Introduction
Diabetes is a thriving disease throughout the world
and evidence from several epidemiological studies suggests that it
is an independent risk factor for cognitive dysfunction (1). Patients with type 1 and type 2
diabetes show performance decrements on measures of executive
functioning (2). Substantial
evidence supports that diabetic cognitive impairment is associated
with dysfunctions of the hippocampus, the major area associated
with learning and memory (3–5).
Experimental studies in animal models and in humans have shown that
hyperglycemia is the main contributor of diabetic cognitive
impairment, which can alter hippocampal synaptic plasticity and
cause decreased neuronal densities in the CA-1 region of the
hippocampus and degeneration and apoptosis of hippocampal neurons
(4,6,7).
Endoplasmic reticulum (ER) stress, often resulting
from the cellular accumulation of misfolded proteins, triggers a
cellular stress response known as the unfolded protein response
(UPR), which is intended to protect the cell (8). UPR is characterized by translational
attenuation, synthesis of ER chaperone proteins and transcriptional
induction of genes including those encoding the three ER
transmembrane receptors, i.e., protein kinase RNA (PKR)-like ER
kinase (PERK), inositol requiring enzyme-1 (IRE1) and activating
transcription factor (ATF6) (8).
If homeostasis cannot be reestablished or ER function is severely
impaired, apoptosis signaling pathways are triggered by ER stress,
including the C/EBP homology protein (CHOP), caspase-12 and c-Jun
N-terminal kinase pathways (9,10).
The CHOP protein, shown to play a critical role in ER
stress-induced apoptosis, can be induced by PERK translation, and
has been involved in promoting neuronal death in numerous
neurodegenerative diseases (11).
Recently, CHOP-dependent ER stress-mediated apoptosis was reported
to be involved in hyperglycemia-induced impairment of hippocampal
synapses and neurons (12).
Regulation of the ER stress-induced execution phase of apoptosis is
not completely understood, but clearly involves glycogen synthase
kinase-3β (GSK3β), which regulates ER stress-induced CHOP
expression in neuronal cells (13).
Ginseng (botanical name: Panax ginseng C.A.
Meyer) is one of the most common herbal remedies and has long been
used in traditional Chinese medicine to restore and enhance
benefits for patients. Ginsenoside Rb1 is generally recognized as
one of the principal bioactive ingredients in ginseng and has been
shown to possess neuroprotective properties in numerous studies.
Ginsenoside Rb1 can increase the proliferation of Schwann cells and
the expression and secretion of nerve growth factor (NGF) and
brain-derived neurotrophic factor (BDNF) from these cells (14), prevent MPP+-induced
apoptosis in PC12 cells (15) and
upregulate cell genesis in hippocampal subregions, thus enhancing
spatial learning and memory in rats (16). However, whether ginsenoside Rb1 has
neuroprotective effects on high glucose-treated hippocampal
neurons, and the mechanism underlying these effects remain unclear.
Therefore, in the present study, we investigated the effects of
ginsenoside Rb1 on high glucose-induced cytotoxicity in primary
cultured rat hippocampal neurons by detecting cell viability, and
further studied the underlying mechanisms by examining
phosphorylated (p)-PERK, PERK, p-GSK3β (Tyr216), GSK3β and CHOP
protein expression, as well as CHOP mRNA expression.
