Introduction
Platelets are essential for the maintenance of
hemostasis through adhesion, aggregation/cohesion and the release
of thrombin and cytokines. Platelet hyperfunction may be involved
in various abnormalities and diseases, including thrombosis,
atherosclerosis, tumor progression/metastasis, stroke and
myocardial infarction induced by arterial thrombosis (1). In addition to thrombosis, platelets
are also a pivotal player in the host defense response (2).
Virus infection is able to stimulate a series of
platelet responses, including platelet binding and engulfment.
Furthermore, activated platelets express P-selectin, which mediates
the elimination of viruses by macrophages (3,4).
Following activation, platelets release granular contents, which
contain biologically active peptides that target the immune system
through RANTES, IL-1β, MCP-1, PF-4 and PAF pathways (5). Thus, platelets are essential in
regulating thrombosis and inflammation.
Human adenovirus (HAdV) is a common cause of human
acute respiratory diseases. HAdVs also cause conjunctivitis,
gastrointestinal and urinary tract infections and occasionally
encephalitis. Platelets are important in the innate immune response
against HAdV infection. Integrin αIIbβ3, which recognizes
arginine-glycine-aspartic acid (RGD) sequences, is expressed on the
surface of platelets (6). RGD
sequences are also present in the penton base of HAdV. HAdV is able
to bind to macrophage cells, which express αIIbβ3, suggesting the
possibility of binding between the RGD sequences of HAdVs and
αIIbβ3 on the surface of platelets (7). Infection of HAdV into the cells
involves RGD sequence mediated adhesion to the cells and virus
type-dependent entry into the cells. The interaction between the
virus and αIIbβ3 leads to the activation of platelets and is
required for the internalization of HAdV (8,9).
However, the effect of HAdV on platelet activation is
controversial: Eggerman et al demonstrated that an HAdV
vector had no effect on platelet aggregation (10), while later Othman et al
demonstrated that an HAdV vector did induce platelet activation
(3). Thus, the platelet response
to HAdV infection requires further examination.
HAdV type 3 (HAdV3) is the most common type of
clinical HAdV infection (11).
HAdV3 infection is particularly severe among young children and
immunocompromised adults (12).
Understanding the platelet response to HAdV infection may aid the
development of measures to prevent severe HAdV infection. In the
present study, we studied platelet-HAdV interactions in
vitro following the establishment of a whole blood and
platelet-rich plasma model infected with HAdV3. We demonstrated
that HAdV at a certain concentration range may potentiate ADP and
ristocetin-induced platelet aggregation. A rapid increase of CD41a
and CD62P expression following incubation with HAdV was also
observed.
Materials and methods
Virus purification
HAdV3 was obtained from ATCC (Manassas, VA, USA).
The virus was propagated in A549 cells and was harvested when the
cytopathic effect (CPE) reached >95% by freezing (−80°C, 10 min)
and thawing (room temperature) the cell culture flasks three times.
The supernatant was stored at −80°C following a low speed
centrifugation (3000 × g for 5 min) (13). The purification of the virus was
performed using the VirTrap™ HAdV purification kit (Biomiga, San
Diego, CA, USA). Purified virus was quantified by
spectrophotometric measurement of the optical density at OD260 and
recorded as virus particles per milliliter (vp/ml).
Blood collection and preparation of
platelets
Ten blood donors (aged from 24 to 47 years) were
selected based on the following criteria: healthy volunteers with
normal platelet aggregation; parameters of coagulation and other
whole blood indicators were normal and no symptoms of flu were
found four weeks prior to blood withdrawal. Written informed
consent was obtained from the donors and the study was approved by
the Ethics Committee of the first clinical college of Harbin
Medical University (Harbin, China). Sodium citrate (109 mM) was
added to the whole blood at the ratio of 1:9 to prevent platelet
clumping prior to centrifuging at 800 × g for 7 min. The
supernatant was platelet-rich plasma (PRP) and the platelet count
was adjusted to 160–380×109/l using corresponding
platelet-free plasma, which was prepared by depleting platelets
with centrifugation at 3000 × g for 10 min. At least 5 replicates
of each of the following experiments were performed.
