Introduction
Glucocorticoids (GCs) are widely used in the local
or systemic treatment of chronic inflammatory or autoimmune
diseases, and of orthopedic disorders. Prolonged treatment with GCs
has various side-effects, including water, salt and in general,
metabolic disorders, and impairment of skeletal development
(1–3). Administration of GCs leads to a
reduction in bone mineral density (BMD), decreased generation of
osteoblasts and osteocytes and prolonged lifespan of osteoclasts,
with fractures occurring in 30–50% of patients chronically treated
with GCs (1). Recent studies have
demonstrated that GCs can induce apoptosis and autophagy in
osteocytes (4–6). The influence of GCs has been rarely
reported on chondrocytes, which play a key role in skeletal
development. Sporadic studies have reported GC effects on the
apoptosis and differentiation of chondrocytes (7,8).
Autophagy is a lysosomal degradation process that is essential for
cell growth, survival, differentiation, development and homeostasis
(9). It is closely regulated that
helps to maintain a balance between synthesis, as well as
degradation and subsequent recycling of cell products. It is
currently unknown whether autophagy occurs in chondrocytes
following GC treatment.
In this study, we examined autophagy upon GC
treatment in the ATDC5 chondrocyte cell line, and further evaluated
the role of autophagy on cell viability. Our findings highlight
autophagy as an important mechanism induced by GCs and potentially
promoting a reduction in chondrocyte cell viability.
Materials and methods
Reagents and antibodies
Dexamethasone (Dex), RU486, rapamycin,
3-methyladenine (3-MA) and polyclonal antibodies for
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and
microtubule-associated protein 1-light chain 3 (MAP1-LC3) were
purchased form Sigma-Aldrich (St. Louis, MO, USA). The polyclonal
antibody targeting beclin 1 was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). The fusion of the coding
sequence of MAP1-LC3 with that of GFP was synthesized and cloned
into the pcDNA3.1+ vector, to construct the
GFP-LC3-expressing plasmid.
Cell culture
The ATDC5 chondrocyte line (Sigma-Aldrich) was
cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s medium
(DMEM): Ham’s F12, supplemented with 2 mM glutamine and 5% fetal
bovine serum (FBS). We mixed sub-confluent cultures (70–80%) with
trypsin or trypsin/EDTA at a 1:4 ratio; the cells were incubated at
37°C in a humidified 5% CO2 incubator.
Assessment of autophagy with LC3 analysis
and electron microscopy
Autophagy was assessed in ATDC5 cells using the
GFP-LC3 construct and confocal microscopy analyses. ATDC5 cells
grown to 80% confluency were transfected for 24 h with the
GFP-LC3-expressing plasmid using Lipofectamine 2000 (Invitrogen
Life Technologies, Carlsbad, CA, USA). Following transfection,
cells were treated with rapamycin (200 nM), Dex (1, 10 or 100 μM)
or RU486 (10 μM) for an additional 24 h and analyzed by
fluorescence microscopy (IX71, Olympus, Tokyo, Japan). In two
additional test groups, cells were pretreated with Dex (10 μM) and
were treated, 2 h later, with 3-MA (100 μM) or RU486 (10 μM) for 24
h before examination under a fluorescent microscope. Electron
microscopy was performed to determine the number of autophagic
vacuoles in ATDC5 cells treated with or without Dex (10 μM).
Briefly, ATDC5 cells were washed three times with 1X
phosphate-buffered saline (PBS), trypsinized and collected by
centrifugation at 1,000 × g for 5 min. The cell pellets were fixed
in 4% paraformaldehyde overnight at 4°C, post-fixed with 1%
OsO4 in cacodylate buffer for 1 h at room temperature,
and gradually dehydrated with ethanol. The dehydrated pellets were
rinsed with propylene oxide for 30 min at room temperature and then
embedded in Spurr resin prior to sectioning. Images of thin
sections were observed under a transmission electron microscope
(JEM 1230; JEOL, Tokyo, Japan).
RNA isolation and reverse
transcription-PCR (RT-PCR)
For semi-quantitative analysis of the expression
levels of mRNAs coding for beclin 1 and Atg 7, total RNA
(1–5×105 cells) was extracted with the RNeasy total RNA
kit (Qiagen, Hilden, Germany), and reverse transcription was
performed on 1 μg of RNA/sample using the OneStep RT-PCR kit
(Qiagen). Primer sequences and conditions are available upon
request. Amplifications were conducted on the resulting cDNAs using
a LightCycler instrument (Roche Applied Science, Penzberg,
Germany). Data were normalized based on the expression of the
GAPDH gene.
