Introduction
Intrauterine adhesions (IUA) have various causes
including, incorrect uterine cavity surgery, induced abortion and
secondary infections. Patients with IUA often exhibit reduced
menstruation, amenorrhea and infertility. The disease was first
described by Asherman in 1948 and is clinically referred to as
Asherman’s syndrome (1).
Histologically, Asherman’s syndrome is a condition in which the
endometrium becomes fibrosed (2),
with the endometrial stroma becoming largely replaced by fibrous
tissue (3). Under hysteroscopy,
patients with Asherman’s syndrome exhibit pale scar tissue
distributed in islands among the normal endometrium, with severe
IUAs forming thick bands (4). At
present, the mechanism underlying intrauterine fibrosis is unclear
and there is a lack of effective means for the diagnosis and
prevention of the disease worldwide. Therefore, basic and clinical
research is required in order to elucidate the mechanism underlying
endometrial fibrosis and develop novel preventative and therapeutic
strategies.
Ang II is a key member of the
renin-angiotensin-aldosterone system (RAAS) and has roles in
several processes, including vasoconstriction, cell proliferation,
fibrinolysis and blood pressure elevation. Ang II has been reported
to be involved in the fibrosis of certain organs, including the
heart (5), kidney (6,7) and
liver (8,9); therefore, Ang II has an important
role in the process of organ fibrosis. Ang II has not only been
reported to promote the proliferation of various mesenchymal cells,
but also to cause the accumulation of extracellular matrix
components, leading to fibrosis (10–12).
Ang-(1–7) is a novel member of the RAAS and has
been shown to be an endogenous antagonist of Ang II. Ang-(1–7) has
been reported to have an important physiological role in dilating
blood vessels, lowering blood pressure, inhibiting vascular smooth
muscle cell and cardiac fibroblast proliferation (13), inhibiting myocardial cell
hypertrophy and reducing ventricular remodeling (14,15).
Ang II has increasingly been reported to have an important role in
the development of tissue fibrosis and the physiological antagonism
of Ang II by Ang-(1–7) in vivo may be important for
inhibiting tissue fibrosis.
The present study aimed to investigate the effect of
Ang-(1–7) and Ang II on human EECs in order to
develop a theoretical basis for the prevention and treatment of
endometrial adhesion.
Materials and methods
Endometrial specimen collection
Endometrial tissues were obtained by hysterectomy
from 20 females with uterine fibroids at The Second Affiliated
Hospital of Hebei Medical University (Shijiazhuang, China) between
September and December 2012. This study was approved by the Ethics
Committee of Hebei Medical University and informed consent was
obtained from all patients prior to hysterectomy. The patients with
uterine fibroids were aged between 30 and 45 years old with
menstrual cycle durations between 24 and 35 days (mean, 28 days).
None of the patients had received any hormonal treatment for at
least three months prior to the hysterectomies. Postoperative
pathological analyses were performed to confirm that the
endometrial tissues exhibited hyperplasia and were disease-free.
Endometrial tissue samples were collected under aseptic conditions
from the uterus and immediately placed in Dulbecco’s modified
Eagle’s medium/Nutrient Mixture F-12 (DMEM/F12; Gibco-BRL, Grand
Island, NY, USA) containing 10% fetal calf serum, penicillin and
streptomycin (100 mg/ml; Gibco-BRL) in an ice bath. Samples were
transported to the laboratory within 2 h.
EEC isolation, purification and
culture
Following several washes with phosphate-buffered
saline (PBS), tissues were cut into 1–2-mm3 pieces using
sterile scissors and incubated in 5 ml DMEM/F12 containing 0.2%
collagenase I (Sigma-Aldrich, St. Louis, MO, USA) in an incubator
with shaking for 60 min at 37°C with 5% CO2. Throughout
the incubation process, the tissue pieces were pipetted gently to
disperse the cells. The whole cell suspension, which contained EECs
and endometrial stromal cells (ESCs), was centrifuged at 500 × g
for 5 min. The supernatant containing the ESCs was discarded, while
the precipitate was resuspended in a culture flask with 3 ml
DMEM/F12 containing 10% fetal bovine serum (FBS; Gibco-BRL) and 1%
penicillin and streptomycin. The EECs attached to the culture flask
were washed several times with serum-free DMEM/F12 to remove the
red blood cells.
The Trypan blue exclusion assay (Zhongshan Biotech
Co., Ltd., Beijing, China) was performed to assess the proportion
of the active cells. A small number of the cells were then seeded
onto 6-well plates containing coverslips and incubated in an
atmosphere of 5% CO2 at 37°C for cell type
characterization and purity analyses.
Morphological observation of EECs
EECs were cultured in the aformentioned medium for 0
and 5 days and were stained with hematoxylin and eosin (H&E).
Endometrial cell morphology and structure were observed using an
inverted phase-contrast microscope (Nikon, Tokyo, Japan) and a
light microscope (Olympus, Tokyo, Japan).
Identification of EECs
To identify EECs and assess their purity,
immunocytochemical staining was performed. PBS was used as a
negative control instead of primary antibodies. Cells that had been
cultured on the cover slides were fixed using 4% paraformaldehyde
and treated with 0.25% Triton X-100. Subsequent to blocking using
5% normal goat serum for 20 min at 37°C, cells were incubated with
rabbit rabbit anti-human cytokeratin (dilution, 1:100) and vimentin
(dilution, 1:100) primary antibodies (Zhongshan Biotech Co., Ltd.)
at 4°C overnight. Cells were then incubated with goat anti-rabbit
immunoglobulin G (IgG; dilution, 1:100; Boster Biological
Technology Co., Ltd., Wuhan, China) for 20 min at 37°C and stained
with 3,3′-Diaminobenzidine (DAB; 5 mg/ml; Sigma-Aldrich) for 5 min
at room temperature. The specimens then underwent three 5-min
washes with PBS and were observed using light microscopy.