Measurement of platelet aggregation,
platelet counting and fibrinogen
PRP (500 μl) was incubated with 10 μl of HAdV at the
concentration range of 109–1011 vp/ml at 37°C
in vitro for 1, 30, 40, 50, 60, 70, 80, 90, 100 and 130 min,
respectively. Adenosine diphosphate (ADP; 10 μM) and ristocetin
(1.0 g/l) were used as inducers of platelet aggregation. The
inducer (2.5 μl) was added to PRP for each experiment. The rate of
platelet aggregation was measured by a 560CA Aggregometer
(Chrono-Log, Havertown, PA, USA). Following incubation of
anticoagulated whole blood with 2 μl (1010 vp/ml) of the
virus, the number of platelets was counted by a MEK-6318 hematology
analyzer (Nihon Kohden, Tokyo, Japan). Fibrinogen in
platelet-depleted plasma, which was obtained following
centrifugation, was analyzed by a Sysmex-1500 automated coagulation
analyzer (Sysmex Corporation, Kobe, Japan). Corresponding amounts
of PRP or anticoagulated whole blood served as controls.
Expression of P-selectin and CD41a in
platelets
P-selectin is a surface marker of activated
platelets. To measure P-selectin expression, 500 μl of PRP was
incubated with 10 μl of 1010 vp/ml HAdV at 37°C in
vitro for 1 and 30 min, respectively, prior to staining with PE
conjugated anti-human CD62P (P-selectin; Becton-Dickinson, Inc.,
Franklin Lakes, NJ, USA) and FITC conjugated anti-human CD41a
(Becton-Dickinson, Inc.) antibodies. Expression of P-selectin and
CD41a was determined by Canto II flow cytometry (Becton-Dickinson,
Inc.). PRP with virus-free buffer solution served as the
control.
Statistical analysis
Continuous variables with a normal distribution were
expressed as the mean ± SEM. The platelet counts, fibrinogen and
platelet aggregation induced by ADP or ristocetin, which were
measured at different time points and at different concentrations,
were subjected to ANOVA analysis. The Greenhouse-Geisser correction
was applied to all the data due to the violation of sphericity.
Independent sample t-tests were applied to compare the rate of
platelet aggregation, CD41a and CD62P between the control and virus
groups. All data were processed by SPSS 18.0 statistical software
and the conditions were as follows: two-sided test, α=0.05 as the
test level and P<0.05 was considered to indicate a statistically
significant difference.
Results
HAdV potentiates ADP-induced platelet
aggregation
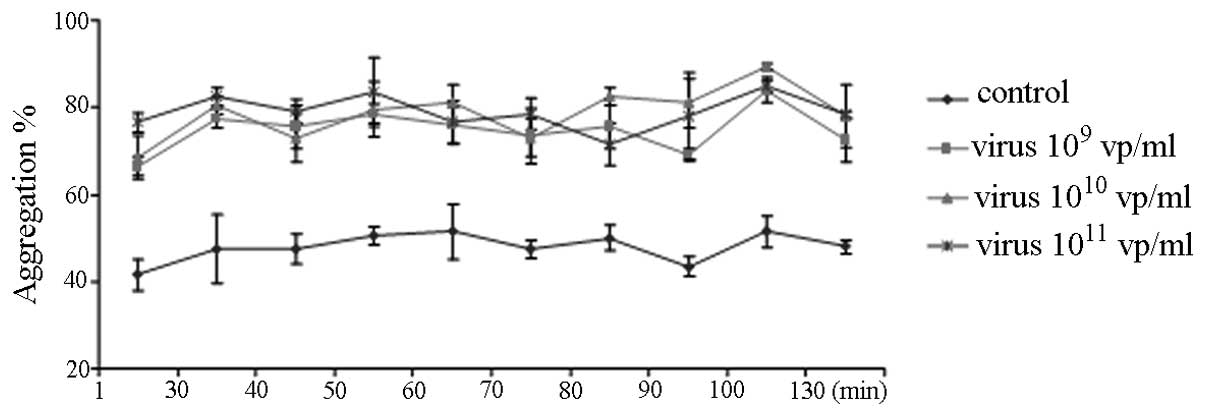
ADP-induced platelet aggregation was measured by the
Chrono-Log Aggregometer (Fig. 1).
