Introduction
Rho proteins are small GTPases of the Ras family
that cycle between an active and an inactive GDP-bound form and
regulate diverse cell functions such as cytoskeletal organization,
smooth muscle contraction, muscle and neuronal differentiation,
cell cycle progression and gene expression (1–4).
RhoA is a key member of the Rho family and is activated downstream
of multiple membrane receptors, such as thrombin and
lysophosphatidic acid (LPA) receptors. RhoA activity is increased
by the guanine nucleotide exchange factor, which promotes the
release of bound GDP and subsequent binding of GTP, and is
decreased by the GTPase-activating protein, which stimulates the
hydrolysis of bound GTP (5). In
addition, previous studies (6)
have suggested that the phosphorylation-dephosphorylation cycle
controls Rho protein activity. For example, the phosphorylation and
inhibition of RhoA by the cAMP-dependent protein kinase (PKA) has
been highlighted as one of the direct antagonizing mechanisms for
RhoA activity. In vitro experiments have indicated that RhoA
phosphorylation at serine 188 (Ser188) increases the ability of the
Rho GDP-dissociation inhibitor to extract RhoA from the membrane
(6–7). Another report further elucidated the
significance of PKA in regulating, through phosphorylation, RhoA
activity (8).
Type II cGMP-dependent protein kinase (PKGII) is a
serine/threonine kinase. It is involved in several physiological
functions including intestinal secretion, bone growth, learning and
memory (9). Findings of previous
studies have suggested that this kinase plays an important role in
regulating additional biological activities such as cell
proliferation, differentiation and apoptosis, especially in tumor
cells (10–13). Previous results from our laboratory
showed that PKGII inhibits the proliferation and migration of
gastric cancer cell lines through inhibition of the EGF-induced
activation of EGFR and of related signal transduction pathways
(14–16). Since our previous results also
showed that PKGII inhibits LPA-induced cell migration (17), whether this kinase exerts an
inhibitory effect on RhoA activity should be elucidated. This study
was designed to investigate the potential inhibition, by PKGII, of
LPA-induced RhoA activation and the underlying mechanism(s)
involved.
Materials and methods
Cell lines and reagents
The human gastric cancer cell line AGS, African
green monkey kidney fibroblast-like cell line COS-7 and human
embryonic kidney (HEK) 293A cell line were provided by the
Institute of Cell Biology (Shanghai, China). Adenoviral vectors
encoding β-galactosidase (pAd-LacZ) and PKGII (pAd-PKGII), the
recombinant pcDNA 3.1 vector bearing the genes encoding PKGII,
wild-type RhoA and mutant RhoA Ser188A, and the expression vector
pGEX-2T-RBD, encoding the cDNA of glutathione S-transferase (GST)
coupled to the Rho-binding domain (RBD) of rhotekin were kind gifts
from Dr Gerry Boss and Dr Renate Pilz (University of California,
San Diego, CA, USA). Dulbecco’s modified Eagle’s medium (DMEM) and
fetal bovine serum (FBS) were from Gibco-BRL (Grand Island, NY,
USA). The antibody against PKGII was from Abgent Biotechnology (San
Diego, CA, USA). Goat anti-β-actin, mouse anti-RhoA and rabbit
anti-phosho (p)-RhoA (Ser188) were from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Mouse anti-FLAG was from Sigma-Aldrich
(St. Louis, MO, USA). Rabbit anti-p-LIMK (Thr505/508) was from
Bioworld Technology Inc. (St. Louis Park, MN, USA). Horseradish
peroxidase-conjugated secondary antibodies were from Jackson
ImmunoResearch Laboratories (West Grove, PA, USA). The cellular
permeable cGMP analog 8-pCPT-cGMP and the ROCK inhibitor Y27632
were from Calbiochem (San Diego, CA, USA). LPA, TRITC-conjugated
phalloidin and the plasmid p3xFLAG-Myc-CMV-24 were purchased from
Sigma-Aldrich. The cell transfection reagent Lipofectamin™ 2000 and
Escherichia coli BL21 (DE3) were from Invitrogen Life
Technologies (Carlsbad, CA, USA). The plasmid pGEX-4T-1 and
glutathione-sepharose beads were purchased from GE Healthcare Life
Sciences (Piscataway, NJ, USA). Electrochemiluminescence (ECL)
reagents were from EMD Millipore (Billerica, MA, USA). Glass bottom
dishes were from Shengyou Biotechnology, Co., Ltd. (Hangzhou,
China). All other reagents used were of analytical grade.
Preparation of adenoviral vectors
HEK 293A cells were transfected with pAd-LacZ and
pAd-PKGII vectors and cultured for ≤10 days until cytopathic
effects were observed. The cells and the culture medium were
harvested and underwent three freezing-thawing cycles. The
supernatants containing adenoviruses (Ad-LacZ and Ad-PKGII) were
used to infect new HEK 293A cells to amplify adenoviruses. The
amplified adenoviral preparations were titrated to determine the
pfu/ml and kept at −80°C until use.
Cell culture, transfection and
infection
AGS cells were cultured in DMEM supplemented with
10% FBS and maintained at 37°C in a humidified incubator with 95%
air and 5% CO2. The medium was changed every two days
and the cells were sub-cultured until they reached confluence. For
transfection of gastric cancer cells with plasmids, the cells were
sub-cultured the day before the process and the transfection was
performed according to the manufacturer’s instructions. On the day
before infection with Ad-LacZ and Ad-PKGII, the cells were cultured
in fresh medium at 70–80% confluence.
Cloning and expression constructs
To generate FLAG-tagged wild-type RhoA and mutant
RhoA Ser188A plasmids, cDNA fragments were amplified from the pcDNA
3.1/full-length wild-type RhoA and /RhoA mutant by PCR. The 5′
primer for amplifying wild-type and mutant RhoA was 5′-CCC AAG CTT
ATG GCT GCC ATC CGG AA-3′. The 3′ primers were: for wild-type RhoA,
5′-CGC GGA TCC CAA GAC AAG GCA CCC AGA TT-3′, and for mutant RhoA,
5′-CGC GGA TCC CAA GAC AAG GCA CCC AGC TT-3′. The PCR products were
cleaved by HindIII and BamHI and the target fragment
was cloned into the expression vector p3xFLAG-Myc-CMV-24.
The cloning vectors for the bimolecular fluorescence
complementation (BiFC) assay, pBiFC-VN155 and pBiFC-VC155, were
kindly provided by Dr Chang-Deng Hu (Purdue University, West
Lafayette, IN, USA). To construct pBiFC-RhoA-VC, a cDNA fragment
encoding the full-length RhoA was amplified by PCR from pcDNA
3.1/wild-type RhoA, using the following oligonucleotides: 5′-GGA
AGA TCT ACA TGG CTG CCA TCC GGA A-3′ (RhoA-VC-N), 5′-CGG GGT ACC
CAA GAC AAG GCA CCC AGA TT-3′ (RhoA-VC-C). The resulting PCR
fragment was digested with BglII and KpnI and then
inserted into the pBiFC-VC155 vector. To construct pBiFC-PKGII-VN,
a cDNA fragment encoding the full-length PKGII was amplified by PCR
using the following oligonucleotides bearing BglII and
KpnI restriction sites: 5′-GGA AGA TCT CTA TGG GAA ATG GTT
CAG TGA AAC-3′ (PKGII-VN-N), 5′-CGG GGT ACC GAA GTC TTT ATC CCA GCC
TGA T-3′ (PKGII-VN-C). The amplified fragment was then cloned into
the pBiFC-VN155 vector.