Experimental groups
EECs were divided into four groups according to the
different treatment interventions. The groups and treatment
conditions were as follows: Control group, treated with serum-free
DMEM/F12; the Ang II group, treated with Ang II and serum-free
DMEM/F12; the Ang-(1–7) group, treated with Ang-(1–7) and
serum-free DMEM/F12; and Ang II + Ang-(1–7)
group, treated with Ang II, Ang-(1–7) and
serum-free DMEM/F12. The final concentrations of Ang-(1–7) and
Ang II were 10−5 and 10−6 mol/l,
respectively.
Cell proliferation assay
EECs (4×104/ml) were seeded on 96-well
plates and cultured in serum-free DMEM/F12 for 24 h, in order to
synchronize their growth. The EECs were divided into the
aformentioned four groups, with six wells/group and cultured for
24, 48 and 72 h. MTT (5 mg/ml) was then added to each well and the
plates were incubated at 37°C for 2 h. The medium was subsequently
replaced with 150 μl dimethylsulfoxide and agitated for 10 min. The
absorbance at 560 nm was measured using a microplate reader
(Packard Instrument Co., Inc., Meriden, CT, USA).
Immunocytochemistry
The four groups of cells were cultured for 72 h. PBS
was used as a negative control instead of primary antibodies. Cells
that had been cultured on the cover slides were fixed using 4%
paraformaldehyde and treated with 0.25% Triton X-100. Subsequent to
blocking with 5% normal goat serum for 20 min at 37°C, cells were
incubated with rabbit anti-human anti-α-SMA and -E-cadherin
monoclonal antibodies (dilution, 1:100; Zhongshan Biotech Co.,
Ltd.) at 4°C overnight. Cells were incubated with goat anti-rabbit
IgG (dilution, 1:100; Boster Biological Technology Co., Ltd.) for
20 min at 37°C and stained with DAB (5 mg/ml; Sigma-Aldrich) for 5
min at room temperature. The specimens then underwent three 5 min
washes with PBS and were observed using light microscopy.
ELISA analysis
The four groups of cells were cultured for 72 h with
the solution provided in the ELISA kit (R&D Systems Inc.,
Minneapolis, MN, USA) according to the manufacturer’s instructions.
The absorbance value of each well was read on the full spectrum of
the spectrophotometer at a wavelength of 450 nm in order to detect
the content of collagen type I (Col I) and fibronectin (FN). Each
group consisted of six wells, with the absorbance values averaged
over the wells.
Western blot analysis
The four groups of cells were grown in 10-cm dishes
and cultured for 72 h. Cells were then washed with PBS and lysed
with lysis buffer (pH 7.4; 1 M Tris-HCl, 1% Triton X-100, sodium
deoxycholate and 10% SDS). Solubilized proteins were centrifuged at
14,000 × g at 4°C for 30 min. Extracted proteins were quantified
using the Coomassie Protein Assay reagent (Sigma-Aldrich). Western
blot analysis of α-SMA and E-cadherin was performed. In brief, 30
μg isolated protein was electrophoresed on 8% sodium dodecyl
sulfate-polyacrylamide gels, then transferred (100 V for 1.5 h)
onto polyvinylidene fluoride membranes (Gibco-BRL). Membranes were
treated with blocking solution [Tris-buffered saline (TBS), pH 7.2;
0.1% Tween-20 and 5% milk] for 1 h, prior to incubation with rabbit
anti-human α-SMA, transforming growth factor (TGF)-β1, insulin-like
growth factor (IGF)-I, Col I and FN monoclonal primary antibodies
(Invitrogen Life Technologies, Carlsbad, CA, USA), diluted 1:1,000
in TBS (pH 7.2) containing 0.1% Tween-20, for 12 h at 4°C.
Subsequent to four 15-min washes with TBS (pH 7.2) containing 0.1%
Tween-20, the membranes were incubated with the goat anti-rabbit
IgG horseradish peroxidase-conjugated antibody (Invitrogen Life
Technologies) diluted 1:1,000 in TBS (pH 7.2) with 0.1% Tween-20
for 1 h. Membranes then underwent four 15-min washes with TBS (pH
7.2) and enhanced chemiluminescence (ECL; Gibco-BRL) was performed
in accordance with the manufacturer’s instructions. Membranes were
then exposed to Kodak X-ray film (R&D Systems Inc.). for 0.5–20
min as required to detect the immunoreactive bands. The relative
intensity of the immunoreactive bands on the exposed films was
quantified using a computer-assisted densitometry program
(SmartView, Major Science, Saratoga, CA, USA). Protein expression
was quantified relative to the internal control, glyceraldehyde
3-phosphate dehydrogenase (GAPDH).
RNA extraction and reverse
transcription
Total RNA was extracted from the four groups of
cells. In brief, the cells were cultured for 72 h with
TRIzol® Reagent (Invitrogen Life Technologies) according
to the manufacturer’s instructions and the RNA was dissolved in
RNase-free water. The integrity of the RNA was assessed using
ethidium bromide agarose gel electrophoresis and the quantity of
RNA was determined by measuring the relative absorbance at 260 vs.
280 nm. Complementary (c)DNA was synthesized in a reaction volume
of 10 μl using a cDNA synthesis kit (Takara Bio, Inc., Shiga,
Japan) according to the manufacturer’s instructions. The cDNA was
stored at −20°C.