Pre-incubation of PRP with 109–1011 vp/ml of
HAdV for up to 130 min caused the rate of ADP-induced platelet
aggregation to markedly increase (Fig.
1). However, the platelet aggregation was not affected by the
length of incubation time with the HAdV (Fig. 1) and was not dose dependent.
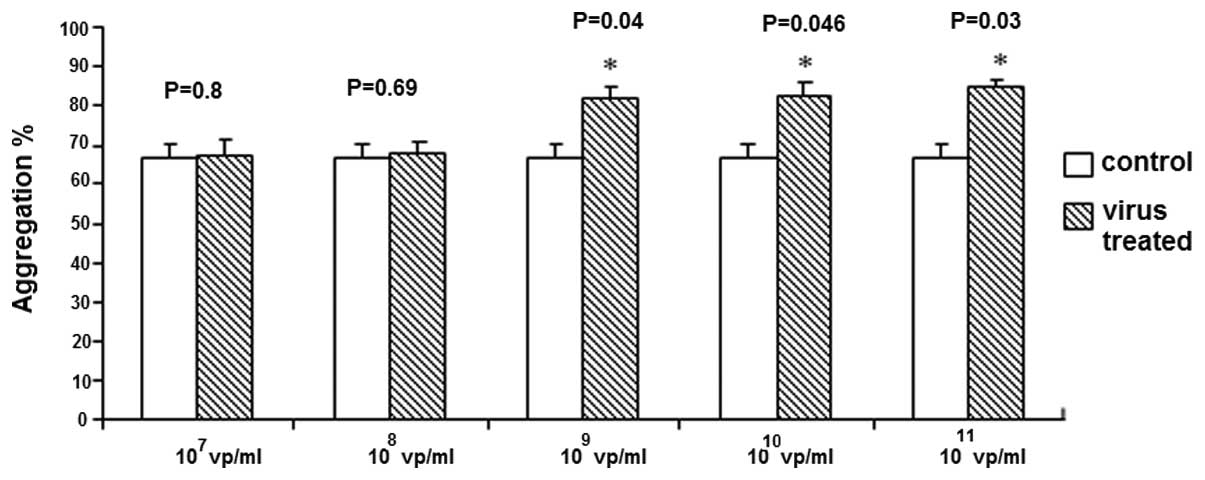
Lower concentrations of HAdV
(107–108 vp/ml) demonstrated no effect
(Fig. 2), however HAdV
concentrations of 109–1011 vp/ml
significantly enhanced ADP-induced platelet aggregation.