Materials and methods
Reagents
Ginsenoside Rb1 standard was purchased from the
National Institute for the Control of Pharmaceutical and Biological
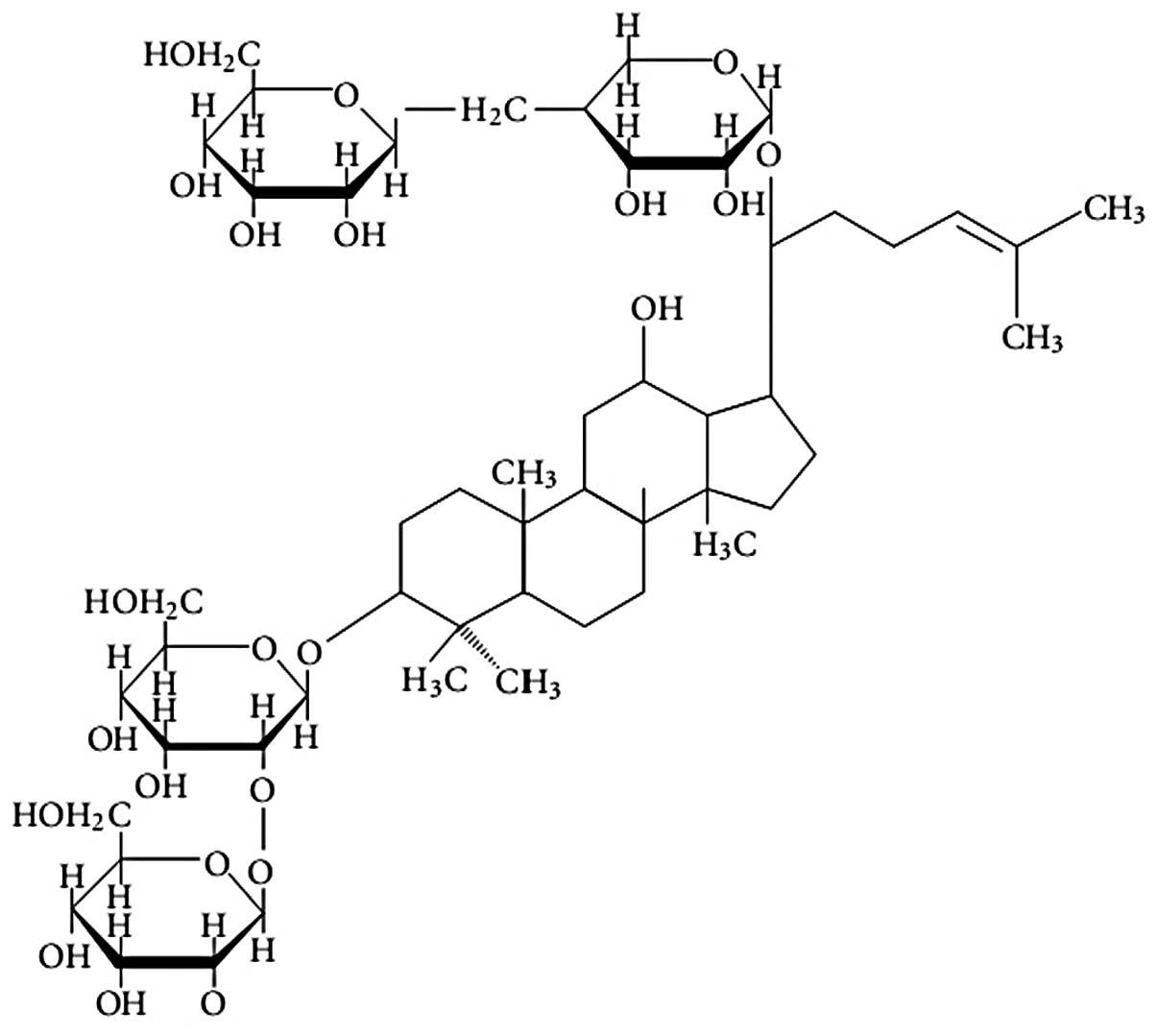
Produces (Beijing, China). The chemical structure of ginsenoside
Rb1 (2-O-β-glucopyranosyl-(3β,
12β)-20-[(6-O-β-D-glucopyranosyl-β-D-glucopyranosyl)
oxy]-12-hydroxydammar-24-en-3-yl β-D-glucopyranoside) is shown in
Fig. 1. All cell culture reagents
were from the Peking Union Cell Resource Center (Beijing, China).
Sprague-Dawley (SD) rats, <24 h old, were purchased from the
Peking University Health Science Center (Beijing, China).
Anti-p-PERK, -PERK, -p-GSK3β, -GSK3β and -CHOP antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA).