Full-length RhoA cDNA and cDNA fragments encoding
RhoA amino acids 1–44 and 1–147 were amplified by PCR from the
full-length pcDNA 3.1/wild-type RhoA vector. The sequence of the
5′-primer was: 5′-GCG GGA TCC ATG GCT GCC ATC CGG AAG-3′. The
sequences of the 3′-primers were: 5′-CGC GTC GAC TCA CAA GAC AAG
GCA CCC AGA TTT-3′, 5′-CGC GTC GAC TCA GCC ACA TAG TTC TCA AAC
AC-3′ and 5′-CGC GTC GAC TCA CAT ATC TCT GCC TTC TTC AGG-3′ for
full-length, RhoA amino acids 1–44 and 1–147, respectively. The PCR
products were cleaved by BamHI-EcoRI and the target
fragment was cloned into pGEX-4T-1. To generate PKGII cDNA
fragments (encoding amino acid residues 1–176, 1–285, 286–762,
286–452, 453–711 and 712–762), cDNA fragments were amplified from
the full-length pcDNA 3.1/PKGII by PCR. The following primers were
used: 5′ primer for PKGII (1–176) and PKGII (1–285), 5′-TCC CCC GGG
TAT GGG AAA TGG TTC AGT G-3′; for PKGII (286–762) and PKGII
(286–452), 5′-TCC CCC GGG TTT GCT GAA GAA TTT ACC TG-3′; for PKGII
(453–711), 5′-TCC CCC GGG TCT TGA GAT TAT TGC AAC ACT GG-3′ and for
PKGII (712–762), 5′-TCC CCC GGG TAA TGG TTT TAA TTG GGA GGG ACT
G-3′. The 3′ primers were: for PKGII (1–176), 5′-CCG CTC GAG CTG
CTG AGG ATC CAG TCT-3′; for PKGII (1–285), 5′-CCG CTC GAG GGA TAC
ACT TCT GAG GAA GTT TCT G-3′; for PKGII (286–452), 5′-CCG CTC GAG
GTT CTG GAA TGG GGA TGA TG-3′; for PKGII (453–711), 5′-CCG CTC GAG
TAA CCA CCT GTG TTT CTT AAT GTC-3′ and for PKGII (712–762) and
PKGII (286–762), 5′-CCG CTC GAG GAA GTC TTT ATC CCA GCC TGA TAG-3′.
The PCR products were cleaved by SmaI-XhoI and the
target fragment was cloned into pGEX-4T-1. The validity of all the
constructs was confirmed by DNA sequencing.
Expression of GST-fusion proteins in E.
coli
RhoA and PKGII fragments fused to GST were expressed
in the E. coli competent cells BL21 (DE3), and transcription
was induced with 0.2 or 1 mM isopropyl-β-D-thiogalactopyranoside.
The expressed GST-fusion proteins were purified on
glutathione-sepharose beads.
Phalloidin staining
Cells were plated on glass coverslips, serum-starved
for 16 h and treated with 250 μM CPT-cGMP for 1 h prior to
stimulation with 10 μM LPA for 5 min. Cells were fixed with freshly
prepared 40 g/l paraformaldehyde in phosphate-buffered saline (PBS)
for 15 min, permeabilized with 0.3% Triton X-100 in PBS for 10 min
and blocked with 3% bovine serum albumin in PBS. The cells were
subsequently incubated with TRITC-labeled phalloidin (1:100) for 1
h at room temperature, and were visualized with fluorescence
microscopy using a Leica DM LB2 microscope digital camera system
(Leica Microsystems, Wetzlar, Germany).
Transwell migration assays
The migratory activity of AGS cells was detected
using a transwell system comprising BD BioCoat™ Control Cell
Culture inserts (8.0-μm PET membrane, 24-well) purchased from BD
Biosciences (Franklin Lakes, NJ, USA). After trypsinization,
5×104 cells were seeded into the upper chamber of a
tissue culture incubator containing culture medium without FBS.
Cell migration to the bottom side of the membrane was induced by
medium containing 10% FBS in the lower chamber for 12 h at 37°C.
The cells remaining in the upper chamber were carefully removed
with cotton swabs. Cells that had migrated to the bottom side of
the membrane were fixed in 4% paraformaldehyde solution for 30 min,
stained in Giemsa solution for 10 min and then rinsed in water. The
stained cells were subjected to microscopic examination under a
light microscope. Migrating cells were counted in five randomly
selected fields per insert and the values were averaged. Each
migration condition experiment was repeated three times.
Confocal laser scanning microscopy
Cos-7 cells (~2×105 cells/dish) were
grown on 35-mm dishes with a 20-mm bottom well, and were
transfected with BiFC pairs. The BiFC fluorescent signals were
detected under a confocal microscope (Zeiss, Oberkochen,
Germany).
Western blot analysis
Protein samples were electrophoresed on an SDS-PAGE
(8–12%) gel, and membrane transfer was performed following the
manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). The
primary antibodies were incubated overnight at 4°C in Tris-buffered
saline with 2% Tween-20 (TBS-T) and the matching secondary
antibodies were incubated for 1 h at room temperature in TBS-T,
with three washes after each incubation. ECL reagents were used to
allow visualizing the positive bands on the membrane. To perform
densitometry analysis, digital images of the positive bands were
obtained with the ChemiDoc XRS acquisition system and analyzed
using the image analysis program Quantity One (Bio-Rad). The
results were expressed as the ratio of target protein/loading
control protein.
In vitro pull-down assay
Cells on 100-mm culture plates at ~90% confluence
were washed three times with cold PBS and harvested in lysis buffer
(25 mM HEPES pH 7.5, 150 mM NaCl, 1% NP40, 10% glycerol, 25 mM NaF,
10 mM MgCl2, 0.25% sodium deoxycholate, 1 mM EDTA, 1 mM
Na3VO4, 10 mg/ml aprotinin and 10 mg/ml
leupeptin) 48 h after transfection or infection. The supernatant
obtained by centrifugation (13,800 × g, 10 min) was then mixed with
GST-fusion protein beads and incubated for 2 h at 4°C with rocking.
After thorough washing with lysis buffer, the bound proteins were
solubilized in 2X SDS buffer and analyzed by western blotting using
antibodies against RhoA or PKGII. To assess the amount of protein
used in each pull-down assay, 5% of the input lysate was loaded as
a control. Beads loaded with GST alone were included as negative
controls in all experiments. RhoA activity was detected by a
similar method, and the supernatant was incubated with
glutathione-sepharose beads and GST-RBD of rhotekin at 4°C for 1
h.
Immunoprecipitation
The cells growing on 100-mm culture plates were
washed twice with cold PBS and lysed with RIPA buffer (50 mM
Tris-HCl pH 7.4, 1% Triton X-100, 1 mM EDTA, 1 mM leupeptin, 1 mM
phenylmethylsulfonyl fluoride, 10 mM NaF and 1 mM
Na3VO4) 48 h after transfection and/or
infection. The supernatant was obtained by centrifugation (13,800 ×
g, 10 min) and then mixed with target antibodies or matched
immunoglobulin G as a negative control for 12 h at 4°C with
rocking. Fresh protein G conjugated to agarose was then added,
followed by a 2–3 h incubation at 4°C with rocking.
Immunoprecipitates were centrifuged at 400 × g for 2 min at 4°C.
The supernatant was discarded and the pellet was washed four times
with binding buffer (50 mM Tris-HCl, 250 mM NaCl, 0.05% Nonidet
P40, 30 mM MgCl2, pH 7.4) and then resuspended with the
same volume of 2X SDS buffer. The precipitates were probed with
antibodies against target proteins.
