Introduction
Macroautophagy (hereafter referred to as autophagy)
is an intracellular catabolic mechanism by which proteins, cellular
organelles and invading microbes are degraded by lysosomal
machinery (1). Autophagy is
associated with aging, autoimmunity, infection, heart disease,
cancer and neurodegenerative disorders (2), and shares certain regulatory pathways
and molecular mechanisms with apoptosis (3).
While a number of studies examined autophagy in
liver disease (4–7), the relationship between autophagy and
liver fibrosis has not been broadly investigated (8–10).
Hernández-Gea et al (11)
demonstrated that autophagy releases lipids that promote activated
hepatic stellate cells (HSCs) to create fibrotic damage. In
addition, Thoen et al (12)
observed that autophagic flow increased following HSC activation
in vitro, and that treatment with an autophagy inhibitor
partially inhibited HSC activation. While there is support for the
theory that autophagy is involved in HSC activation, the underlying
molecular mechanisms remain elusive. Autophagy in liver cells may
produce distinct physiological and pathological outcomes under
different conditions. In the majority of types of liver disorders,
including liver ischemia/reperfusion injury and fatty liver
disease, and following liver transplantation, autophagy mainly
serves a protective role in which it promotes lipid droplet
degradation, decreases protein deposition and guards against cell
apoptosis (13,14). However, these protective actions of
autophagy may activate HSC proliferation by blocking apoptosis,
leading to accelerated fibrotic progression (15).
HSC activation and proliferation is an essential
mechanism for promoting liver fibrosis (16,17).
TGF-β1 is important in HSC activation and proliferation, and TGF-β1
inhibitors have been demonstrated to reduce liver fibrosis in
rodents. However, since TGF-β1 is a prototypical cytokine that
regulates a plethora of cellular pathways, non-specific inhibition
of TGF-β1 is likely to produce multiple unpredictable adverse
effects, thereby limiting its application in humans (18).
TGF-β1 induces autophagy activation, which regulates
cell proliferation and apoptosis in a cell type-dependent manner
(19). In epithelial cells,
TGF-β1-induced autophagy inhibited cell growth and promoted
apoptosis. However, TGF-β1-induced autophagy inhibited apoptosis in
mesenchymal cells (20). In the
present study, the effect of TGF-β1 stimulation on autophagy
activation in HSC-T6 cells and its regulatory role in HSC-T6
proliferation and apoptosis was investigated.
Materials and methods
Cell culture
The activated rat HSC-T6 cell line with SV40
transfection was provided as a gift by Professor Lie-Ming Xu
(Division of Liver Diseases, Shanghai University of Traditional
Chinese Medicine, Shanghai, China). Cells were cultured in
Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA)
supplemented with 100 U/ml penicillin, 100 U/ml streptomycin, and
10% fetal bovine serum (FBS; HyClone). Cells were incubated at 37°C
with 5% CO2 in a humidified incubator and medium was
replaced every two days.
Plasmid and cell transfection
HSC-T6 cells were seeded in six-well plates
(2×105/well) and transfected with PLVX-AcGFP-N1-LC3B
(Shanghai ExCell Biology, Inc., Shanghai, China) or vector alone.
The cells were used for subsequent experiments 48 h post
transfection.
Cell proliferation assay
Cell viability was measured using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay (Promega, Madison, WI, USA). Cells were plated at a
density of 1.0×105/well in 96-well plates and incubated
with MTS for 4 h at 37°C with 5% CO2 in a humidified
incubator. Absorbance at 570 nm was measured on a SpectraMax
Plus384 microplate reader (Molecular Devices, Sunnyvale, CA,
USA).
Western blot analysis
Protein concentrations were determined using the
bicinchoninic acid (BCA) Protein Assay kit (Pierce, Rockford, IL,
USA). Samples of 30 μg total protein were used for western blots.