Cell culture and treatments
All experimental procedures were performed in
accordance with the experimental standards of the Chinese Academy
of Medical Sciences, as well as with international guidelines on
the ethical treatment of animals. Primary hippocampal neuronal
cultures were prepared as previously described (17,18)
with some modifications. The Animal Welfare Committee of Peking
Union Medical College Hospital and Chinese Academy of Medical
Sciences (Beijing, China) approved the animal protocols. Hippocampi
were dissected from the brain on ice and minced in sterile ice-cold
D-Hanks’ solution, with the blood vessels and meninges carefully
removed. The tissues were digested with 0.25% trypsin for 15 min at
37°C, and the digestion reaction was terminated by adding 5 ml
fetal bovine serum (FBS). The cell suspension was passed through a
200 mesh cell strainer and separated by density gradient
centrifugation at 702.4× g for 20 min. Then, the cell suspension,
containing the desired cell fractions in D-Hanks’ solution, was
centrifuged for 5 min at 395.1 × g and resuspended in 15–20 ml
Dulbecco’s modified Eagle’s medium (DMEM). The cells were then
plated on poly-D-lysine (Sigma-Aldrich, St. Louis, MO, USA)-coated
glass coverslips, 96-well plates or 24-well plates at a density of
5×105 cells/ml. The cell density was previously
determined using a hemacytometer. The cells were maintained at 37°C
in a humidified 5% CO2 incubator (Thermo Scientific
Forma, Waltham, MA, USA). Neurons were cultured in DMEM medium and
supplemented with 10% FBS, 20 mmol/l sodium pyruvate, 1 mmol/l
glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10 ng/ml
NGF (Millipore, Billerica, MA, USA). Ginsenoside Rb1 standard (1
μM), high glucose (final concentration, 50 mM) and Licl (1 mM;
Sigma-Aldrich) were added to the neurons for 72 h.
Cell viability assay
After exposure to 50 mM glucose and ginsenoside or
LiCl for 72 h, cell viability was determined using the MTT assay
system (Peking Union Cell Resource Center). Briefly, the cells were
plated on 96-well culture plates at a density of 5×105
cells/ml. Then, MTT reagent (100 μl of a 5 mg/ml solution) was
added to each well (1:10) and incubated for 4 h at 37°C. Following
incubation, the medium was removed and the precipitated dye was
dissolved in 100 μl of dimethylsulfoxide solution for another 30
min at 37°C. The OD value of each sample was measured at 570 nm
using a Multiskan 3 ELISA Reader (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Cell viability (%) was calculated as follows:
ODtreated group/OD24 h control group
×100.
Western blot analysis
Following treatment as described above, total
protein was extracted from cells lysed with RIPA buffer (50 mM
Tris-HCl at pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1 mM EDTA,
0.1% SDS, 5 μg/ml aprotinin and 5 μg/ml leupeptin). The protein
concentration of the supernatants was determined with the DC
protein assay kit 1 (Bio-Rad, Hercules, CA, USA) and then adjusted
to the same concentration with 0.9% NaCl. A total of 40 μg of
protein was resolved by 15% SDS-polyacrylamide gel electrophoresis
(PAGE) and then transferred to a nitrocellulose membrane. The
membrane was blocked with 5% skim milk in Tris-buffered saline with
0.1% Tween-20 (TBST) for 1 h at room temperature before overnight
incubation with primary antibodies specific to p-PERK (1:100), PERK
(1:100), p-GSK3β (1:200), GSK3β (1:200) and CHOP (1:200) in 5% skim
milk-TBST. The membrane was then rinsed three times in TBST and
incubated with horseradish peroxidase-conjugated secondary IgG
(1:1,000) in TBST for 1.5 h, followed by a final series of rinses
in TBST. Protein bands were visualized using an enhanced
chemiluminescence (ECL) kit (Roche Diagnostics GmbH, Penzberg,
Germany) and were quantified using the Quantity One software
(Bio-Rad).
Real-time PCR analysis
Total RNA was extracted from cells using the TRIzol
reagent according to the manufacturer’s instructions. The
concentration and purity of RNA were determined
spectrophotometrically at 260 and 280 nm. Single-strand cDNA was
prepared from 2 μg of total RNA according to the protocol of the
kit. The following primers were designed using Primer Premier 5.0
software (http://www.premierbiosoft.com/): CHOP,
5′-CCTAGCTTGGCTGACTGAGG-3′, 5′-CTGCTCCTTCTCCTTCATGC-3′; GAPDH,
5′-TGG TATCGTGGAAGGACTCA-3′, 5′-CCAGATGAGGCAGGGA TGAT-3′. Briefly,
the PCR reaction was carried out in a 20 μl final volume
containing: 10 μl of 2× SYBR®-Green I master mix, 1 μl
of each primer (10 μM), 2 μl of 1 μg/μl cDNA template, and 6 μl of
0.1% diethylpyrocarbonate-treated water. The PCR conditions were as
follows: initial denaturation at 95°C for 120 sec, followed by 40
cycles of denaturation at 95°C for 20 sec, annealing at 60°C for 20
sec, and extension at 72°C for 30 sec. A melting curve was obtained
after amplification by holding the temperature at 65°C for 20 sec,
followed by a gradual increase in temperature to 95°C at a rate of
0.5°C/sec. The GAPDH mRNA level was used as an internal
quantitative control, allowing to normalize the level of each
target gene transcript.