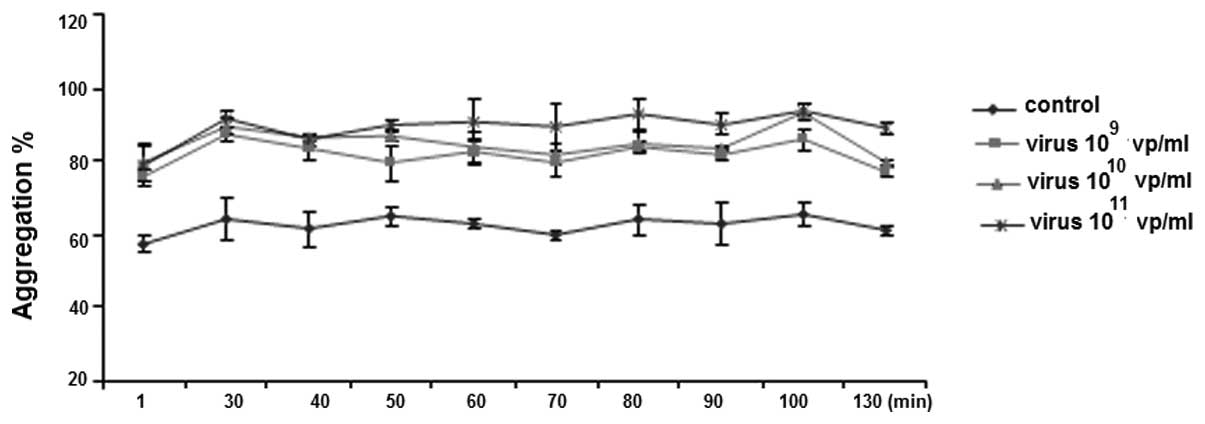
HAdV potentiates ristocetin-induced
platelet aggregation
Similar to ADP-induced platelet aggregation,
pre-incubation of PRP with 109–1011 vp/ml of
HAdV significantly increased ristocetin-induced platelet
aggregation (Fig. 3). However,
there was no difference among the three dosages (109,
1010 and 1011 vp/ml) or incubation time
(Fig. 3).
HAdV did not alter the platelet count or
fibrinogen
HAdV (1010 vp/ml) was incubated with
whole blood for the indicated time periods (1–130 min) prior to
platelet counting. Variance analysis demonstrated no statistical
significance in platelet counts among all groups (df=1, F=0.15,
P=0.74) or among different time points (df=1.88, F=1.25,
P=0.38).
1010 vp/ml of HAdV was incubated with
whole blood for the indicated time periods (1–130 min) prior to
fibrinogen measurement. Variance analysis of fibrinogen suggested
no significance among groups (df=1, F=7.74, P=0.11) or among time
points (df=1.37, F=0.94, P=0.45).
We demonstrated that incubation of whole blood with
1010 vp/ml of HAdV did not alter platelet count and
fibrinogen in anticoagulated whole blood. There was no significant
difference in platelet count and fibrinogen among the control and
virus groups at different time points (P>0.05; data not
shown).
HAdV induced P-selectin and CD41a
expression on platelets
Pre-incubation of PRP with HAdV for 30 min increased
the percentage of CD62P-positive platelets from 0.38±0.07 to
0.70±0.02 (P=0.03), indicating activation of the platelets.
Similarly, 30 min of treatment with HAdV increased the mean channel
fluorescence intensity of CD41a surface staining from 247.6±105.7
to 382.3±71.7 (P=0.04) compared with the control group (Fig. 4).