Statistical analysis
Data are expressed as means ± standard deviation
(SD). Statistical significance was assessed with analysis of
variance (ANOVA) analyses using the SPSS statistical software (IBM,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
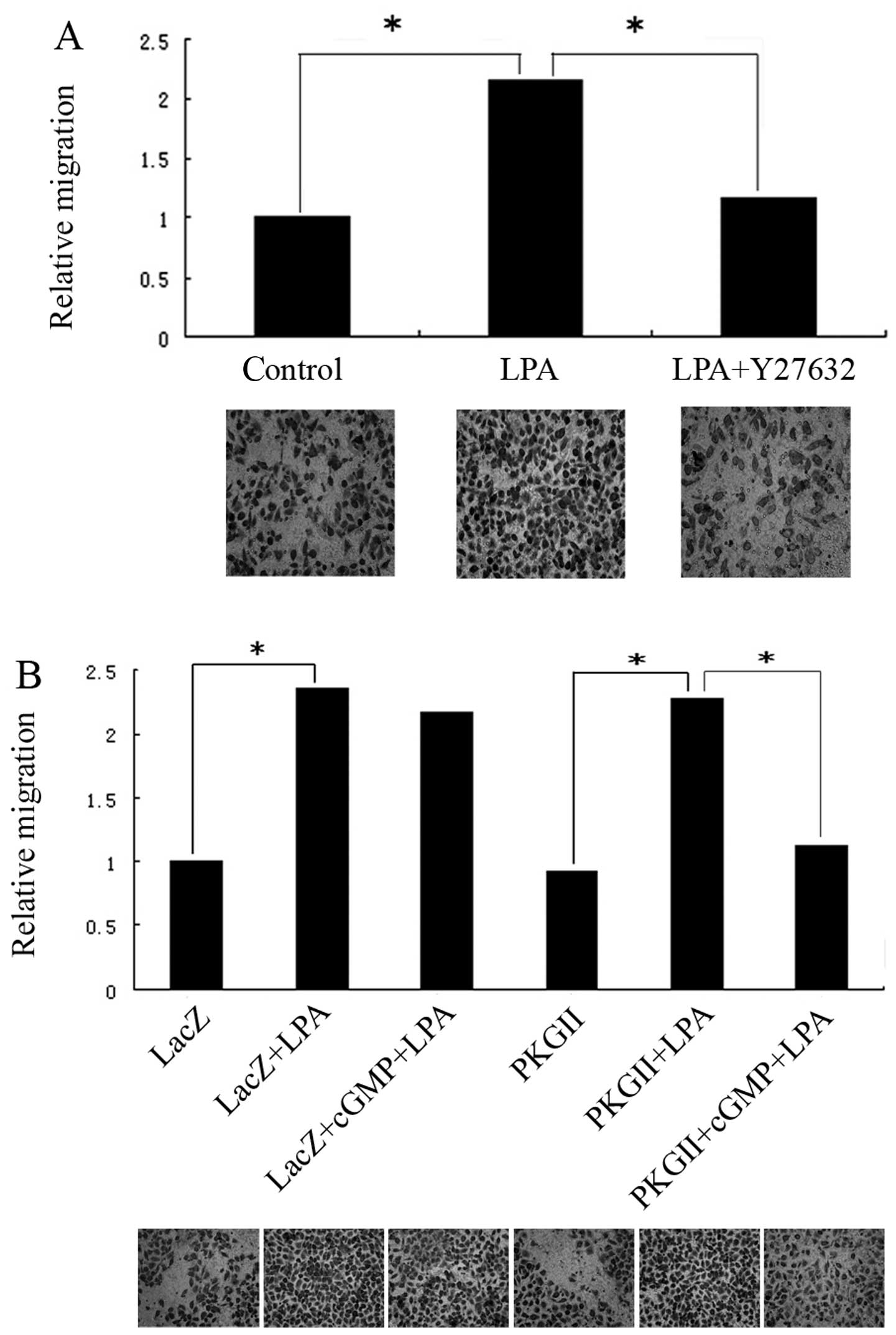
PKGII inhibits the LPA-induced migration
and cytoskeletal changes in AGS cells
LPA is a potent mediator in the modulation of the
cell migration process. A previous study suggested that the
G12/G13-RhoA signaling pathway contributes to
efficient LPA-stimulated cell migration (18). To detect the role of RhoA in
LPA-induced migration of gastric cancer cells, AGS cells were
treated with 10 μM Y27632 (ROCK inhibitor) for 1 h before LPA
stimulation. A transwell assay was used to measure the migratory
activity of the cells. The results showed that LPA stimulation
resulted in ~2-fold increase in cell migration compared to that of
unstimulated cells (Fig. 1A).
Treatment with Y27632 inhibited the LPA-stimulated cell migration,
indicating that LPA-induced cell migration is controlled by the
activation of the RhoA/ROCK pathway (Fig. 1A). To investigate the effect of
PKGII on LPA-induced migration, AGS cells were infected with
Ad-LacZ or Ad-PKGII and then treated with 250 μM 8-pCPT-cGMP for 1
h prior to treatment with LPA. Treatment with 8-pCPT-cGMP markedly
and significantly inhibited LPA-induced cell migration in the cells
infected by Ad-PKGII but not in those infected by Ad-LacZ,
indicating that PKGII activity is important, not only for impeding
the LPA-induced migration, but also for inhibition of the
LPA-induced activation of RhoA (Fig.
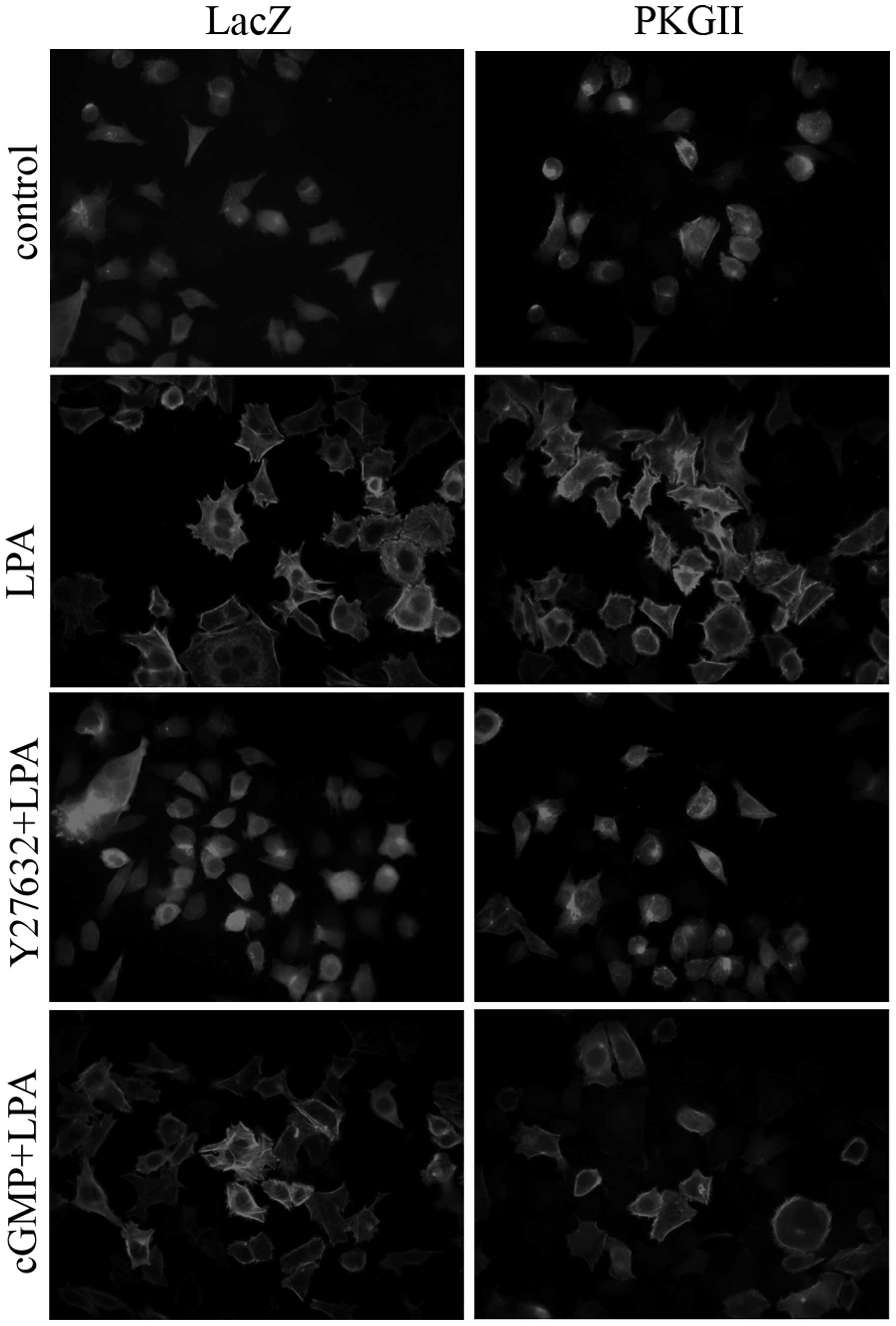
1B). To confirm that PKGII functions through cross-talk with
the RhoA/ROCK pathway, we examined the effect of PKGII on
LPA-induced stress fiber formation, which is well-known to be
dependent on RhoA activation. The results showed that pre-infection
with Ad-PKGII and treatment with 8-pCPT-cGMP reduces the formation
of stress fibers caused by LPA stimulation (Fig. 2).