Statistical analysis
Data are expressed as mean ± SD. Differences between
groups were examined for statistical significance using a one-way
ANOVA and Dunnett’s tests. P<0.05 was considered to indicate
significant differences.
Results
Neuronal morphological changes
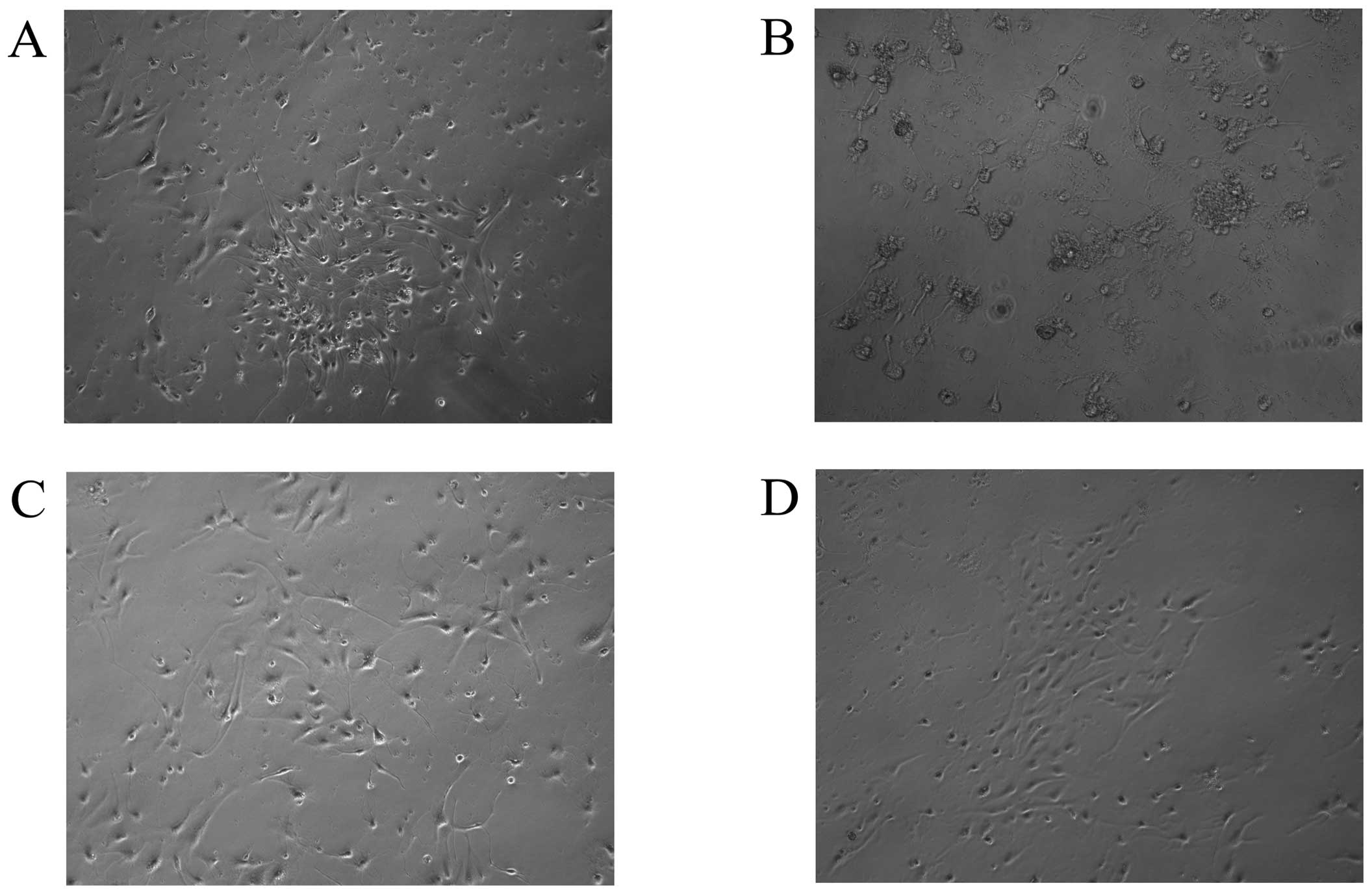
Neuronal morphological changes were assessed under
an inverted microcope. As shown in Fig. 2, hippocampal neurons treated with
normal medium (control) exhibited large, vacuole-free cell bodies,
with elaborate networks of neurites. However, exposure to 50 mM
glucose for 72 h resulted in obvious cell loss, with the
disappearance of neurites, appearance of disrupted membranes and
shrinkage of cell bodies. By contrast, cultures exposed to the same
concentration of glucose in the presence of ginsenoside Rb1 (1 μM)
appeared markedly preserved, indicating that Rb1 has significant
cytoprotective effects against high glucose insult. Licl (1 mM),
which is a GSK3β inhibitor, also showed cytoprotective effects
against high glucose treatment.