Primary antibodies were as follows: Rabbit polyclonal anti-LC3B
(1/1,000; Cell Signaling, Danvers, MA, USA), rabbit polyclonal
anti-GAPDH (1/1,000) and rabbit polyclonal anti-cleaved caspase-3
(1/1,000) (Shanghai ExCell Biology, Inc.). Goat anti-rabbit
immunoglobulin (Ig) G-horseradish peroxidase (1/10,000; Shanghai
ExCell Biology, Inc.) was used as the secondary antibody for
detection. Protein bands were visualized using SuperSignal West
Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham,
MA, USA).
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA from cultured cells was extracted using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA).
RNA was reverse-transcribed using SuperScript II Reverse
Transcriptase (Invitrogen, Life Technologies) at 65°C for 5 min,
42°C for 50 min, and 70°C for 15 min. Gene-specific primers
(MAP1LC3) used in the present study were purchased from Invitrogen
and had the following sequences: Map1LC3, reverse
ACCAAGCCTTCTTCCTCC and forward GCTCTTCTATTTCAAGTCCCTA; GADPH,
reverse ATGGTGGTGAAGACGGTA and forward GGCACAGTCAAGGCTGAGAATG.
Maxima® SYBR Green qPCR Master Mix (Thermo Fisher
Scientific, Waltham, MA, USA), cDNA template, and primer were mixed
at a final volume of 20 μl and subjected to qPCR in an ABI 7500
Real-Time PCR system (Applied Biosystems, Foster City, CA, USA).
Data were analyzed using the ΔΔ threshold (Ct) method, and Ct
values were normalized to GAPDH, which served as an internal
control.
Flow cytometric analysis of
apoptosis
HSC-T6 cells were seeded in 12-well plates
(0.5×105/well) and incubated at 37°C with 5%
CO2. After 24 h, medium was removed and serum-free
Hank’s balanced salt solution (137.93 mM NaCl, 5.33 mM KCl, 4.17 mM
NaHCO3, 0.441 mM KH2PO4, 0.338 mM
Na2HPO4, 5.56 mM D-Glucose) was added. Cells
were then treated with 10 ng/ml TGF-β1 (PeproTech, Rocky Hill, NJ,
USA). Following TGF-β1 treatment, cells were harvested, washed in
cold phosphate-buffered saline, double-stained with fluorescein
isothiocyanate (FITC)-conjugated Annexin V and propidium iodide
(Merck, Darmstadt, Germany) and analyzed by flow cytometry (BD
Biosciences, San Jose, CA, USA).
Immunofluorescence
HSC-T6 cells were seeded in six-well plates
(2×105/well) on cover slips. Cells were formalin-fixed
for 30 min, immersed in 0.2% Triton X-100 and rabbit polyclonal
anti-LC3B for 10 min and incubated with FITC-conjugated AffiniPure
Goat Anti-rabbit IgG (1/50) and DAPI (Shanghai ExCell Biology,
Inc.). Cells were subsequently observed under a fluorescence
microscope (LSM 510 META, Zeiss, Jena, Germany).
Statistical analysis
All experiments were repeated a minimum of three
times. All data are expressed as the mean ± standard error.
Differences between groups were assessed using one-way analysis of
variance or independent sample t-test. All statistical analyses
were performed using SPSS, version 17.0 (SPSS, Inc., Chicago, IL,
USA), and P<0.05 was considered to represent a statistically
significant difference.