Protein isolation and western blot
analysis
Whole-cell extracts were prepared by a standard
protocol, and proteins were detected by western blot analysis using
mouse anti-beclin 1, mouse anti-Atg 7 and rabbit anti-LC3 or GAPDH
polyclonal antibodies (Sigma-Aldrich). Anti-mouse or anti-rabbit
goat IgG (Pierce Biotechnology, Inc., Rockford, IL, USA) conjugated
to horseradish peroxidase were used as the secondary antibody, and
the SuperSignal West Femto system (Pierce Biotechnology, Inc.,
Rockford, IL, USA) was used for enhanced chemiluminescence
detection (ECL).
Cell viability assay
Cell viability was determined by the MTT assay.
ATDC5 cells were seeded in 96-well plates, and after 24 h, the
medium was changed with α-minimum essential medium (MEM) containing
1% FBS, Dex (0, 0.1, 1 or 10 μM) and/or 3-MA (0, 50, 100 or 200
μM). Following incubation for 0, 12, 24 or 48 h, the medium was
replaced with 50 μl of 1X MTT solution, and the cells were
incubated for 2 h at 37°C. The MTT solution was then discarded, and
150 μl dimethylsulphoxide (DMSO) was added at room temperature to
completely dissolve the precipitate. The optical density of the
final solution was measured at 570 nm using a spectrophotometer
(Crystaleye; Olympus). Cell viability was expressed as a percentage