Primer preparation
For quantitative polymerase chain reaction (qPCR)
amplification, primers (Invitrogen Life Technologies) were derived
from the GenBank database. GAPDH was used as the housekeeping gene.
The primer sequences were as follows: Forward: 5′-GATGGGCATCTATCA
GATAC-3′ and reverse: 5′-AAGCATTTCTGATGGTGATG-3′ for α-SMA;
forward: 5′-ATAGAGAACGCATTGC CACATACA-3′ and reverse:
5′-TTCTGATCGGTTACCGTG ATCA-3′ for E-cadherin; forward:
5′-AGGGCCAAGACGAAG ACATC-3′ and reverse: 5′-GTCGGT
GGGTGACTCTGAGC-3′ for Col I; forward: 5′-TGGAGGAAGCCGAGGTTT-3′ and
reverse: 5′-CAGCGGTTTGCGATGGTA-3′ for FN; and forward:
5′-TGCACCACCAACTGCTTAGC-3′ and reverse: 5′-GGCATGGACTGTGGTCATGAG-3′
for GAPDH.
qPCR analysis
qPCR reactions were performed using a Brilliant
SYBR® Green qRT-PCR Master mix according to the
manufacturer’s instructions (Invitrogen Life Technologies). α-SMA,
Col I, E-cadherin and FN RNA was amplified using the ABI Prism
7500® Real-Time PCR system (Applied Biosystems, Foster
City, CA, USA). qPCR analysis was performed under the following
reaction conditions: 95°C for 10 min followed by 40 cycles of 95°C
for 15 sec, 60°C for 30 sec, and 72°C for 32 min. The amplified
products were subjected to a stepwise increase in temperature from
60°C to 95°C and dissociation curves were constructed.
Target mRNA was quantified by measuring the
threshold cycle and reading against a calibration curve. The
relative quantity of the target gene mRNA was normalized to that of
the housekeeping gene, GAPDH. Results were analyzed using the
relative standard curve method of analysis/ΔCt method of
analysis.
Statistical analysis
Data are presented as bar graphs with the mean ±
standard deviation of six independent experiments with samples from
independent subjects. Data were analyzed using SPSS 15.0 (SPSS,
Inc., Chicago, IL, USA) for Windows. Statistical analysis was
performed using one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
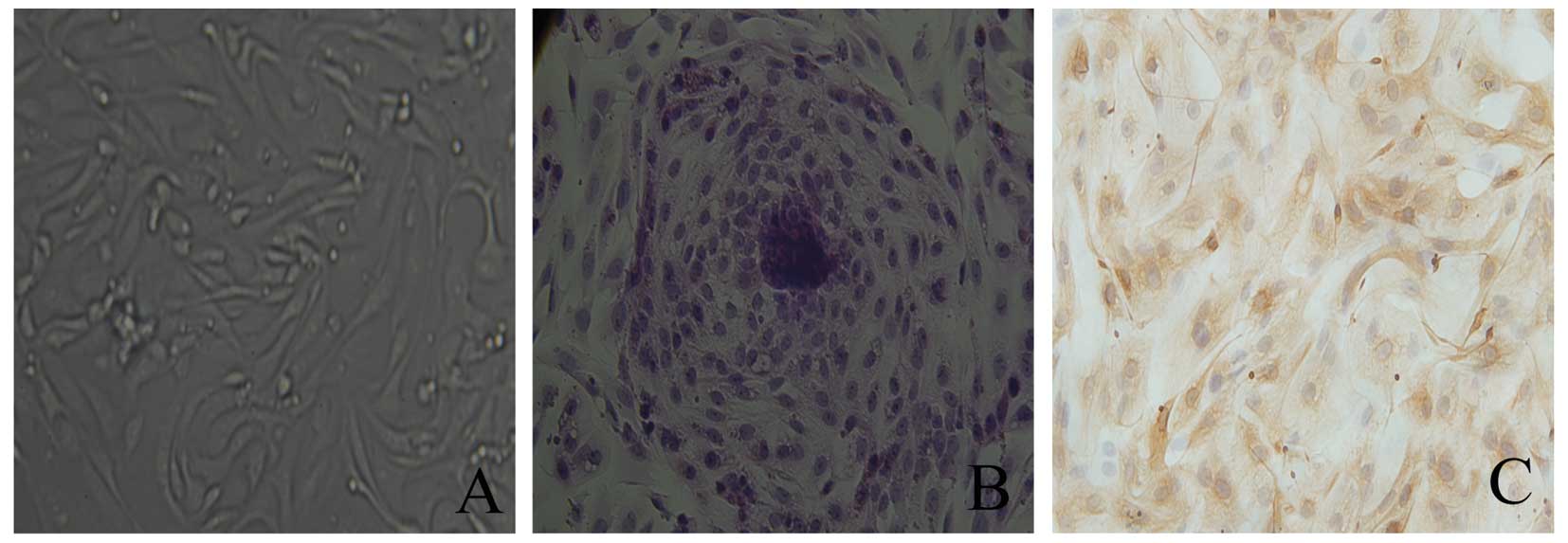
Morphological observation of EECs
The EECs were observed using an inverted
phase-contrast microscope. The isolated EECs exhibited a
round-shaped morphology. The differential centrifugation method
revealed that the purity of the EECs was ~90%. Trypan blue staining
showed that the viability of the EECs was 92–96%. The cells were
found to attach and grow after 24 h of culture and the majority of
the EECs formed tightly packed whorls and grew to confluence after
3 days of culture. The EECs exhibited round or elliptical
morphology with single large, round nuclei in the center of each
cell with a nucleolus (Fig. 1A).
The EECs were stained using H&E after 5 days of culture
(Fig. 1B).