Neuroprotective effects of ginsenoside
Rb1 on cytotoxicity induced by high glucose
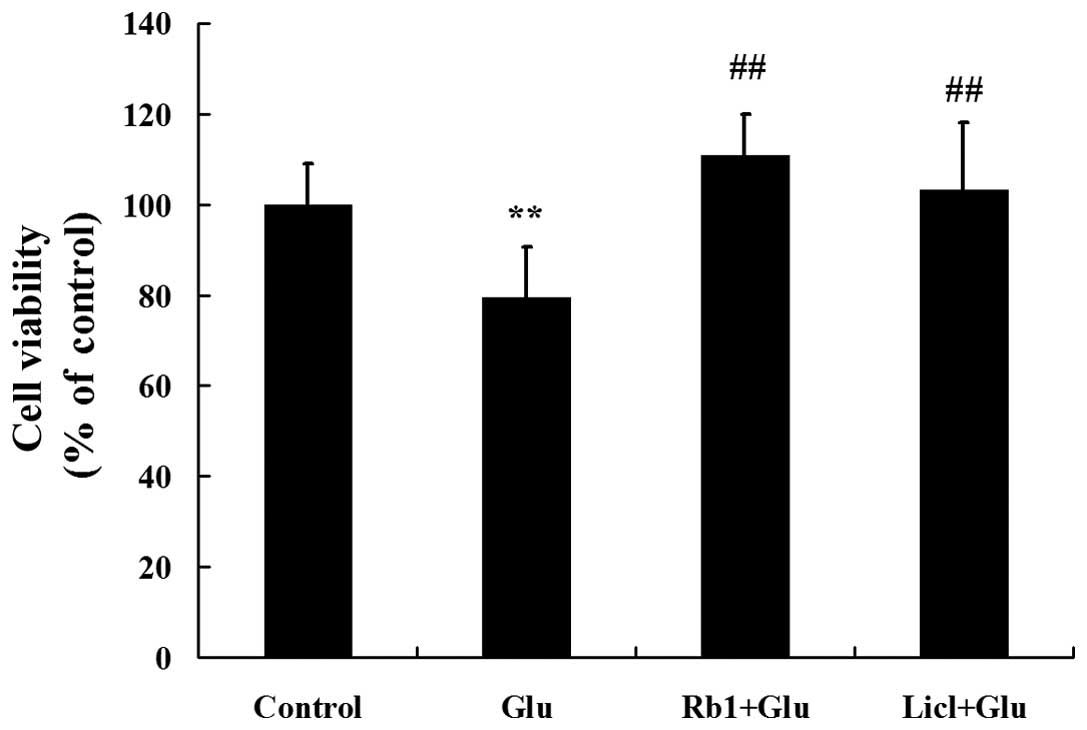
To verify the protective effects of ginsenoside Rb1
on hippocampal neurons exposed to a high level of glucose, cell
viability was evaluated by determining the percentage of MTT
reduction. As shown in Fig. 3, 50
mM of glucose induced a significant decrease in cell viability
compared to the control group. However, treatment with 1 μM of
ginsenoside Rb1 significantly improved cell viability in high
glucose-treated hippocampal neurons.