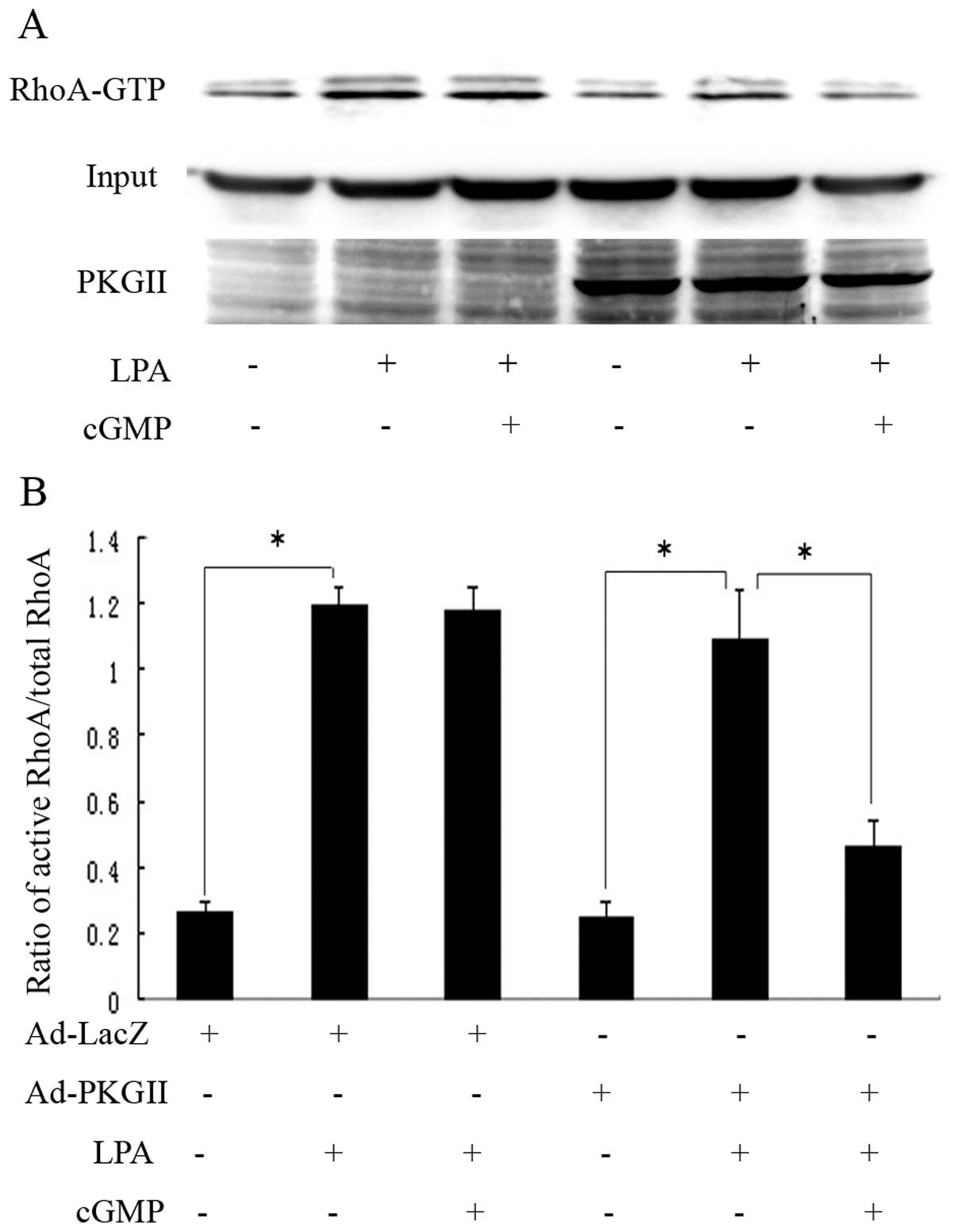
PKGII inhibits RhoA activation by
phosphorylating RhoA at Ser188
We used a pull-down assay to assess whether RhoA
activation is affected by PKGII. Treatment of Ad-PKGII-infected
cells with 8-pCPT-cGMP marginally decreased the basal RhoA
activity, but inhibited LPA-induced RhoA activation by >50%
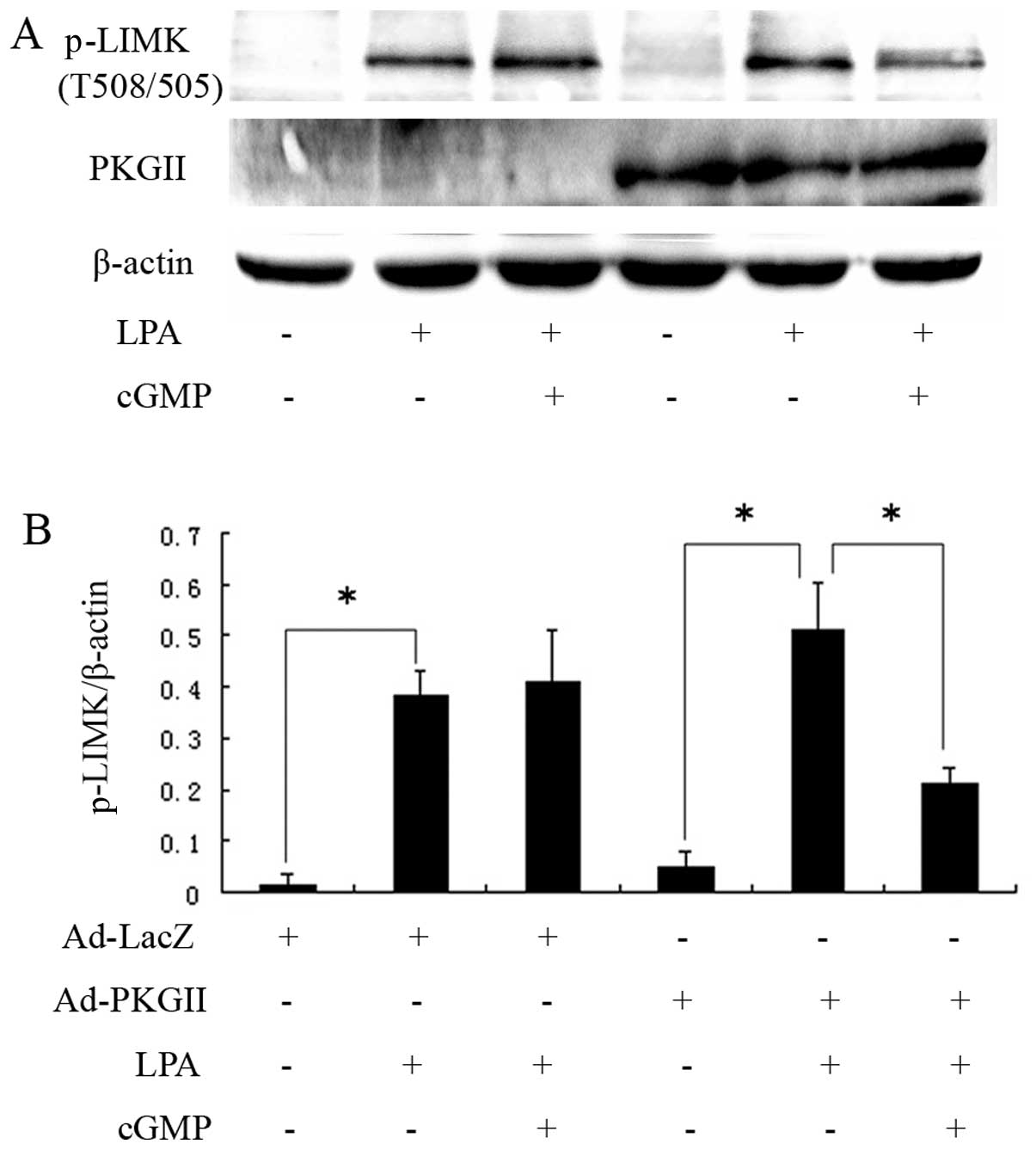
(Fig. 3). LIMK is a signaling
component downstream of RhoA, participating in regulating the
formation of stress fibers. We detected Thr505/508
phosphorylation/activation of LIMK and found that PKGII reduced the
levels of phosphorylated LIMK which was likely to have been induced
by LPA (Fig. 4). These results
indicated that inhibition of LPA-induced migration and stress fiber
formation by PKGII is associated with inhibition of LPA-induced
RhoA activity and its downstream signaling.