Identification of EECs
EECs were detected using immunocytochemical
staining. The cytoplasm of EECs, which were positively stained for
cytokeratin and negatively stained for vimentin, was stained
claybank (Fig. 1C).
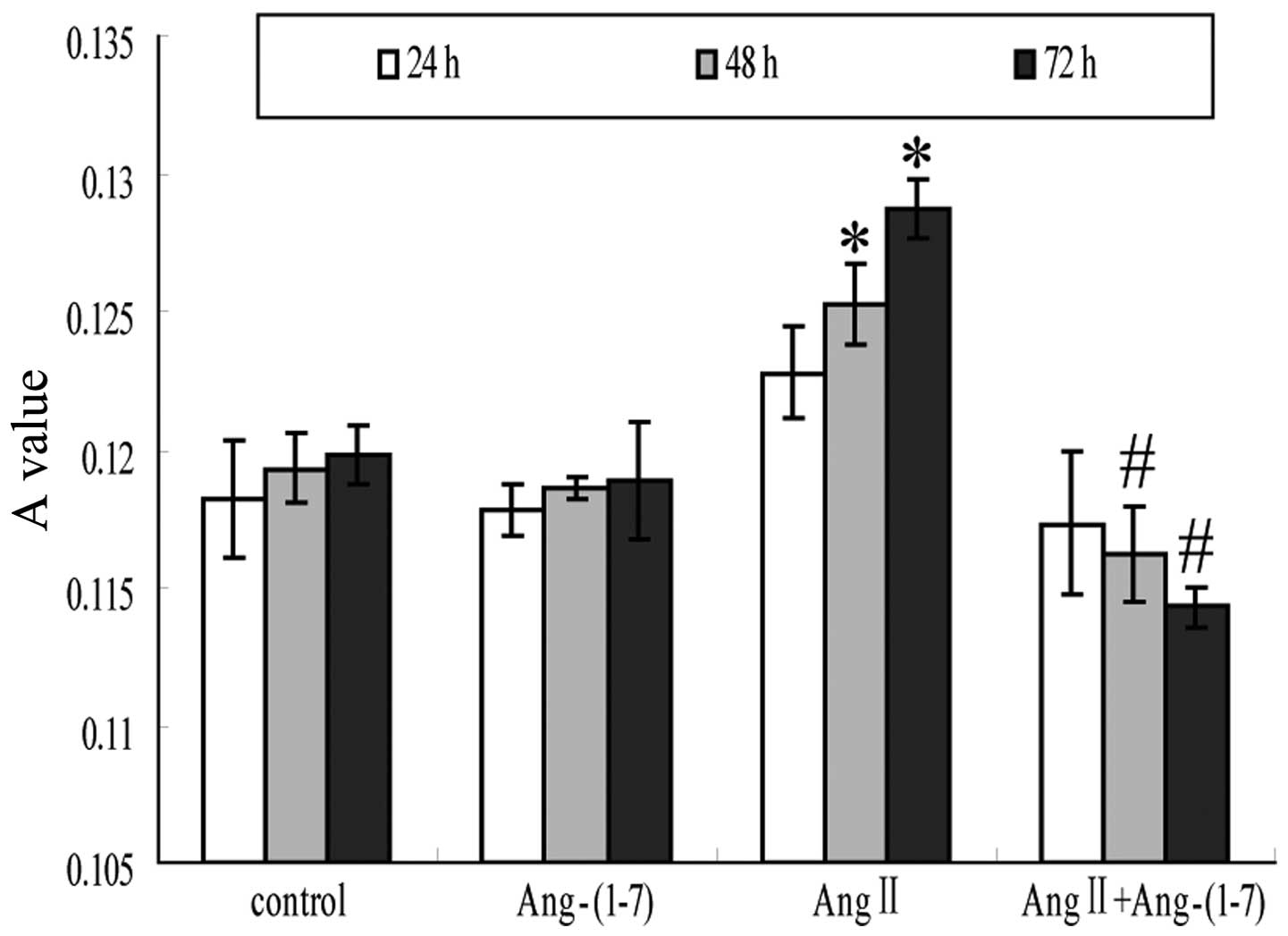
Ang-(1–7)
attenuates Ang II-stimulated cell proliferation
The four groups of EECs were cultured for 24, 48 and
72 h, and the number of EECs in each well were observed by
measuring the the absorbance value (A) at 550 nm of the purple
crystals in each well. Compared with the control group, the number
of EECs in the Ang-(1–7) group was not identified to be
significantly different (P>0.05), while the number of EECs in
the Ang II group was significantly increased in a time-dependent
manner compared with that in the control group (P<0.05).
Furthermore, the number of EECs in the Ang II+Ang-(1–7) was
significantly reduced compared with that in the Ang II group,
suggesting that Ang-(1–7) significantly inhibits Ang II-induced
EEC proliferation (Fig. 2).