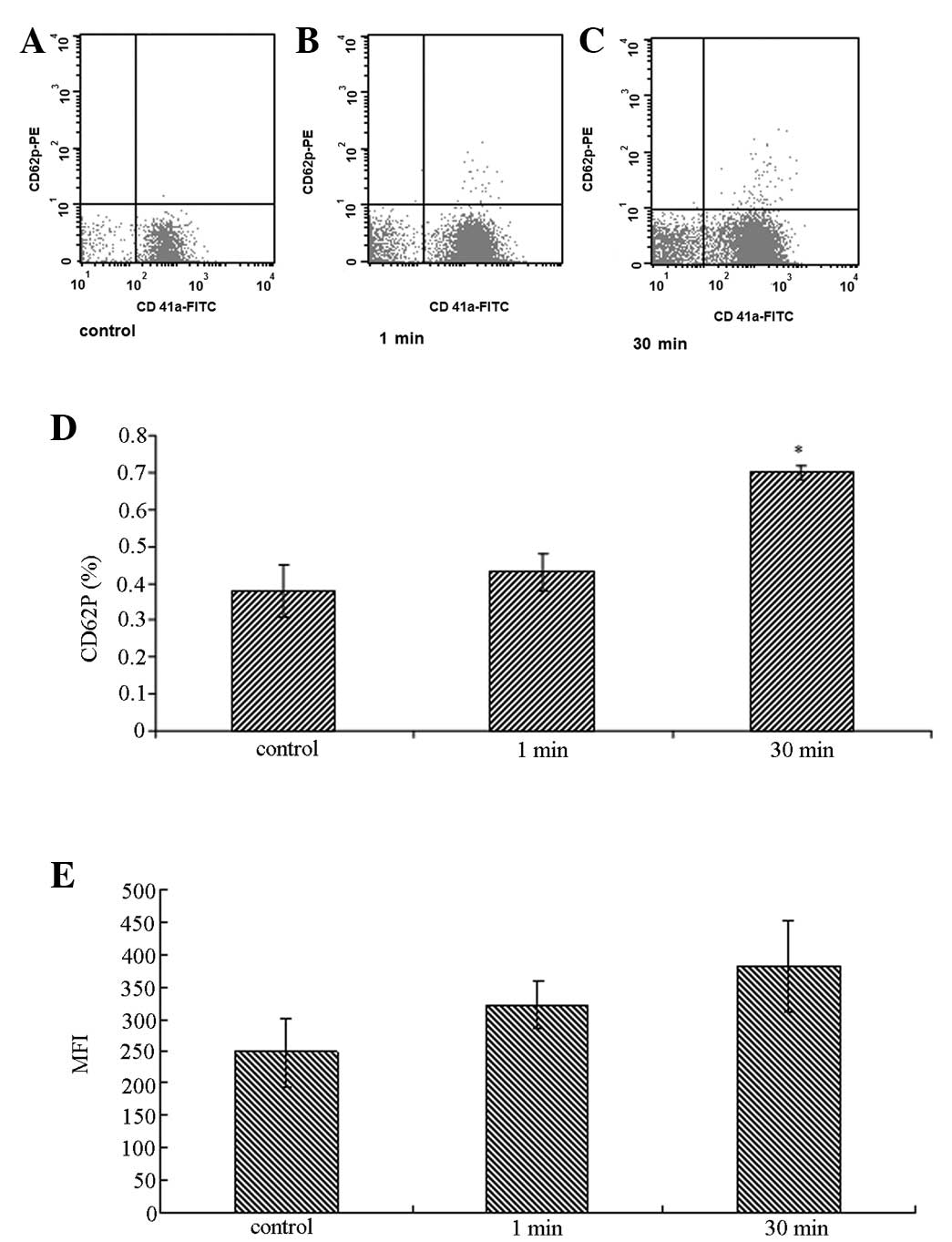 | Figure 4Detection of CD62P and CD41a on the
surface of platelets by flow cytometry. PRP was incubated with 10
μl of 1010 vp/ml adenovirus at 37°C in vitro for
1 and 30 min, respectively, prior to staining with PE-conjugated
anti-human CD62P (P-selectin; BD Biosciences, Franklin Lakes, NJ,
USA) and FITC-conjugated anti-human CD41a (Becton-Dickinson, Inc.,
Franklin Lakes, NJ, USA) antibodies. PRP with virus-free buffer
solution served as the control. * P<0.05, compared
with the 1 min control group. PRP, platelet-rich plasma; MFI, mean
fluorescence intensity. |
Discussion
HAdV is the second most common pathogen, which
causes adult community-acquired pneumonia (14). HAdV3 is one of the most prevalent
serotypes detected globally and its variants have been associated
with outbreaks of severe disease (12). Recently, HAdV3 infection
demonstrated an increasing trend (11). However, the majority of studies are
focused on the interaction between platelets and HAdV type 5 (often
used as a gene vector), whereas studies on the wild-type HAdV are
scattered (6,9). In particular, platelet function
during HAdV infection is unclear (15). This study provided evidence that
HAdV infection may activate platelets and increase platelet
aggregation.