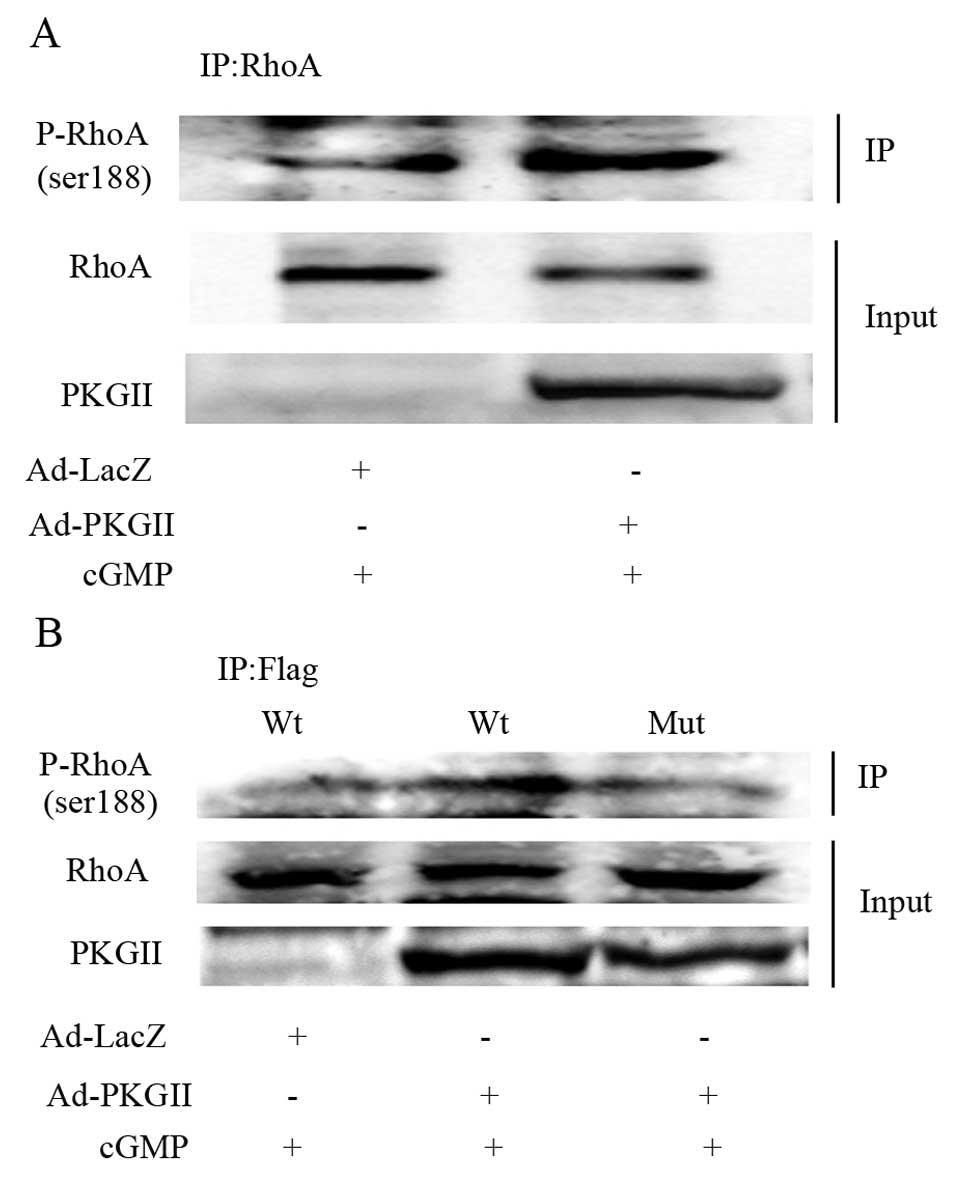
It was previously shown that Ser188 is the
phosphorylation site through which protein kinases exert their
inhibitory effect on RhoA activity (8). We used western blotting with an
antibody against RhoA phosphorylated at Ser188 to detect the
PKGII-induced phosphorylation of RhoA in AGS cells. The cells were
treated as described above. The results showed that the level of
phosphorylated RhoA Ser188 was increased in cells infected by
Ad-PKGII and treated with 8-pCPT-cGMP (Fig. 5A). In the cells transfected with
the plasmid expressing wild-type RhoA, the PKGII activity was
sufficient to phosphorylate exogenous RhoA at Ser188. By contrast,
in the cells transfected with the plasmid expressing mutant RhoA
Ser188A, the phosphorylation of exogenous RhoA at Ser188 did not
increase after treatment with 8-pCPT-cGMP (Fig. 5B). These results indicated that
PKGII phosphorylates RhoA at Ser188.