Effects of ginsenoside Rb1 on
GSK3β-mediated ER stress in high glucose-treated hippocampal
neurons
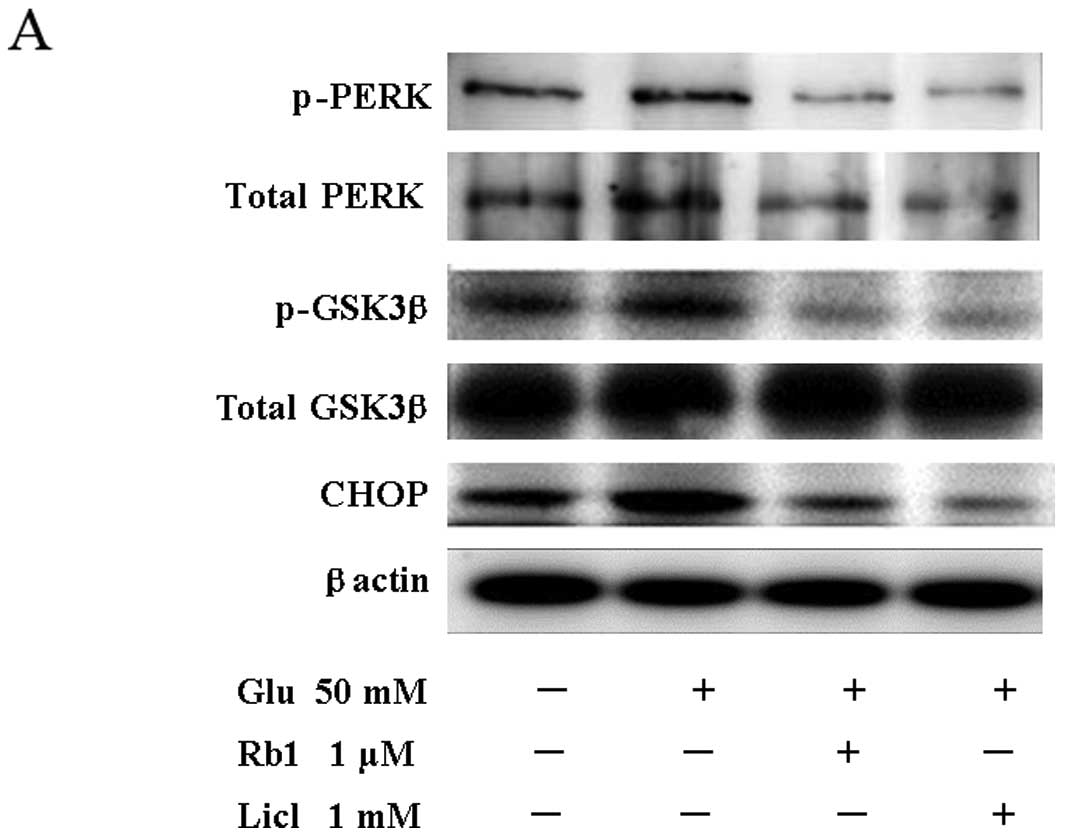
To examine the activation of ER stress in high
glucose-induced cell damage conditions, the phosphorylation level
of PERK and GSK3β and the expression of CHOP were assessed by
western blot analysis (Fig. 4A).
Compared to the control, cells exposed to a high level of glucose
for 72 h showed significantly increased p-PERK/PERK and
p-GSK3β/GSK3β ratios and an increased level of the CHOP protein
(Fig. 4B). By contrast, treatment
with ginsenoside Rb1 effectively attenuated the high
glucose-induced phosphorylation of PERK and GSK3β without affecting
the levels of the unphosphorylated forms of the enzymes, and
reduced the level of CHOP (Fig. 4A and
B).
We then investigated the effect of ginsenoside Rb1
on CHOP gene expression using quantitative RT-PCR analysis.
As shown in Fig. 4C, exposure to
high glucose for 72 h resulted in a significant increase in
CHOP gene expression compared to the control. However, when
cells were treated with ginsenoside Rb1, the CHOP mRNA level
was significantly decreased compared to the high glucose group.
To investigate the hypothesis that active GSK3β is
required for the increase in CHOP following ER stress, hippocampal
neurons were treated with the inhibitor of GSK3β (Licl) in the
presence of a high level of glucose for 72 h. Western blot analysis
and RT-PCR measurements confirmed that Licl inhibits high
glucose-induced phosphorylation of PERK and GSK3β, as well as CHOP
protein and mRNA expression (Fig.
4). The MTT assay indicated that cell viability is also
significantly improved by treatment with Licl in high
glucose-treated hippocampal neurons (Fig. 3).
Discussion
Ginseng has served as an important component of
numerous traditional Chinese medicine prescriptions for thousands
of years and is now popular as a natural medicine worldwide.
Ginsenoside Rb1 is the major pharmacologically active ingredient of
ginseng, and its anti-aging and neuroprotective effects have been
often reported. Cell-based studies showed that ginsenoside Rb1
protects hippocampal neurons from glutamate-induced
neurodegeneration (19) and
promotes cell survival, enhanced neurite outgrowth and synaptic
marker expression during differentiation of human neural stem cells
(20). However, the precise
biological functions and underlying mechanisms of ginsenoside Rb1
are still largely unknown.