The platelet aggregation test is routinely used to
detect the state of platelet activation in the blood. αIIbβ3
integrin on platelets is important in platelet aggregation and
thrombus formation. The major platelet receptor, αIIbβ3 integrin is
inactive in quiescent cells, however inducers, including ADP and
thrombin or platelet activators, may lead to its persistent
activation and binding to fibrinogen and other ligands (16,17).
Thus, a large number of micro-organisms are able to enter into the
cells mediated by integrin (18).
We demonstrated that the HAdV at a certain concentration range
(109–1011 vp/ml) is able to induce platelet
aggregation. It is likely that the binding of the RGD sequence of
HAdV and αIIbβ3 integrin on platelets leads to the activation of
platelets. αIIbβ3 on the activated platelet surface may also bind
to fibrinogen to facilitate the aggregation of platelets (19).
In the present study, the rate of platelet
aggregation did not change with the length of incubation time (up
to 130 min). This may be explained by the short incubation periods
we selected since it is advised to finish platelet aggregation
tests in vitro within 2 h. However, in vivo
experiments have demonstrated that HAdV type 5 induces platelet
activation 6 h after the virus is injected into the body (9).
Among various concentrations of HAdV we examined,
only 109 vp/ml and above potentiated the significant
aggregation of platelets, suggesting that platelet activation is
concentration dependent. It is possible that less HAdV binds to
glycoprotein (GP) IIb–IIIa (αIIbβ3-integrin) at lower
concentrations. Thus, certain amounts of activated integrin may be
required to form the bridge among platelets and HAdVs. Although no
direct evidence suggests the expression of αIIbβ3 integrin is able
to directly induce platelet aggregation, pharmacokinetic studies of
an αIIbβ3 integrin antagonist demonstrate a marked correlation
between platelet aggregation and the quantity of free platelet GP
IIb–IIIa receptors (20).
Recently, studies have demonstrated that the
injection of HAdV3 causes thrombocytopenia in mice and clinical
studies have also demonstrated a decreased platelet count following
infection with HAdV in humans (21,22).
However, our study demonstrated a slight decrease in platelet
counts without statistical significance following HAdV infection.
This may be explained by the activation of the anti-inflammatory
defense system stimulated by platelets infected with HAdV, which
subsequently activate phagocytosis by monocytes/macrophages to
engulf HAdV infected platelets (9,19).
However, there is no such mechanism in vitro, which may
explain the insignificant decrease of platelets.
In order to confirm the role of HAdV in platelet
activation, we next measured the CD62P expression on the membrane
of platelets. P-selectin is a cell adhesion molecule expressed in
platelets and endothelial cells. It is usually stored in secretory
granules and rapidly expressed on the plasma membrane following the
activation of platelets (23).
P-selectin plays two major roles: firstly, P-selectin
re-distributes on the surface of activated endothelial cells (to
mediate the rolling of the leukocyte) during inflammation (24,25);
secondly, P-selectin expression on activated platelets (in the
thrombus) enhances leukocyte recruitment in the process of
thrombosis (26). Thus, P-selectin
expressed on platelets is involved in the processes of
inflammation, thrombosis and coagulation and may serve as an
indicator for the hypercoagulable state in vivo(26,27).
In the present study, we observed rapid exposure of P-selectin on
the platelet surface following the addition of HAdV, suggesting
that the interaction between HAdV and platelets leads to platelet
activation. In addition, our in vitro data demonstrated a
significantly increased expression of CD41a on the surface of
platelets incubated with HAdV but not on the resting platelets.
Since CD41a is important in platelet aggregation (19), our observation indicated possible
platelet aggregation induced by the HAdV.