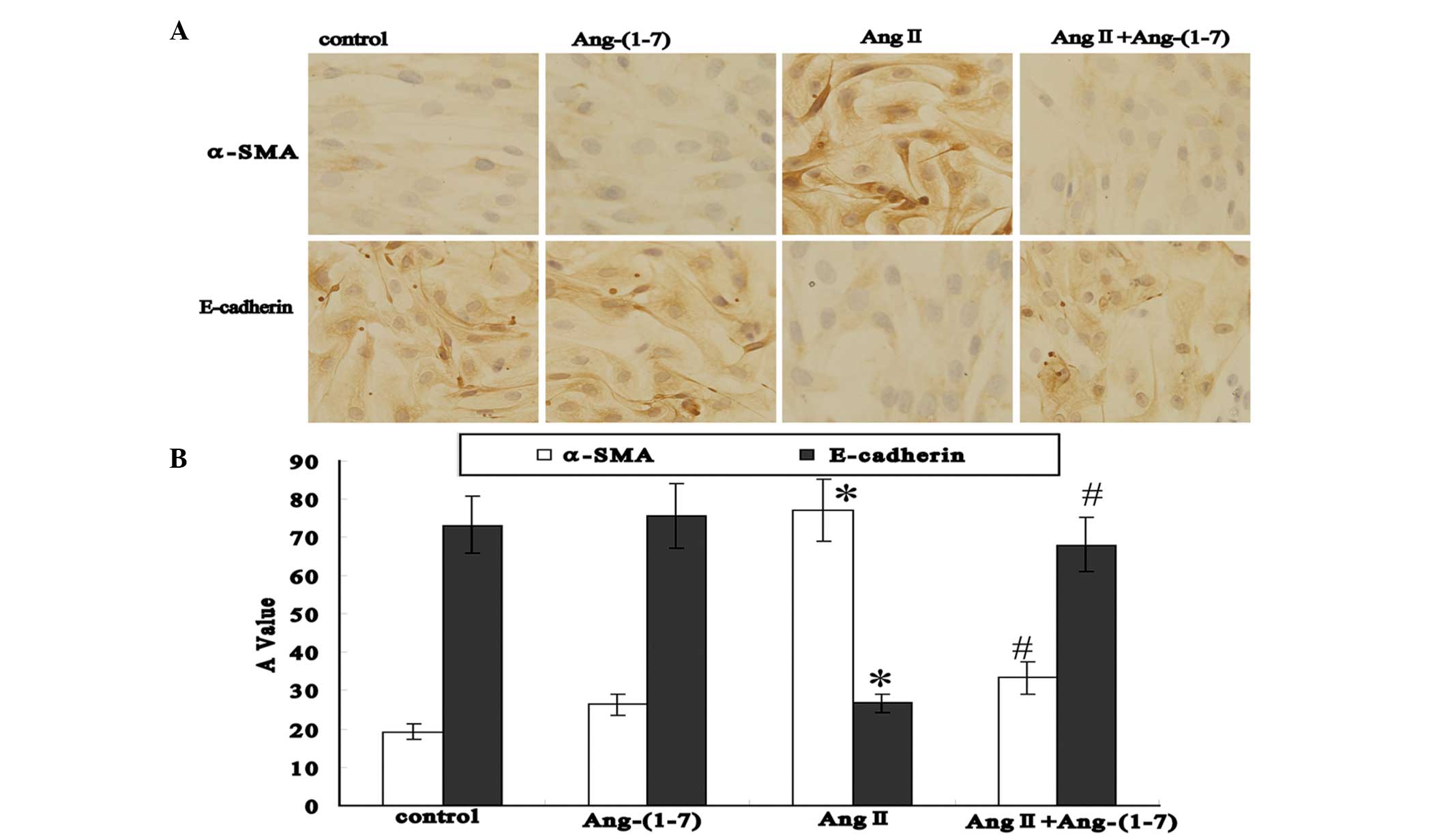
Effect of Ang II and Ang-(1–7) on
α-SMA and E-cadherin expression in EECs detected using
immunocytochemistry
After the EECs had been cultured for 72 h,
immunocytochemistry revealed that the EECs in the control group
exhibited low positive expression for α-SMA and high positive
expression for E-cadherin. In the Ang-(1–7)
group, α-SMA and E-cadherin expression was similar to that in the
control group (P>0.05). However, α-SMA expression was observed
to be significantly increased in the Ang II group compared with the
control group (P<0.05) and significantly decreased in the Ang
II+Ang-(1–7) group compared with the Ang II group
(P<0.05). Furthermore, E-cadherin expression was found to be
significantly decreased in the Ang II group compared with the
control group (P<0.05), but significantly increased in the Ang
II+Ang-(1–7) group compared with the Ang II group
(P<0.05) (Fig. 3).
Effect of Ang II and Ang-(1–7) on
α-SMA and E-cadherin expression in EECs detected using western blot
analysis
After the EECs had been cultured for 72 h, western
blot analysis revealed that the EECs in the control group exhibited
low α-SMA expression and high E-cadherin expression. α-SMA and
E-cadherin expression in the Ang-(1–7)
group was found to be similar to that in the control group
(P>0.05). However, α-SMA expression was observed to be increased
significantly increased in the Ang II group compared with the
control group (P<0.05), while significantly decreased in the Ang
II+Ang-(1–7) group compared with that in the Ang II
group (P<0.05). Furthermore, E-cadherin expression was found to
be significantly decreased in the Ang II group compared with the
control group (P<0.05), but significantly increased in the Ang
II+Ang-(1–7) group compared with that in the Ang II
group (P<0.05) (Fig. 4A and
B).