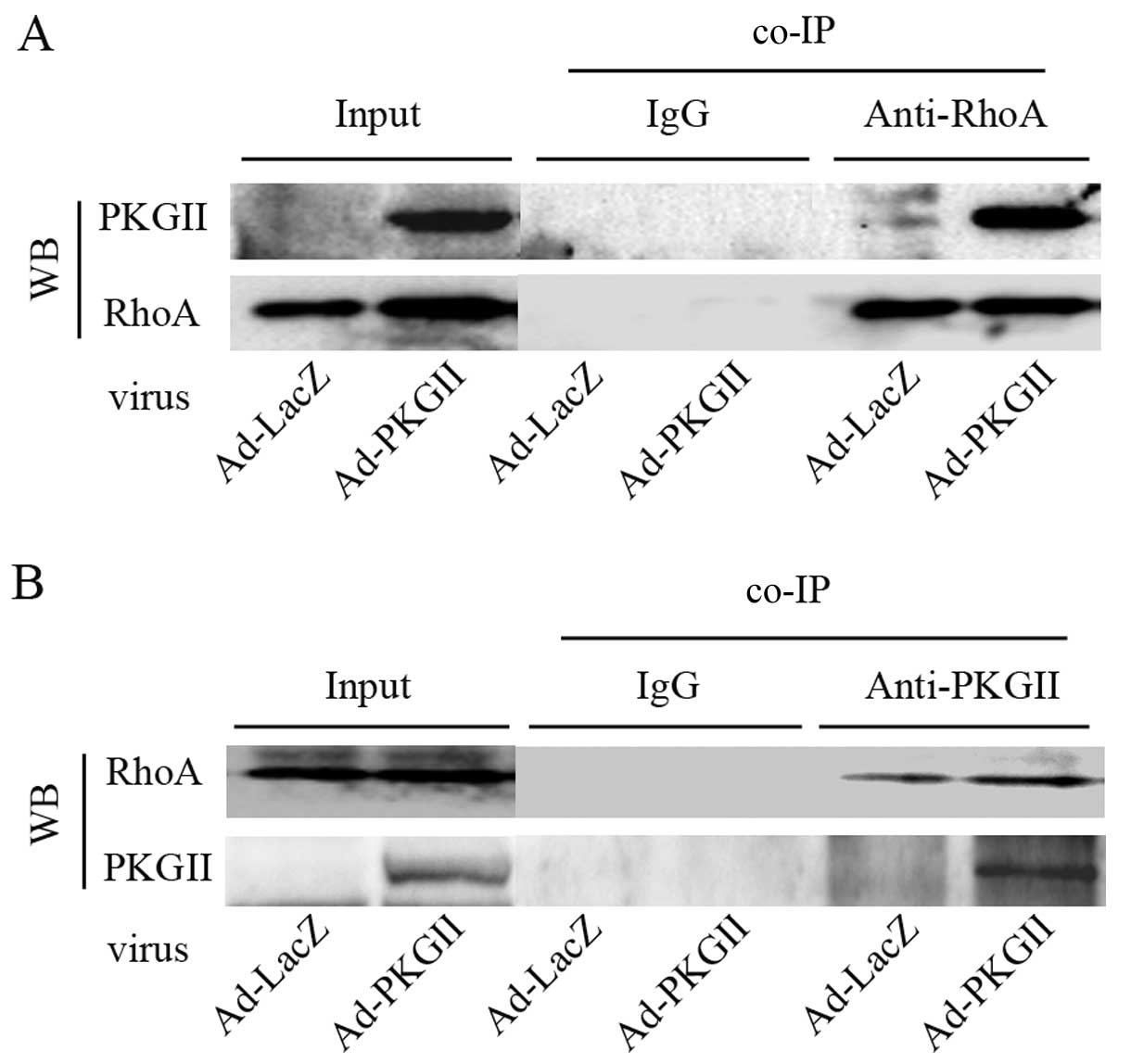
PKGII interacts directly with RhoA
The prerequisite for a kinase to phosphorylate its
substrate protein is its ability to directly bind the substrate. To
determine whether RhoA is the substrate of PKGII, we used a
co-immunoprecipitation (co-IP) approach to detect the potentially
direct binding of PKGII to RhoA. A binding complex between PKGII
and RhoA was detected in AGS cells (Fig. 6). To confirm this result, we
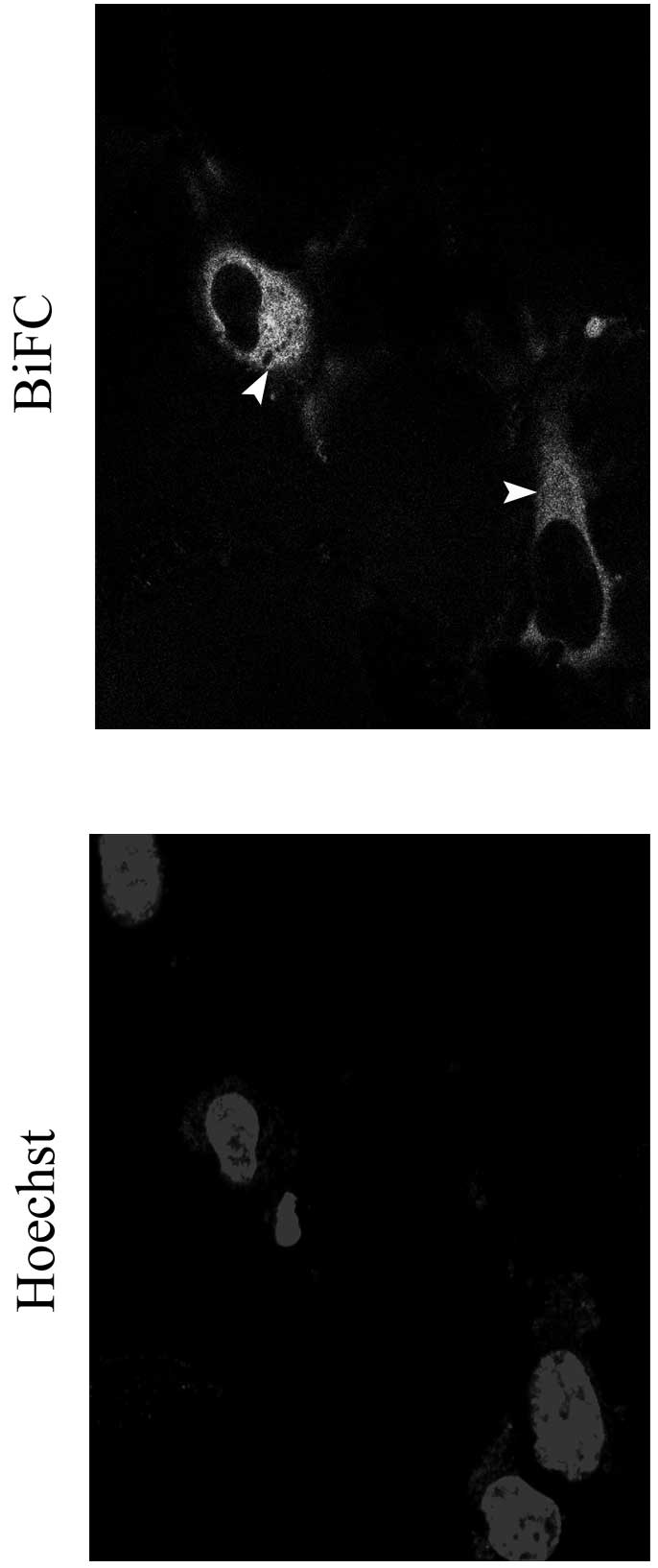
performed a BiFC assay, based on the formation of a fluorescent
complex by two non-fluorescent fragments of the Venus protein,
VN155 (VN) and VC155 (VC), brought together by the association of
proteins fused with each Venus fragment (19). We found that diffuse cytoplasmic
signals with a few aggregates were present in cells expressing
PKGII-VN and RhoA-VC (Fig. 7).
This result indicated that PKGII can bind RhoA protein in
vitro.