of viable cells relative to control cells.
Statistical analysis
LC3 dot numbers, expression levels of beclin 1 and
Atg 7 and MTT measurements are presented as mean ± SE. Statistical
evaluations of differences in the data were analyzed using the
Student’s t- or -Newman-Keuls test. P<0.05 was considered to
indicate statistical significance.
Results
Dex induces autophagy and
autophagy-associated protein expression in ATDC5 cells
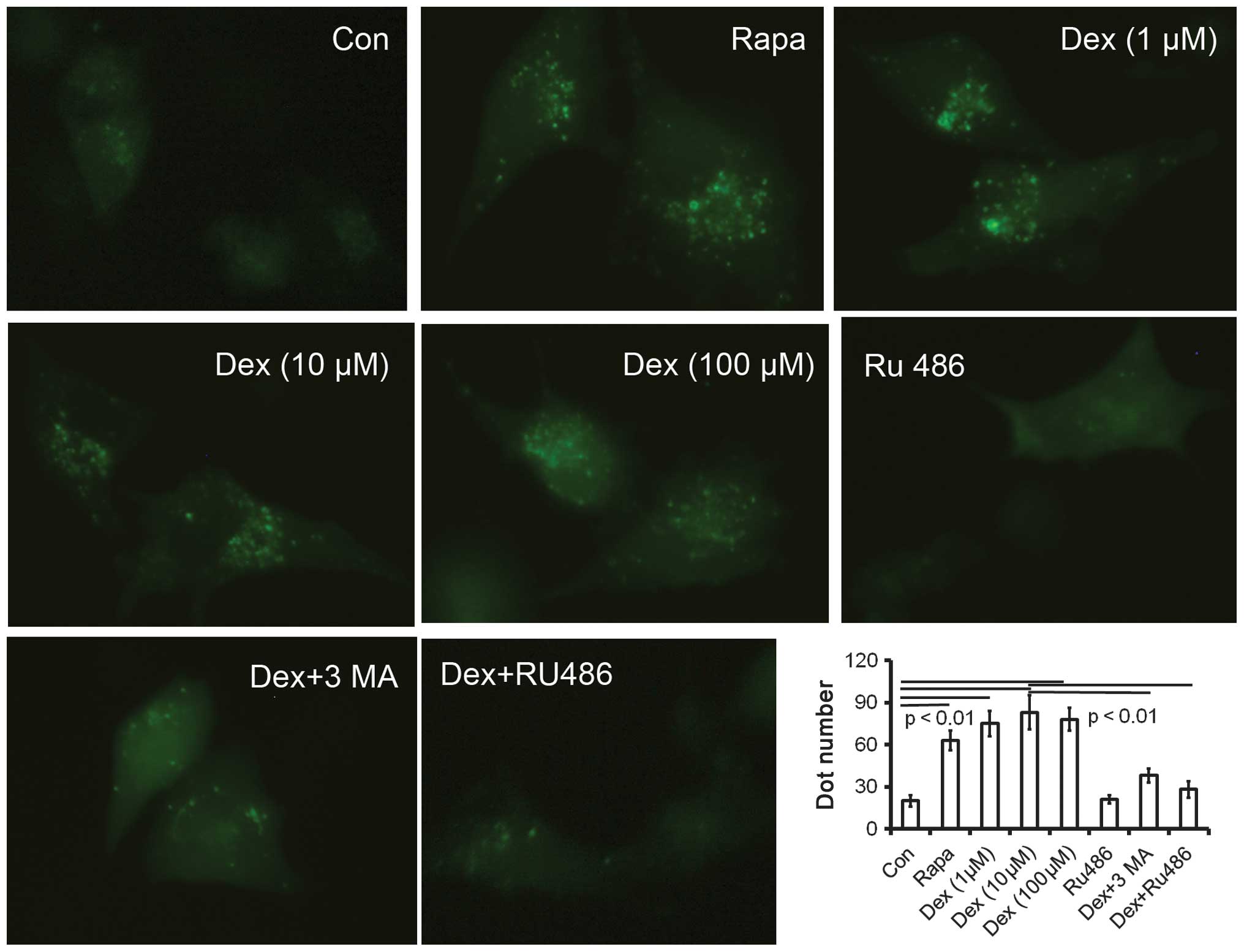
Autophagy is characterized by the formation of
numerous acidic vesicles known as acidic vesicular organelles
(AVOs) (10). Microphotographs of
AVOs were obtained by examining the expression of the GFP-LC3
vector under the fluorescence microscope. Untreated ATDC5 cells
showed limited numbers of AVOs in the cytoplasm. Cells treated with
rapamycin (200 nM) or Dex (1, 10 and 100 μM) demonstrated a
significant increase in AVOs (Fig.
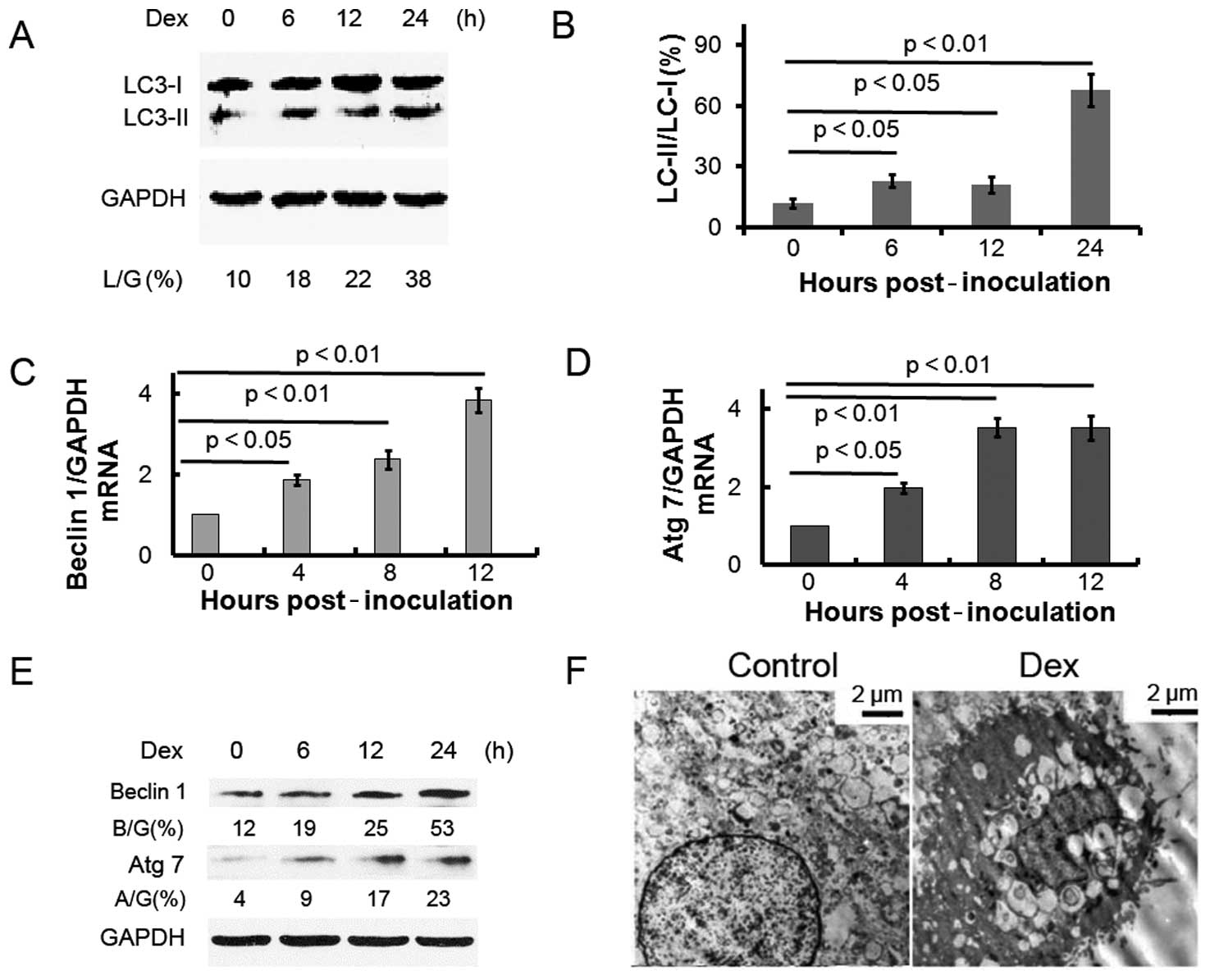
1). In addition, it has been reported (11) that the microtubule-associated
protein 1-light chain 3 form II (noted here as LC3-II) level
increases compared to the LC3-I level during autophagy. To detect
the expression of LC3-II, we performed western blot analysis with
lysates from ATDC5 cells treated with Dex. The LC3-II/LC3-I