Antiplatelet therapy reduces the inflammatory
response by inhibiting platelet activation and aggregation
(28). Our results suggested that
HAdV3 may induce the activation of platelets and lead to a
prothrombotic state. Our results may assist in the development of
measures to prevent severe HAdV infection.
Acknowledgements
This study is supported by the National Natural
Science Foundation of China (grant no. 30771909 and grant no.
81172725).
Abbreviations:
|
PRP
|
platelet-rich plasma
|
|
RGD
|
arginine-glycine-aspartic acid
|
|
ADP
|
adenosine diphosphate
|
|
HAdV3
|
human adenovirus type 3
|
References
|
1
|
Picker SM: In-vitro assessment of platelet
function. Transfus Apher Sci. 44:305–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klinger MH and Jelkmann W: Role of blood
platelets in infection and inflammation. J Interferon Cytokine Res.
22:913–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Othman M, Labelle A, Mazzetti I, Elbatarny
HS and Lillicrap D: Adenovirus-induced thrombocytopenia: the role
of von Willebrand factor and P-selectin in mediating accelerated
platelet clearance. Blood. 109:2832–2839. 2007.PubMed/NCBI
|
|
4
|
Youssefian T, Drouin A, Massé JM, Guichard
J and Cramer EM: Host defense role of platelets: engulfment of HIV
and Staphylococcus aureus occurs in a specific subcellular
compartment and is enhanced by platelet activation. Blood.
99:4021–4029. 2002. View Article : Google Scholar
|
|
5
|
Senzel L, Gnatenko DV and Bahou WF: The
platelet proteome. Curr Opin Hematol. 16:329–333. 2009. View Article : Google Scholar
|
|
6
|
Zhang Y and Bergelson JM: Adenovirus
receptors. J Virol. 79:12125–12131. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Faraday N, Rade JJ, Johns DC, Khetawat G,
Noga SJ, DiPersio JF, Jin Y, Nichol JL, Haug JS and Bray PF: Ex
vivo cultured megakaryocytes express functional glycoprotein
IIb–IIIa receptors and are capable of adenovirus-mediated transgene
expression. Blood. 94:4084–4092. 1999.PubMed/NCBI
|
|
8
|
Triantafilou K, Triantafilou M, Takada Y
and Fernandez N: Human parechovirus 1 utilizes integrins
alphavbeta3 and alphavbeta1 as receptors. J Virol. 74:5856–5862.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni
S and Lieber A: Adenovirus-platelet interaction in blood causes
virus sequestration to the reticuloendothelial system of the liver.
J Virol. 81:4866–4871. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eggerman TL, Mondoro TH, Lozier JN and
Vostal JG: Adenoviral vectors do not induce, inhibit, or potentiate
human platelet aggregation. Hum Gene Ther. 13:125–128. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gray GC, McCarthy T, Lebeck MG, et al:
Genotype prevalence and risk factors for severe clinical adenovirus
infection, United States 2004–2006. Clin Infect Dis. 45:1120–1131.
2007.PubMed/NCBI
|
|
12
|
Lebeck MG, McCarthy TA, Capuano AW,
Schnurr DP, Landry ML, Setterquist SF, Heil GL, Kilic S and Gray
GC: Emergent US adenovirus 3 strains associated with an epidemic
and serious disease. J Clin Virol. 46:331–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shang L, Qu Z, Sun L, Wang Y, Liu F, Wang
S, Gao H and Jiang F: Astragaloside IV inhibits adenovirus
replication and apoptosis in A549 cells in vitro. J Pharm
Pharmacol. 63:688–694. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Angeles Marcos M, Camps M, Pumarola T,
Antonio Martinez J, Martinez E, Mensa J, Garcia E, Peñarroja G,
Dambrava P, Casas I, Jiménez de Anta MT and Torres A: The role of
viruses in the aetiology of community-acquired pneumonia in adults.