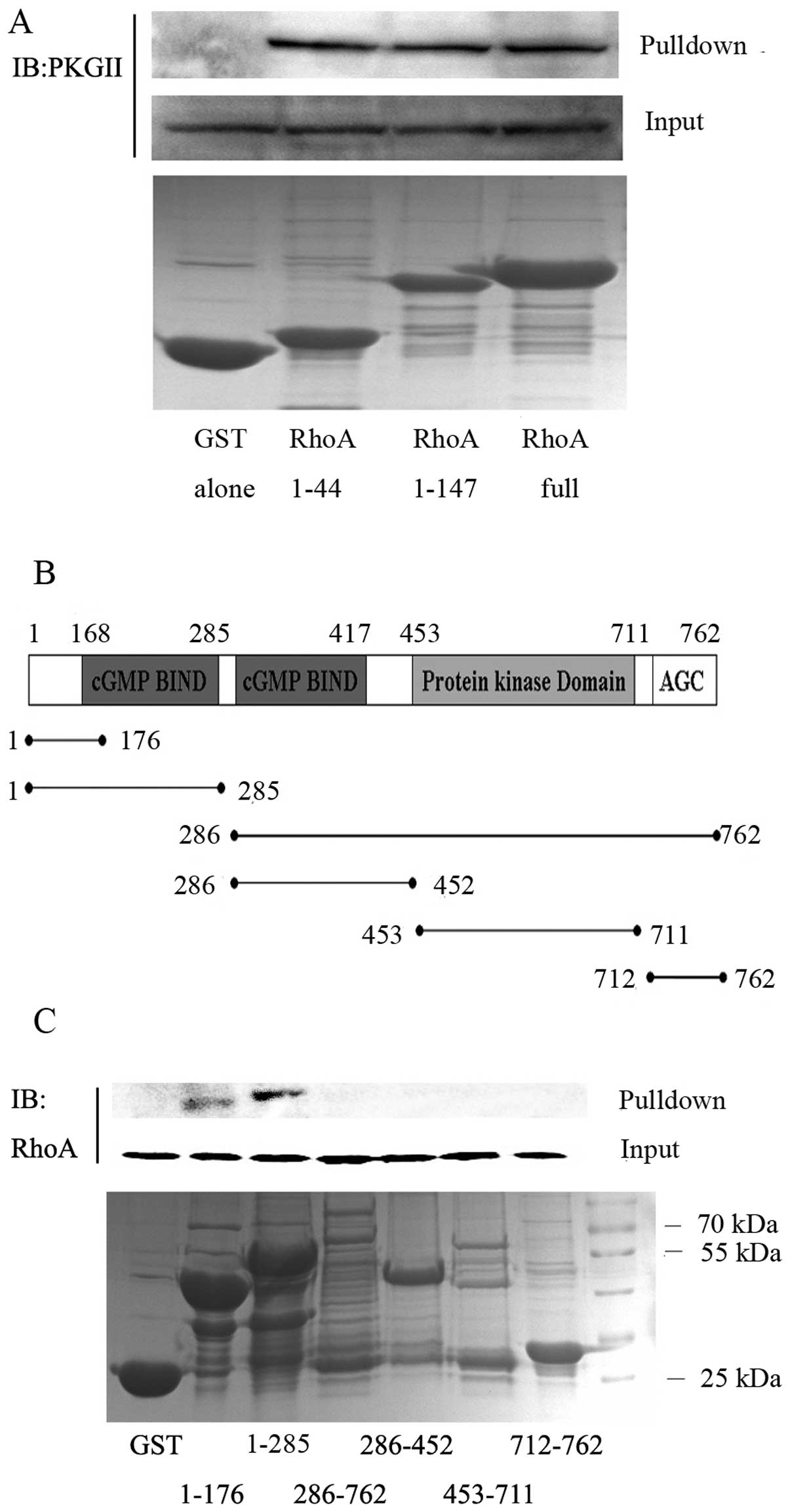
To define the specific domains of RhoA required for
binding to PKGII, different RhoA fragments were fused with the GST
protein according to Kato et al (20): the RhoA switch I domain (residues
1–44), the RhoA switch I and switch II domains (residues 1–147),
and the full-length RhoA (residues 1–192). The fragments were
expressed in E. coli, purified and added to cell extracts to
test PKGII precipitation. The results showed that all three
GST-RhoA constructs, but not GST alone, precipitated PKGII,
suggesting that amino acid residues 1–44 of RhoA, constituting the
switch I domain, are sufficient for the interaction with PKGII
(Fig. 8A). We also investigated
which domains of PKGII mediate the PKGII-RhoA interaction.
GST-fusion proteins containing specific domains of PKGII (Fig. 8B) were generated and incubated with
cell lysate containing abundant RhoA protein. Co-IP results showed
that RhoA is detected in pull-downs containing the PKGII N-terminal
portion, either using the fragments containing amino acid residues
1–176 or residues 1–285 (Fig. 8C).
This indicated that the fragment with amino acid residues 1–176 of
PKGII is important for the interaction with RhoA.
Discussion
PKGs are serine/threonine kinases that are widely
distributed in eukaryotes. Two genes (prkg1 and
prkg2) code for PKGs, the type I cGMP-dependent protein
kinase (PKGI) and PKGII. In mammals, PKGI is widely expressed in
vascular smooth muscle cells, platelets, lung, certain endothelial
cells, fibroblasts, heart and the cerebellum (21). PKGI has been identified as a tumor
suppressor protein based on its profound anti-tumor effect
(22,23). Unlike PKGI, PKGII is a
tissue-specific enzyme that is expressed mainly in the brush border
of intestinal mucosa and in specific regions of the brain (24,25).
PKGII is involved in regulating electrolyte and water secretion,
renin and aldosterone secretion and in the adjustment of the
biological clock (9,25). In recent years, new insights on the
roles of PKGII on cell proliferation, migration and apoptosis were
reported. For example, Swartling et al (11) found that PKGII has
anti-proliferative effects on human glioma cells. Wang et al
(12) reported a
pro-differentiation effect of PKGII in colon cancer cells. Results
from our laboratory have shown that PKGII inhibits proliferation
and MAPK-mediated signal transduction in human gastric cancer cells
(14,15). Furthermore, we demonstrated that
PKGII inhibits migration of gastric cancer cells by blocking EGFR
activation and consequently, PLCγ1- and MAPK/ERK-mediated signal
transduction pathways (16).
Overall, these studies indicate that PKGII plays important roles in
regulating a number of tumor cell biological activities, and that
this kinase, similarly to PKGI, is a potential tumor suppressor
protein.
The small G protein RhoA, a member of the Rho
subfamily of the Ras superfamily, is important for
actin/myosin-based cortical contractility, migration, invasion,
transformation, proliferation and survival of tumor cells (1–4). In
the GTP-bound active state, RhoA translocates to the plasma
membrane, where it interacts with effectors to transduce downstream
signals. It has been confirmed that RhoA activates ROCK1 and then
LIMK proteins to regulate the formation of stress fibers and induce
the migration of cancer cells (26–28).
Numerous signaling molecules may cross-talk with RhoA. Among them,
protein kinases exert their effect through
phosphorylation-dephosphorylation of the protein. In most cases,
phosphorylation occurs at a serine residue located at the
C-terminal domain of RhoA protein, and modifies its cellular
localization. The phosphorylation and inhibition of RhoA by PKA is
well established (6,7). It was shown that PKGI can also
phosphorylate RhoA at Ser188 and thereby, inactivate RhoA signaling
(29,30). However, no study thus far has
provided evidence that RhoA is the direct substrate of PKGII.
In the present study, we confirmed the inhibitory
effect of PKGII on RhoA and elucidated the underlying mechanism
in vitro. Previous studies have shown that the
G12/G13-RhoA pathway is important for
LPA-induced cell migratory activity, and the majority of studies
available on stress fiber signaling pathways have identified RhoA
as the major regulator of stress fiber formation under most
physiological conditions (18,31).
To confirm this in gastric cancer cells, we used Y27632, an
inhibitor of ROCK, to identify the role of the RhoA/ROCK pathway in
the process. The results showed that Y27632 partially blocked
LPA-induced migration and stress fiber formation in AGS cells,
indicating that the RhoA/ROCK pathway is involved in these
processes. To determine whether RhoA is the key target of PKGII, we
examined the inhibitory effect of PKGII on RhoA activation. The
results showed that PKGII inhibits the LPA-induced activation of
RhoA. Since the inhibition of RhoA is associated with its
phosphorylation status, we measured the phosphorylation level of
RhoA in cells with high activity of PKGII. The results confirmed
that PKGII phosphorylates RhoA at Ser188, and that this
phosphorylation partially contributes to the decrease in RhoA
activation.