expression ratio gradually increased over time in ATDC5 cells
treated with Dex (Fig. 2A and B).
The autophagy protein beclin 1 is part of a type III
phosphatidylinositide (PI) 3 kinase complex, required for the
formation of autophagic vesicles (12). In addition, autophagy-related gene
(Atg) products, such as Atg 7, play essential roles in autophagy.
Fig. 2C–E shows that the
expression of beclin 1 and Atg 7 increased post-Dex treatment, at
the mRNA and protein levels. These results indicate that Dex
treatment induces autophagy in ATDC5 cells. The ultrastructure of
ATDC5 cells treated with or without Dex was examined by electron
microscopy (Fig. 2F). Prominent
features of autophagy, i.e., cytoplasmic vacuoles and autolysosomes
were observed in the cells treated with Dex.
Dex-induced autophagy is inhibited by the
GC antagonist RU486 in ATDC5 cells
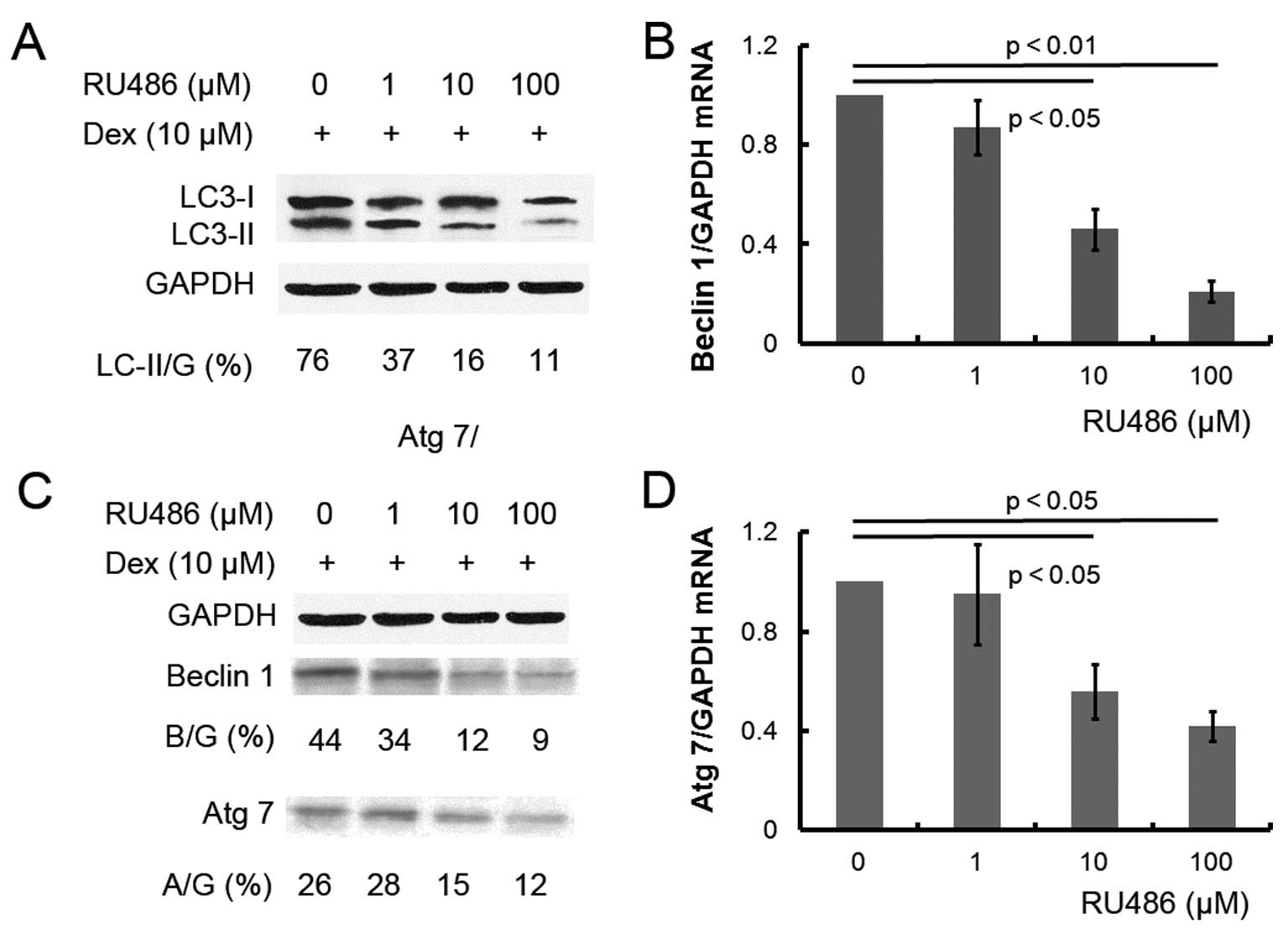
To further confirm the effect of Dex on autophagy,
ATDC5 cells were pretreated with 10 μM of the GC antagonist RU486
prior to Dex treatment. Pretreatment with RU486 completely reversed
the effect of Dex: microphotographs of the GFP-LC3 reporter vector
revealed a decreased number of AVOs in the cells subsequently
treated with RU486 or with the autophagy inhibitor 3-MA (Fig. 1), while beclin 1 and Atg 7 mRNA and