Antivir Ther. 11:351–359. 2006.
|
|
15
|
Maurice A, Marchand-Arvier M, Edert D, Le
Faou A, Gondrexon G and Vigneron C: The virucidal effect of
platelet concentrates: preliminary study and first conclusions.
Platelets. 13:219–222. 2002.PubMed/NCBI
|
|
16
|
Basani RB, French DL, Vilaire G, Brown DL,
Chen F, Coller BS, Derrick JM, Gartner TK, Bennett JS and Poncz M:
A naturally occurring mutation near the amino terminus of alphaIIb
defines a new region involved in ligand binding to alphaIIbbeta3.
Blood. 95:180–188. 2000.
|
|
17
|
Shimaoka M and Springer TA: Therapeutic
antagonists and conformational regulation of integrin function. Nat
Rev Drug Discov. 2:703–716. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Plow EF, Haas TA, Zhang L, Loftus J and
Smith JW: Ligand binding to integrins. J Biol Chem.
275:21785–21788. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yakushkin VV, Zyuryaev IT, Khaspekova SG,
Sirotkina OV, Ruda MY and Mazurov AV: Glycoprotein IIb–IIIa content
and platelet aggregation in healthy volunteers and patients with
acute coronary syndrome. Platelets. 22:243–251. 2011.
|
|
20
|
Mazurov AV, Pevzner DV, Antonova OA,
Byzova TV, Khaspekova SG, Semenov AV, Vlasik TN, Samko AN,
Staroverov II and Ruda MY: Safety, inhibition of platelet
aggregation and pharmacokinetics of Fab’2 fragments of the
anti-glycoprotein IIb–IIIa monoclonal antibody FRaMon in high-risk
coronary angioplasty. Platelets. 13:465–477. 2002.
|
|
21
|
Appledorn DM, Kiang A, McBride A, Jiang H,
Seregin S, Scott JM, Stringer R, Kousa Y, Hoban M, Frank MM and
Amalfitano A: Wild-type adenoviruses from groups A-F evoke unique
innate immune responses, of which HAd3 and SAd23 are partially
complement dependent. Gene Ther. 15:885–901. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klinger JR, Sanchez MP, Curtin LA, Durkin
M and Matyas B: Multiple cases of life-threatening adenovirus
pneumonia in a mental health care center. Am J Respir Crit Care
Med. 157:645–649. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McEver RP, Beckstead JH, Moore KL,
Marshall-Carlson L and Bainton DF: GMP-140, a platelet
alpha-granule membrane protein, is also synthesized by vascular
endothelial cells and is localized in Weibel-Palade bodies. J Clin
Invest. 84:92–99. 1989. View Article : Google Scholar
|
|
24
|
Denis CV, André P, Saffaripour S and
Wagner DD: Defect in regulated secretion of P-selectin affects
leukocyte recruitment in von Willebrand factor-deficient mice. Proc
Natl Acad Sci USA. 98:4072–4077. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sundd P, Pospieszalska MK, Cheung LS,
Konstantopoulos K and Ley K: Biomechanics of leukocyte rolling.
Biorheology. 48:1–35. 2011.PubMed/NCBI
|
|
26
|
Palabrica T, Lobb R, Furie BC, Aronovitz
M, Benjamin C, Hsu YM, Sajer SA and Furie B: Leukocyte accumulation
promoting fibrin deposition is mediated in vivo by P-selectin on
adherent platelets. Nature. 359:848–851. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blann AD, Nadar SK and Lip GY: The
adhesion molecule P-selectin and cardiovascular disease. Eur Heart
J. 24:2166–2179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iannacone M, Sitia G, Narvaiza I, Ruggeri
ZM and Guidotti LG: Antiplatelet drug therapy moderates
immune-mediated liver disease and inhibits viral clearance in mice
infected with a replication-deficient adenovirus. Clin Vaccine
Immunol. 14:1532–1535. 2007. View Article : Google Scholar
|