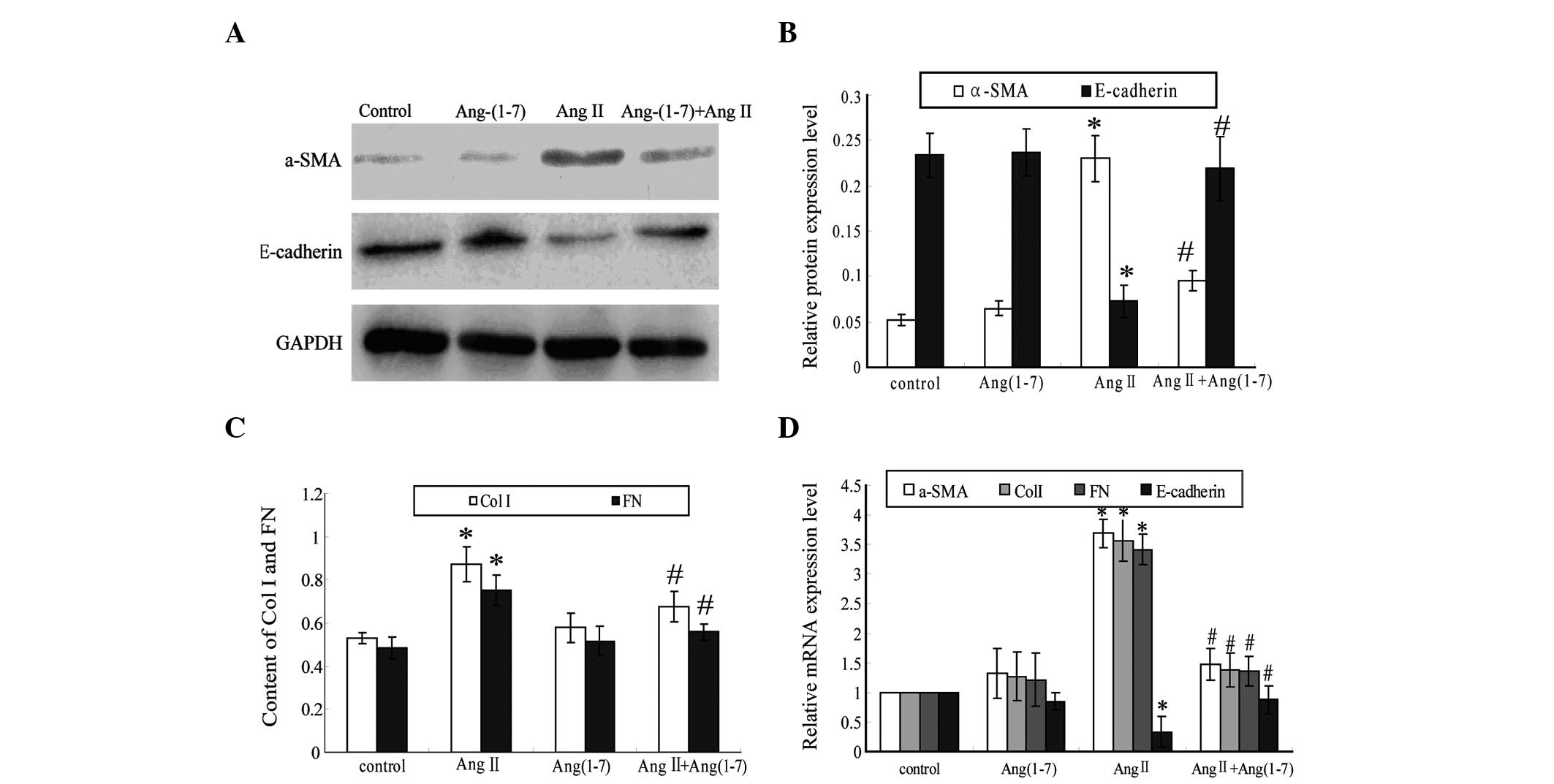 | Figure 4Ang-(1–7)
inhibits Ang II-induced EEC transdifferentiation. EECs were
incubated with different intervention factors for 72 h. (A) The
protein expression of α-SMA and E-cadherin was analyzed using
western blot analysis. (B) The protein expression of α-SMA and
E-cadherin were quantified using densitometry. (C) The levels of
Col I and FN protein in the EEC culture supernatant detected using
ELISA. (D) Col I, FN, α-SMA and E-cadherin mRNA expression analyzed
using quantitative polymerase chain reaction. The groups were as
follows: Control, EECs incubated with serum-free DMEM/F12; Ang II,
EECs incubated with 10-6 mol/l Ang II and serum-free DMEM/F12;
Ang-(1–7), EECs incubated with 10-5 mol/l
Ang-(1–7) and serum-free DMEM/F12; Ang
II+Ang-(1–7), EECs incubated with 10-6 mol/l Ang II
and 10−5 mol/l Ang-(1–7) and
serum-free DMEM/F12. Values are presented as the mean ± standard
deviation (n=6). *P<0.05 vs. the control group and
#P<0.05 vs. the Ang II group. α-SMA, α-smooth muscle;
EEC, endometrial epithelium cell; Ang, angiotensin; col I, collagen
type I; FN, fibronectin. |
Effect of Ang II and Ang-(1–7) on
Col I and FN expression in cultured EEC supernatants detected using
ELISA
After the EECs had been cultured for 72 h, ELISA
revealed that Col I and FN levels in the EEC culture supernatant in
the Ang-(1–7) group showed no significant difference
compared with that in the control group (P>0.05). Furthermore,
the levels of Col I and FN in the EEC culture supernatant were
observed to be significantly increased in the Ang II group compared
with the control group (P<0.05), while significantly decreased
in the Ang II+Ang-(1–7) group compared with the Ang II group
(P<0.05) (Fig. 4C).
Effect of Ang II and Ang-(1–7) on
α-SMA, E-cadherin, Col I and FN expression in EECs detected using
qPCR analysis
After the EECs had been cultured for 72 h, qPCR
analysis revealed that compared with those of the control group,
the mRNA levels of α-SMA, E-cadherin, Col I and FN in the
Ang-(1–7) group showed no significant change.
However, compared with the control group, in the Ang II group,
α-SMA, Col I and FN expression were found to be significantly
increased and E-cadherin expression was observed to be
significantly decreased (P<0.05). Furthermore, compared with the
Ang II group, in the Ang II+Ang-(1–7)
group, α-SMA, Col I and FN expression were significantly decreased,
while E-cadherin expression was significantly increased (P<0.05)
(Fig. 4D).