To elucidate whether RhoA is the direct substrate of
PKGII, we investigated and confirmed the binding between RhoA and
PKGII by using co-IP and BiFC assays. Kato et al (20) found that PKGI binds directly to
RhoA via a direct interaction between the amino-terminus of RhoA
(residues 1–44), containing the switch I domain and the
amino-terminus of PKGI (residues 1–59), which includes a leucine
zipper heptad repeat motif. PKGII has a domain structure similar to
PKGI, consisting of an N-terminal regulatory domain, which contains
a dimerization and an auto-inhibitory region, two cGMP-binding
domains and a C-terminal catalytic domain. However, the position of
the high- and low-affinity cGMP-binding domains in PKGII is
reversed in comparison to PKGI (32). Moreover, the two PKG iso-enzymes
exhibit a different affinity towards various membrane permeable
cGMP-analogs, which allows their differentiation (24). To identify the domains of PKGII and
RhoA that are involved in their interaction, we expressed fusion
proteins with GST and RhoA or PKGII fragments and assessed their
binding ability by GST pull-down assays. These experiments
demonstrated that the N-terminal of PKGII and the switch I domain
of RhoA are required for PKGII-RhoA binding. Taken together, these
data support that PKGII directly binds to RhoA.
Since RhoA is involved in the biological activity of
tumor cells, new and more effective methods blocking the RhoA/ROCK
signal transduction pathway are expected to be useful in cancer
therapy. Therefore, our finding that PKGII can inhibit RhoA and
downstream signal transduction molecules is of high significance.
It provides the evidence that PKGII is a potential tumor inhibitor
and may also provide information on the development of therapeutic
strategies against cancer.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81272755, 31040002 and
81201959) and the Specialized Research Fund for Senior Personnel
Program of Jiangsu University (no. 11JDG032). We thank Dr Gerry
Boss and Dr Renate Pilz at the University of California, San Diego,
CA, USA for the kind gifts of adenoviral constructs.
References
|
1
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaffe AB and Hall A: RHO GTPases:
biochemistry and biology. Annu Rev Cell Dev Biol. 21:247–269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar
|
|
4
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bos JL, Rehmann H and Wittinghofer A: GEFs
and GAPs: critical elements in the control of small G proteins.
Cell. 129:865–877. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Forget MA, Desrosiers RR, Gingras D and
Beliveau R: Phophorylation states of Cdc42 and RhoA regulate their
interactions with Rho GDP dissociation inhibitor and their
extraction from biological membranes. Biochem J. 361:243–254. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lang P, Gesbert F, Delespine-Carmagnat M,
Stancou R, Pouchelet M and Bertoglio J: Protein kinase A
phosphorylation of RhoA mediates the morphological and functional
effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 15:510–519.
1996.PubMed/NCBI
|
|
8
|
Ellerbroek SM, Wennerberg K and Burridge
K: Serine phosphorylation negatively regulates RhoA in vivo. J Biol
Chem. 278:19023–19031. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hofmann F: The biology of cyclic
GMP-dependent protein kinases. J Biol Chem. 280:1–4. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cook AL and Haynes JM: Protein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cells. Cell Signal. 16:253–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swartling FJ, Ferletta M, Kastemar M,
Weiss WA and Westermark B: Cyclic GMP-dependent protein kinase II
inhibits cell proliferation, Sox9 expression and Akt
phosphorylation in human glioma cell lines. Oncogene. 28:3121–3131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang R, Kwon IK, Thangaraju M, Singh N,
Liu K, Jay P, Hofmann F, Ganapathy V and Browning DD: Type 2
cGMP-dependent protein kinase regulates proliferation and
differentiation in the colonic mucosa. Am J Physiol Gastrointest
Liver Physiol. 303:G209–G219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fallahian F, Karami-Tehrani F, Salami S
and Aghaei M: Cyclic GMP induced apoptosis via protein kinase G in
oestrogen receptor-positive and -negative breast cancer cell lines.
FEBS J. 278:3360–3369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen YC, Ren F, Sang JR, Tao Y and Xu WR:
Type II cGMP-dependent protein kinase inhibits proliferation of the
gastric cancer cell line BGC-823. Mol Med Rep. 3:361–366.
2010.PubMed/NCBI
|
|
15
|
Wu Y, Chen Y, Qu R, Lan T and Sang J: Type
II cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2012.PubMed/NCBI
|
|
16
|
Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu
M, Tao Y, Wang Y, Qian H and Gu L: PKG II inhibits EGF/EGFR-induced
migration of gastric cancer cells. PLoS One. 8:e616742013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren F, Gao Q, Tao Y, Sang J, Yang X, Fan S
and Chen Y: Impacts of cGMP dependent protein kinase II on gastric
cancer cell BGC-823 migration. J Jiangsu Univ (Med Ed). 20:113–116.
2010.
|
|
18
|
Bian D, Mahanivong C, Yu J, Frisch SM, Pan
ZK, Ye RD and Huang S: The G12/13-RhoA signaling pathway
contributes to efficient lysophosphatidic acid-stimulated cell
migration. Oncogene. 25:2234–2244. 2006.
|
|
19
|
Kodama Y and Hu CD: An improved
bimolecular fluorescence complementation assay with a high
signal-to-noise ratio. Biotechniques. 49:793–805. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kato M, Blanton R, Wang GR, Judson TJ, Abe
Y, Myoishi M, Karas RH and Mendelsohn ME: Direct binding and
regulation of RhoA protein by cyclic GMP-dependent protein kinase
Iα. J Biol Chem. 287:41342–41351. 2012.
|
|
21
|
Keilbach A, Ruth P and Hofmann F:
Detection of cGMP dependent protein kinase isozymes by specific
antibodies. Eur J Biochem. 208:467–473. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deguchi A, Thompson WJ and Weinstein IB:
Activation of protein kinase G is sufficient to induce apoptosis
and inhibit cell migration in colon cancer cells. Cancer Res.
64:3966–3973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou Y, Gupta N, Schoenlein P, Wong E,
Martindale R, Ganapathy V and Browning D: An anti-tumor role for
cGMP-dependent protein kinase. Cancer Lett. 240:60–68. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lohmann SM, Vaandrager AB, Smolenski A,
Walter U and De Jonge HR: Distinct and specific functions of
cGMP-dependent protein kinases. Trends Biochem Sci. 22:307–312.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hofmann F, Feil R, Kleppisch T and
Schlossmann J: Function of cGMP-dependent protein kinases as
revealed by gene deletion. Physiol Rev. 86:1–23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maekawa M, Ishizaki T, Boku S, Watanabe N,
Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K and Narumiya S:
Signaling from Rho to the actin cytoskeleton through protein
kinases ROCK and LIM-kinase. Science. 285:895–898. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yiu G and He Z: Glial inhibition of CNS
axon regeneration. Nat Rev Neurosci. 7:617–627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caramel J, Quignon F and Delattre O:
RhoA-dependent regulation of cell migration by the tumor suppressor
hSNF5/INI1. Cancer Res. 68:6154–6161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sawada N, Itoh H, Yamashita J, Doi K,
Inoue M, Masatsugu K, Fukunaga Y, Sakaguchi S, Sone M, Yamahara K,
et al: cGMP-dependent protein kinase phosphorylates and inactivates
RhoA. Biochem Biophys Res Commun. 280:798–805. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sauzeau V, Le Jeune H, Cario-Toumaniantz
C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P and
Loirand G: Cyclic GMP-dependent protein kinase signaling pathway
inhibits RhoA-induced Ca2+ sensitization of contraction
in vascular smooth muscle. J Biol Chem. 275:21722–21729. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pellegrin S and Mellor H: Actin stress
fibres. J Cell Sci. 120:3491–3499. 2007. View Article : Google Scholar
|
|
32
|
Vaandrager AB, Hogema BM and de Jonge HR:
Molecular properties and biological functions of cGMP-dependent
protein kinase II. Front Biosci. 10:2150–2164. 2005. View Article : Google Scholar : PubMed/NCBI
|