The present study evaluated ginsenoside Rb1 as a
protective agent against high glucose-induced neurotoxicity.
Ginsenoside Rb1 attenuated the neuronal loss and improved cell
viability in high glucose-treated hippocampal neurons. We further
provide evidence that this neuroprotective effect may involve the
inhibition of GSK3β-mediated ER stress induced by high glucose.
Accumulating evidence suggests that diabetes is
associated with cognitive deficits, and previous studies have shown
that hyperglycemia has toxic effects and can lead to functional and
structural abnormalities in the brain, especially the hippocampus
(2,21). Hippocampal atrophy is evident early
in the course of the disease, and has even been documented in
elderly individuals with prediabetes (22). In animal models, diabetes is
associated with changes in the hippocampal synaptic plasticity,
molecular changes in hippocampal neurons, including pre- and
post-synaptic structures, and degeneration of hippocampal neurons
(23–25). Therefore, we used hippocampal
neurons exposed to high glucose as an in vitro model to
investigate the neuroprotective effects of ginsenoside Rb1 on high
glucose-induced neurotoxicity, as well as the underlying mechanism.
Consistent with a previous study (26), the results from morphological
observations and the cell viability assay showed that a high level
of glucose induces cell damage. Treatment of hippocampal neurons
with ginsenoside Rb1 significantly attenuated the high
glucose-induced morphological changes in the neurons and the
decrease in cell viability, suggesting that ginsenoside Rb1 can
revert the neurotoxicity induced by high glucose.
Hyperglycemia plays a critical role in hippocampus
dysfunction, however, the precise cellular mechanisms underlying
this effect have not yet been fully elucidated. A recent study on a
murine model of type 2 diabetes suggested that ER stress is
activated in the hippocampus (27), while another study indicated that
CHOP-dependent ER stress-mediated apoptosis may be involved in high
glucose-induced neuronal damage (12). In rats it has also been reported
that diabetes exacerbates ischemic brain injury by increasing ER
stress and apoptosis, associated with enhanced CHOP induction
(28). CHOP is a pro-apoptotic
protein that plays a major role in ER stress-induced apoptosis. In
neuronal cells, GSK3β was shown to regulate ER stress-induced CHOP
expression, and inhibition of GSK3β effectively reduced the
expression of CHOP (13).
Therefore, we hypothesized that GSK3β-mediated CHOP induction may
be involved in high glucose-induced cell injury. In this study, we
observed a significantly higher expression of p-PERK, p-GSK3β and
CHOP in high glucose-treated hippocampal neurons, which was
effectively decreased by the GSK3β inhibitor, indicating that
GSK3β-mediated CHOP signaling is involved in mediating the effects
of high glucose on cell morphology and structure. The results of
this study showed that high glucose-induced activation of CHOP is
significantly inhibited by treatment with the ginsenoside Rb1 and
by the GSK3β inhibitor, which suggests that ginsenoside Rb1 can
protect from high glucose-induced neurotoxicity via inhibition of
the GSK3β pathway.
In summary, our study demonstrated that ginsenoside
Rb1 protects hippocampal neurons from high glucose-induced
cytotoxicity. The mechanisms underlying this neuroprotective effect
may involve, at least in part, the inhibition of GSK3β-mediated
CHOP induction. To better understand diabetic cognitive impairment,
the contribution of proteins downstream of CHOP in high
glucose-induced neurotoxicity merit further investigation.
Additional investigations on ginsenoside Rb1 is expected to further
clarify the mechanisms by which this substance protects the neurons
from damage, which may contribute to the development of potential
therapeutic applications for this potent neuroprotective agent.
Acknowledgements
We are grateful to Professor Hongchao Yin and Bei Gu
for their helpful technical assistance and scientific
suggestions.
References
|
1
|
Cukierman T, Gerstein HC and Williamson
JD: Cognitive decline and dementia in diabetes - systematic
overview of prospective observational studies. Diabetologia.
48:2460–2469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McCrimmon RJ, Ryan CM and Frier BM:
Diabetes and cognitive dysfunction. Lancet. 379:2291–2299. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Revsin Y, Saravia F, Roig P, et al:
Neuronal and astroglial alterations in the hippocampus of a mouse
model for type 1 diabetes. Brain Res. 1038:22–31. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li ZG, Zhang W, Grunberger G and Sima AA:
Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res.