Discussion
IUAs are caused by numerous factors and are
characterized by the proliferation of ESCs and the excessive
accumulation of extracellular matrix components. Mechanisms
involved in the development of IUAs include: (i) Inflammatory,
immune and toxic stimuli-induced ESC and EEC activation, causing
proliferation and transdifferentiation into myofibroblast cells;
(ii) increases in extracellular matrix components and decreases in
extracellular matrix degradation; and (iii) the involvement of
certain vascular active substances, including Ang II, as well as
TGF-β1 and cytokines. The disappearance of a large number of EECs
during the development of IUA, may be associated with EEC
transdifferentiation into myofibroblasts.
EEC transdifferentiation is important during the
development and progression of IUA. During EEC transdifferentiation
under certain pathological conditions, EECs lose the expression of
epithelial markers, including cytokeratin and E-cadherin, and begin
to express mesenchymal cell markers, including α-SMA and vimentin.
The transformation of the cell phenotype often loses control and
leads to excessive proliferation or hypertrophy. Furthermore,
secretion and degradation of extracellular matrix components often
loses balance and excessive secretion of cytokines, inflammatory
chemokines or cell adhesion molecules occurs. These events are
directly involved in the process of endometrial fibrosis.
As EECs and stromal cells originate from the same
embryonic stem cell, under certain pathological conditions, the
transdifferentiation of EECs to mesenchymal cells can occur, with
EECs transdifferentiating into myofibroblasts with increased α-SMA
expression and migrating through the basement membrane into the
lamina propria (16).
Myofibroblasts have a role in tissue fibrosis and have a higher
capacity for proliferation and the secretion of ECM components
compared with ordinary fibroblasts. Myofibroblasts initially
secrete FN, which provides support for the sedimentation of other
ECM components and the formation of collagen fibers. Myofibroblasts
then secrete collagen, primarily type I and type III collagen, as
well as laminin and proteoglycans. Thus, ECM generation is greater
than ECM degradation, eventually leading to endometrial
fibrosis.
Ang II not only promotes the proliferation of
various types of mesenchymal cell, but also causes the accumulation
of extracellular matrix components, leading to fibrosis. A study
demonstrated that Ang II is capable of inducing cardiomyocyte
hypertrophy and myocardial interstitial fibrosis (5). Ang II has also been reported to
stimulate renal tubular epithelial cells to produce TGF-β and to
promote the expression of fibrotic factors, including connective
tissue growth factor, basic fibroblast growth factor, PAl-1, as
well as promote the expression of platelet-derived growth factor
(PDGF), which aggravate renal interstitial fibrosis (17). The present study showed that in
vitro, Ang II promotes proliferation and activation of EECs,
and significantly increases the expression of α-SMA protein and
α-SMA mRNA (P<0.05), while Ang II significantly decreases the
expression of E-cadherin protein and mRNA (P<0.05). These
findings suggest that Ang II promotes cells with EEC phenotypes to
transdifferentiate into cells with myofibroblast phenotypes.
Furthermore, in the present study, Ang II was observed to
significantly increase the expression of Col I and FN mRNA in EECs
and EEC culture supernatant (P<0.05), suggesting that Ang II
promotes the secretion of extracellular matrix proteins, including
FN and Col I, consistent with the results. Therefore, Ang II may
have an important role in promoting endometrial fibrosis.
Ang-(1–7) is a novel member of the RAAS and is an
endogenous antagonist of Ang II, which may exhibit an anti-fibrotic
effect by inhibiting cell proliferation and extracellular matrix
accumulation. Le Tran and Forster (18) were the first to identify that
Ang-(1–7) is capable of inhibiting the effect of
Ang II, FBS and PDGF on the proliferation of smooth muscle cells.
Tallant et al (14) also
showed that Ang-(1–7) inhibits the effect of FBS and
endothelin-1 on the proliferation of newborn rat cardiac
fibroblasts and on myocyte hypertrophy. Furthermore, Zhang et
al (19) identified that
Ang-(1–7) inhibits the effect of Ang II on the
proliferation of vascular smooth muscle cells. The rat model of
carotid artery injury was treated with Ang (1–7) (24
pg/kg/h) for 12 days, and the results demonstrated that Ang
(1–7) can reduce the neointimal area and
significantly reduce DNA synthesis of membrane cells (mainly in
smooth muscle cells). These findings show that Ang-(1–7) is
capable of inhibiting the proliferation of smooth muscle cells
in vivo and may prevent restenosis following angioplasty
(20). Ang-(1–7) has
also been shown to inhibit the effect of Ang II on fibrosis of
skeletal muscle cells, as well as reduce the expression of TGF-β1
induced by Ang-II in a dose-dependent manner (21). TGF-β1 is one of the predominant
cytokines involved in causing fibrosis, thus the inhibitory effect
of Ang-(1–7) on TGF-β1 inhibition may be an
important anti-fibrotic mechanism in the body. Burns et al
(7) further showed that
Ang-(1–7) inhibits Ang II-induced
transdifferentiation of renal tubular epithelial cells and the
expression of extracellular matrix proteins, using the joint
intervention of Ang II and Ang-(1–7) in
rat renal tubular epithelial cells. The present study identified
that in vitro, Ang-(1–7) had
an inhibitory effect on Ang II-induced EEC proliferation and
Ang-(1–7) inhibited Ang II-induced α-SMA
expression (P<0.05). Furthermore, Ang-(1–7) was
observed to inhibit the Ang II-induced decrease in E-cadherin mRNA
and protein expression (P<0.05). These findings suggest that
Ang-(1–7) is capable of inhibiting Ang II-induced
EEC transdifferentiation at the protein and gene level. Compared
with the Ang II group, the expression of Col I and FN mRNA and the
levels of FN and Col I in the EEC culture supernatant were found to
decrease significantly in the Ang-(1–7)+Ang
II group (P<0.05). This indicates that Ang-(1–7)
inhibits the Ang II-induced secretion of FN and Col I in EECs,
suggesting Ang-(1–7) is capable of reducing the synthesis of
ECM components, inhibiting the occurrence and development of
endometrial fibrosis, thus delaying the process of endometrial
fibrosis.
