
for displaying the hydropathic character of a protein. J Mol Biol.
157:105–132. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou X, Liu J, Li B, Pillai S, Lin D, Liu
J and Zhang Y: Assembly of glucagon (proto)fibrils by longitudinal
addition of oligomers. Nanoscale. 3:3049–3051. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arvinte T, Cudd A and Drake AF: The
structure and mechanism of formation of human calcitonin fibrils. J
Biol Chem. 268:6415–6422. 1993.PubMed/NCBI
|
|
24
|
Steckmann T, Awan Z, Gerstman BS and
Chapagain PP: Kinetics of peptide secondary structure conversion
during amyloid β-protein fibrillogenesis. J Theor Biol. 301:95–102.
2012.
|
|
25
|
Kodali R, Williams AD, Chemuru S and
Wetzel R: Abeta(1–40) forms five distinct amyloid structures whose
beta-sheet contents and fibril stabilities are correlated. J Mol
Biol. 401:503–517. 2010.
|
|
26
|
Macchi F, Hoffmann SV, Carlsen M, et al:
Mechanical stress affects glucagon fibrillation kinetics and fibril
structure. Langmuir. 27:12539–12549. 2011. View Article : Google Scholar : PubMed/NCBI
|