protein expression levels were significantly decreased (Fig. 3B–D). The results confirmed that Dex
can induce autophagy in ATDC5 cells.
ATDC5 cell viability decreases due to
Dex-induced autophagy
Dex treatment induced autophagy and increased the
expression of autophagy-associated proteins in ATDC5 cells. To
further determine the effect of Dex-induced autophagy on cell
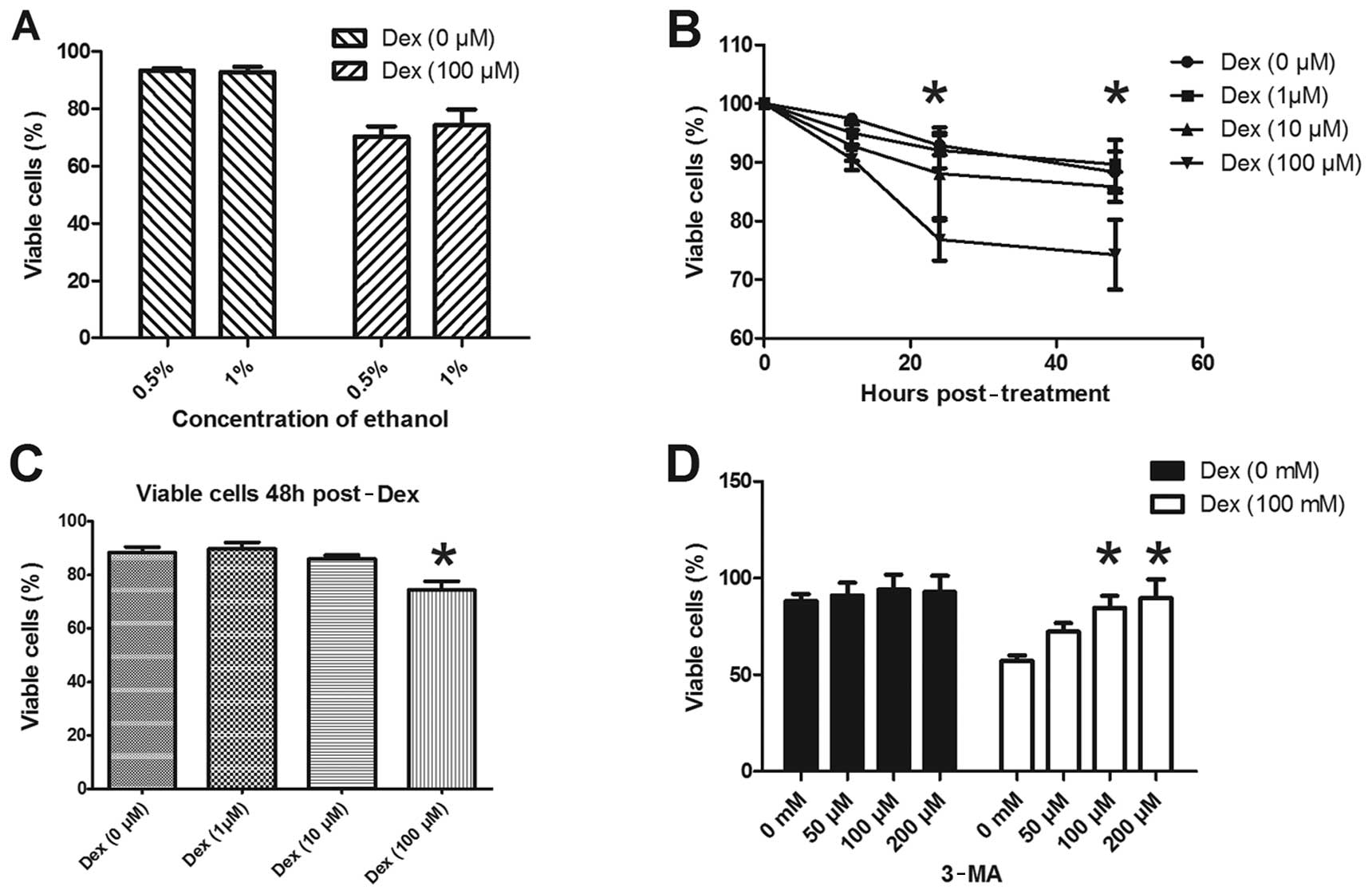
viability, the MTT assay was conducted in ATDC5 cells. Since Dex
was dissolved in 0.5% ethanol in our experiments, we also evaluated
the effect of ethanol on cell viability as a control. Fig. 4A shows that no significant
difference in cell viability was observed between the cells treated
with or without Dex dissolved in 0.5% and 1% ethanol. The ATDC5
cell viability was assayed following treatment with various
concentrations of Dex. No significant difference in the viability
of ATDC5 cells was detected after treatment with 1 or 10 μM Dex for
24–48 h, while the percentage of viable cells following treatment
with 100 μM Dex was significantly decreased compared to cells not
treated with Dex (Fig. 4B and C).
Furthermore, to evaluate whether autophagy is involved in the cell
viability reduction induced by Dex, the autophagy inhibitor 3-MA
was used. Fig. 4D shows that there
was no significant difference in ATDC5 cell viability among groups
treated with various doses of 3-MA alone; however, the reduction in
cell viability induced by 100 μM Dex was significantly reverted
subsequent to treatment with 100 or 200 μM of 3-MA. These results
suggest that the viability reduction induced by Dex in ADTC5 cells
has been most probably caused by autophagy.
Discussion
Clinical studies have shown that growth and skeletal
development are impaired during treatment with GCs (1–3). In
children, maintenance of growth is a complex process that is
affected by a number of distinct mechanisms, such as alterations in
growth hormone (GH) secretion and in GH/insulin-like growth factor
1 (IGF-1) sensitivity (3,13). Although compensatory growth often
follows the cessation of GC therapy, children with systemic chronic
inflammatory diseases subjected to long-term GC treatment may have
reduced final height (14,15). Normalizing height in these
children, even with concomitant therapy with high-dose recombinant
GH, is not straightforward (16–18).
Moreover, permanent growth impairment has been reported in children
receiving alternate-day GC treatment (19,20).
It is well known that the growth plate is responsible for
longitudinal bone growth (21).