946:221–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gold SM, Dziobek I, Sweat V, et al:
Hippocampal damage and memory impairments as possible early brain
complications of type 2 diabetes. Diabetologia. 50:711–719. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamal A, Biessels GJ, Urban IJ and Gispen
WH: Hippocampal synaptic plasticity in streptozotocin-diabetic
rats: impairment of long-term potentiation and facilitation of
long-term depression. Neuroscience. 90:737–745. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stranahan AM, Arumugam TV, Cutler RG, Lee
K, Egan JM and Mattson MP: Diabetes impairs hippocampal function
through glucocorticoid-mediated effects on new and mature neurons.
Nat Neurosci. 11:309–317. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Bio. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu D, Zhang M and Yin H: Signaling
pathways involved in endoplasmic reticulum stress-induced neuronal
apoptosis. Int J Neurosci. 123:155–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang W and Paschen W: The endoplasmic
reticulum and neurological diseases. Exp Neurol. 219:376–381. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang X, Xu L, He D and Ling S:
Endoplasmic reticulum stress-mediated hippocampal neuron apoptosis
involved in diabetic cognitive impairment. Biomed Res Int.
2013:9243272013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meares GP, Mines MA, Beurel E, et al:
Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER)
stress-induced CHOP expression in neuronal cells. Exp Cell Res.
317:1621–1628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang W, Ge S, Yang L, et al: Ginsenosides
Rb1 and Rg1 promote proliferation and expression of neurotrophic
factors in primary Schwann cell cultures. Brain Res. 1357:19–25.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hashimoto R, Yu J, Koizumi H, Ouchi Y and
Okabe T: Ginsenoside Rb1 prevents MPP+-induced apoptosis
in PC12 cells by stimulating estrogen receptors with consequent
activation of ERK1/2, Akt and inhibition of SAPK/JNK, p38 MAPK.
Evid Based Complement Alternat Med. 2012:6937172012.PubMed/NCBI
|
|
16
|
Liu L, Hoang-Gia T, Wu H, et al:
Ginsenoside Rb1 improves spatial learning and memory by regulation
of cell genesis in the hippocampal subregions of rats. Brain Res.
1382:147–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaech S and Banker G: Culturing
hippocampal neurons. Nat Protoc. 1:2406–2415. 2007. View Article : Google Scholar
|
|
18
|
Brewer GJ and Torricelli JR: Isolation and
culture of adult neurons and neurospheres. Nat Protoc. 2:1490–1498.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Radad K, Gille G, Moldzio R, Saito H and
Rausch WD: Ginsenosides Rb1 and Rg1 effects on mesencephalic
dopaminergic cells stressed with glutamate. Brain Res. 1021:41–53.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L and Kisaalita WS: Administration of
BDNF/ginsenosides combination enhanced synaptic development in
human neural stem cells. J Neurosci Methods. 194:274–282. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gispen WH and Biessels GJ: Cognition and
synaptic plasticity in diabetes mellitus. Trends Neurosci.
23:542–549. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Convit A, Wolf OT, Tarshish C and de Leon
MJ: Reduced glucose tolerance is associated with poor memory
performance and hippocampal atrophy among normal elderly. Proc Natl
Acad Sci USA. 100:2019–2022. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alvarez EO, Beauquis J, Revsin Y, et al:
Cognitive dysfunction and hippocampal changes in experimental type
1 diabetes. Behav Brain Res. 198:224–230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo J, Yu C, Li H, et al: Impaired neural
stem/progenitor cell proliferation in streptozotocin-induced and
spontaneous diabetic mice. Neurosci Res. 68:329–336. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Artola A, Kamal A, Ramakers GM, Biessels
GJ and Gispen WH: Diabetes mellitus concomitantly facilitates the
induction of long-term depression and inhibits that of long-term
potentiation in hippocampus. Eur J Neurosci. 22:169–178. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gaspar JM, Castilho A, Baptista FI,
Liberal J and Ambrosio AF: Long-term exposure to high glucose
induces changes in the content and distribution of some exocytotic
proteins in cultured hippocampal neurons. Neuroscience.
171:981–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sims-Robinson C, Zhao S, Hur J and Feldman
EL: Central nervous system endoplasmic reticulum stress in a murine
model of type 2 diabetes. Diabetologia. 55:2276–2284. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Srinivasan K and Sharma SS: Augmentation
of endoplasmic reticulum stress in cerebral ischemia/reperfusion
injury associated with comorbid type 2 diabetes. Neurol Res.
33:858–865. 2011. View Article : Google Scholar : PubMed/NCBI
|