In conclusion, Ang-(1–7) is
capable of inhibiting Ang II-induced transdifferentiation of EECs
into myofibroblasts and Ang-(1–7)
inhibits the Ang II-induced secretion of extracellular matrix
components, including FN and Col I. This has important significance
for the prevention and treatment of endometrial fibrosis in the
clinic.
Acknowledgements
The authors would like to thank Dr Su-qing Chen at
the Second Affiliated Hospital, Hebei Medical University
(Shijiazhuang, China) for the endometrial tissue collection.
References
|
1
|
Deans R and Abbott J: Review of
intrauterine adhesions. J Minim Invasive Gynecol. 17:555–569. 2010.
View Article : Google Scholar
|
|
2
|
Buckley CH: Normal endometrium and
non-proliferative conditions of the endometrium. Obstetrical and
gynaecological pathology. Fox H and Wells M: 5th edition. Churchill
Livingstone 66; London: pp. 391–442. 2002
|
|
3
|
Yu D, Wong YM, Cheong Y, et al: Asherman
syndrome - one century later. Fertil Steril. 89:759–779. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Inany H: Intrauterine adhesions. An
update. Acta Obstet Gynecol Scand. 80:986–993. 2001.PubMed/NCBI
|
|
5
|
Kawano H, Do YS, Kawano Y, et al:
Angiotensin II has multiple profibrotic effects in human cardiac
fibroblasts. Circulation. 101:1130–1137. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mezzano SA, Aros CA, Droguett A, et al:
Renal angiotensin II up-regulation and myofibroblast activation in
human membranous nephropathy. Kidney Int Suppl. 64:S39–S45. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burns WC, Velkoska E, Dean R, et al:
Angiotensin II mediates epithelial-to-mesenchymal transformation in
tubular cells by ANG 1–7/MAS-1-dependent pathways. Am J Physiol
Renal Physiol. 299:F585–F593. 2010.PubMed/NCBI
|
|
8
|
Powell EE, Edwards-Smith CJ, Hay JL, et
al: Host genetic factors influence disease progression in chronic
hepatitis C. Hepatology. 31:828–833. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bataller R, Ginès P, Nicolás JM, et al:
Angiotensin II induces contraction and proliferation of human
hepatic stellate cells. Gastroenterology. 118:1149–1156. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson RJ, Alpers CE, Yoshimura A, et al:
Renal injury from angiotensin II-mediated hypertension.
Hypertension. 19:464–474. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen L, Liu BC, Zhang XL, et al: Influence
of connective tissue growth factor antisense oligonucleotide on
angiotensin II-induced epithelial mesenchymal transition in HK2
cells. Acta Pharmacol Sin. 27:1029–1036. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rupérez M, Ruiz-Ortega M, Esteban V, et
al: Angiotensin II increases connective tissue growth factor in the
kidney. Am J Pathol. 163:1973–1947. 2003.PubMed/NCBI
|
|
13
|
Gallagher PE, Chappell MC, Ferrario CM and
Tallant EA: Distinct roles for ANG II and ANG-1–7 in the regulation
of angiotensin-converting enzyme 2 in rat astrocytes. Am J Physiol
Cell Physiol. 290:C420–C426. 2006.
|
|
14
|
Tallant EA, Ferrario CM and Gallagher PE:
Angiotensin-1–7 inhibits growth of cardiac myocytes through
activation of the mas receptor. Am J Physiol Heart Circ Physiol.
289:H1560–H1566. 2005.
|
|
15
|
Liu J, Ma LH, Wang MY, et al: Influence of
angiotensin-1–7 on proliferation and secretion of cultured rat’s
glomerular mesangial cells induced by angiotensin II. Chin J Modern
Med. 15:1789–1802. 2005.(In Chinese).
|
|
16
|
Bi WR, Xu GT, Lv LX and Yang CQ: The ratio
of transforming growth factor-β1/bone morphogenetic protein-7 in
the progression of the epithelial-mesenchymal transition
contributes to rat liver fibrosis. Genet Mol Res. 13:1005–1014.
2014.
|
|
17
|
Ng YY, Huang TP, Yang WC, et al: Tubular
epithelial-myofibroblast transdifferentiation in progressive
tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int.
54:864–876. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Le Tran Y and Forster C: Angiotensin-1–7
and the rat aorta: modulation by the endothelium. J Cardiovasc
Pharmacol. 30:676–682. 1997.
|
|
19
|
Zhang F, Hu Y, Xu Q and Ye S: Different
effects of angiotensin II and angiotensin-1–7 on vascular smooth
muscle cell proliferation and migration. PLoS One.
5:e123232010.
|
|
20
|
Ferreira AJ and Santos RA: Cardiovascular
actions of angiotensin-1–7. Braz J Med Biol Res. 38:499–507.
2005.
|
|
21
|
Morales MG, Abrigo J, Meneses C, et al:
Angiotensin 1–7/Mas-1 axis attenuates the expression and signaling
of TGF-β1 induced by Angiotensin II in skeletal muscle. Clin Sci
(Lond). Mar 3–2014.(Epub ahead of print).
|