Decreased growth is accompanied by morphological changes in the
growth plate in that it becomes thinner (2,22),
an effect attributed to decreased proliferation of the chondrocytes
(22,23). Detailed studies on this process
have revealed that decreased proliferation (22–24)
and increased apoptosis (25,26)
of epiphyseal chondrocytes contribute to the thinner cartilage.
Autophagy is an intracellular lysosomal (vacuolar)
degradation process that is characterized by the formation of
double-membrane vesicles in cytoplasm. Since autophagy is involved
in cell growth, survival, development and death, the levels of
autophagy need to be closely regulated to maintain a balance
between synthesis and degradation, and subsequent recycling, of
cellular products (27). Autophagy
has been proposed to be a ‘double-edged sword’: it can protect the
cells from apoptosis by removing oxidatively damaged organelles,
but excess autophagy can destroy cellular components. Autophagy can
therefore preserve viability, but also act as a self-destructive
process that leads to cell death (28).
In this study, we detected increased autophagic
activity in ATDC5 chondrocytes induced by Dex. Several approaches
were adopted to examine autophagy, including counting of
fluorescent GFP-LC3 dots, electron microscope imaging and
quantification of the relative mRNA and protein expression levels
of autophagy-associated molecules. A significant increase in the
autophagy-specific AVOs was observed in rapamycin-treated (200 nM)
cells or cells treated with 1, 10 or 100 μM Dex (Fig. 1); the fact that no significant
differences in the number of AVOs were found among cells treated
with different Dex doses (Fig. 1)
may be due to the limits of the used detection method.
Alternatively, the time point chosen for examining AVO formation,
i.e., 24 h post-transfection of cells with the GFP-LC3 report
vector, may not be the most suitable to discriminate differences
among groups. Analyses of the levels of autophagy-associated
molecules further confirmed that autophagy is induced by Dex: the
LC3-II/LC3-I protein ratio increased in cells post-Dex treatment;
beclin 1 and Atg 7 were also more highly expressed in ATDC5 cells
post-Dex treatment, at both the both mRNA and protein levels.
To confirm the role of Dex in autophagy in ATDC5
cells, RU486, which is a GC antagonist, was applied to the cells so
as to reverse Dex-induced autophagy. As expected, RU486 reduced the
Dex-induced autophagy induction (Fig.
1), and the same effect was observed for treatment with the
autophagy inhibitor 3-MA. The reversal in autophagy caused by RU486
was confirmed by the reduction in the expression of
autophagy-associated molecules (Fig.
3). Taken together, Dex induces autophagy in ATDC5 cells in
vitro.
Autophagy was induced in ATDC5 cells by the three
doses of Dex tested here, but cell viability was not evenly
affected by these doses. Doses of 1 or 10 μM Dex appeared to
ameliorate, and not reduce, cell viability, although no significant
difference was observed compared to the control cells. The
‘double-edged sword’ feature of the autophagic process was clearly
demonstrated by applying the higher dose of Dex: 100 μM Dex
significantly inhibited ATDC5 cell viability (Fig. 4), and this effect could be reversed
by 3-MA. The autophagy induced by the high dose of Dex may thus be
sufficient to destroy cellular components, promoting a
self-destructive process that eventually leads to cell death.
Therefore, autophagy may constitute a novel mechanism involved in
the decreased proliferation of chondrocytes following sustained
treatment with high GC doses.
In summary, results from this study suggest that GCs
can induce autophagy in ATDC5 chondrocytes. In low doses, GCs can
exert protective effects, but high doses of GC can induce excess
autophagy and reduce cell viability.
Acknowledgements
This study was supported by grants from funds of the
Jilin University Second Hospital and the China-Japan Union
Hospital.
References
|
1
|
Canalis E and Delany AM: Mechanisms of
glucocorticoid action in bone. Ann N Y Acad Sci. 966:73–81. 2002.
View Article : Google Scholar
|
|
2
|
Altman A, Hochberg Z and Silbermann M:
Interactions between growth hormone and dexamethasone in skeletal
growth and bone structure of the young mouse. Calcif Tissue Int.
51:298–304. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Allen DB: Growth suppression by
glucocorticoid therapy. Endocrinol Metab Clin North Am. 25:699–717.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xia X, Kar R, Gluhak-Heinrich J, et al:
Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res.
25:2479–2488. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weinstein RS, Nicholas RW and Manolagas
SC: Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis
of the hip. J Clin Endocrinol Metab. 85:2907–2912. 2000.PubMed/NCBI
|
|
6
|
Jia J, Yao W, Guan M, et al:
Glucocorticoid dose determines osteocyte cell fate. FASEB J.
25:3366–3376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Owen HC, Roberts SJ, Ahmed SF and
Farquharson C: Dexamethasone-induced expression of the
glucocorticoid response gene lipocalin 2 in chondrocytes. Am J
Physiol Endocrinol Metab. 294:E1023–E1034. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mushtaq T, Farquharson C, Seawright E and
Ahmed SF: Glucocorticoid effects on chondrogenesis, differentiation
and apoptosis in the murine ATDC5 chondrocyte cell line. J
Endocrinol. 175:705–713. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
11
|
Cherra SJ 3rd, Kulich SM, Uechi G, et al:
Regulation of the autophagy protein LC3 by phosphorylation. J Cell
Biol. 190:533–539. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun Y and Peng ZL: Programmed cell death
and cancer. Postgrad Med J. 85:134–140. 2009. View Article : Google Scholar
|
|
13
|
Mushtaq T and Ahmed SF: The impact of
corticosteroids on growth and bone health. Arch Dis Child.
87:93–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allen DB, Mullen M and Mullen B: A
meta-analysis of the effect of oral and inhaled corticosteroids on
growth. J Allergy Clin Immunol. 93:967–976. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Simon D, Lucidarme N, Prieur AM, Ruiz JC
and Czernichow P: Treatment of growth failure in juvenile chronic
arthritis. Horm Res. 58(Suppl 1): 28–32. 2002. View Article : Google Scholar
|
|
16
|
Touati G, Prieur AM, Ruiz JC, Noel M and
Czernichow P: Beneficial effects of one-year growth hormone
administration to children with juvenile chronic arthritis on
chronic steroid therapy. I Effects on growth velocity and body
composition. J Clin Endocrinol Metab. 83:403–409. 1998.
|
|
17
|
Bechtold S, Ripperger P, Muhlbayer D, et
al: GH therapy in juvenile chronic arthritis: results of a two-year
controlled study on growth and bone. J Clin Endocrinol Metab.
86:5737–5744. 2001.PubMed/NCBI
|
|
18
|
Allen DB, Julius JR, Breen TJ and Attie
KM: Treatment of glucocorticoid-induced growth suppression with
growth hormone. National Cooperative Growth Study. J Clin
Endocrinol Metab. 83:2824–2829. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sadeghi-Nejad A and Senior B: Adrenal
function, growth, and insulin in patients treated with corticoids
on alternate days. Pediatrics. 43:277–283. 1969.PubMed/NCBI
|
|
20
|
Lai HC, FitzSimmons SC, Allen DB, et al:
Risk of persistent growth impairment after alternate-day prednisone
treatment in children with cystic fibrosis. N Engl J Med.
342:851–859. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baron J, Klein KO, Colli MJ, et al:
Catch-up growth after glucocorticoid excess: a mechanism intrinsic
to the growth plate. Endocrinology. 135:1367–1371. 1994.PubMed/NCBI
|
|
22
|
Annefeld M: Changes in rat epiphyseal
cartilage after treatment with dexamethasone and
glycosaminoglycan-peptide complex. Pathol Res Pract. 188:649–652.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kember NF and Walker KV: Control of bone
growth in rats. Nature. 229:428–429. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mehls O, Himmele R, Homme M, Kiepe D and
Klaus G: The interaction of glucocorticoids with the growth
hormone-insulin-like growth factor axis and its effects on growth
plate chondrocytes and bone cells. J Pediatr Endocrinol Metab.
14(Suppl 6): 1475–1482. 2001.PubMed/NCBI
|
|
25
|
Silvestrini G, Ballanti P, Patacchioli FR,
et al: Evaluation of apoptosis and the glucocorticoid receptor in
the cartilage growth plate and metaphyseal bone cells of rats after
high-dose treatment with corticosterone. Bone. 26:33–42. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chrysis D, Ritzen EM and Savendahl L:
Growth retardation induced by dexamethasone is associated with
increased apoptosis of the growth plate chondrocytes. J Endocrinol.
176:331–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neufeld TP: Autophagy and cell growth -
the yin and yang of nutrient responses. J Cell Sci. 125:2359–2368.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Differ. 12(Suppl
2): 1528–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|