Results
TGF-β1 ameliorates HSC-T6 apoptosis
induced by serum deprivation
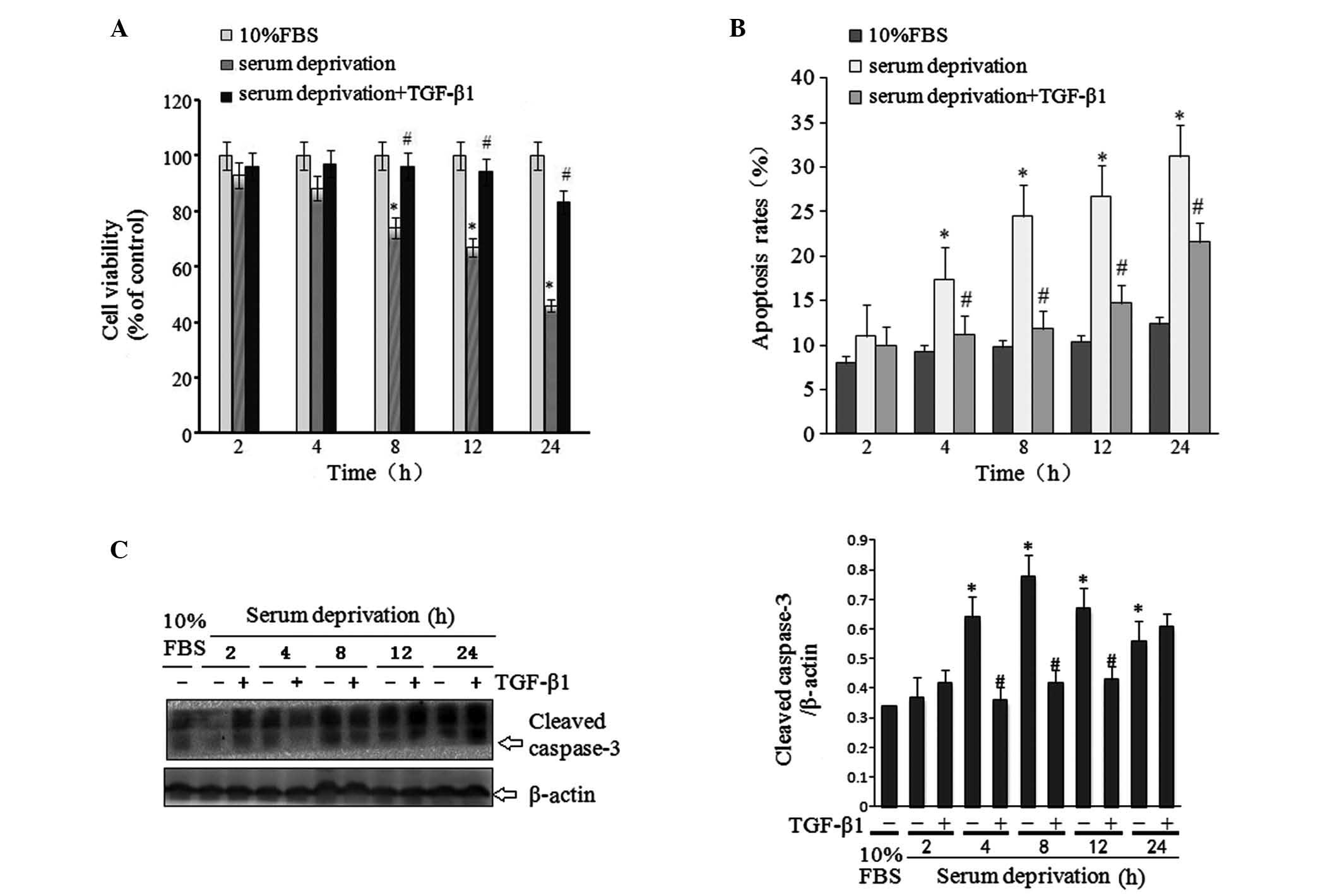
The MTS assay was utilized to determine the
viability of HSC-T6 cells under various treatment conditions. The
viability of cells in medium containing 10% FBS was stable for the
24-h experimental period, while the viability of cells in
serum-free medium was significantly reduced in a time-dependent
manner. Of note, treatment with TGF-β1 (10 ng/ml) significantly
ameliorated this serum deprivation-induced reduction in cell
viability (Fig. 1A). Western blots
were then conducted to determine protein expression levels of
caspase-3, a key regulator of apoptosis. The results indicated that
TGF-β1 treatment significantly reduced serum deprivation-induced
caspase-3 expression (Fig. 1B), in
line with the effect of TGF-β1 on cell viability. In addition, flow
cytometry was used to study the effect of TGF-β1 on cell apoptosis.
The results indicated that serum deprivation induced significant
cell apoptosis, which was ameliorated by TGF-β1 treatment (Fig. 1C).
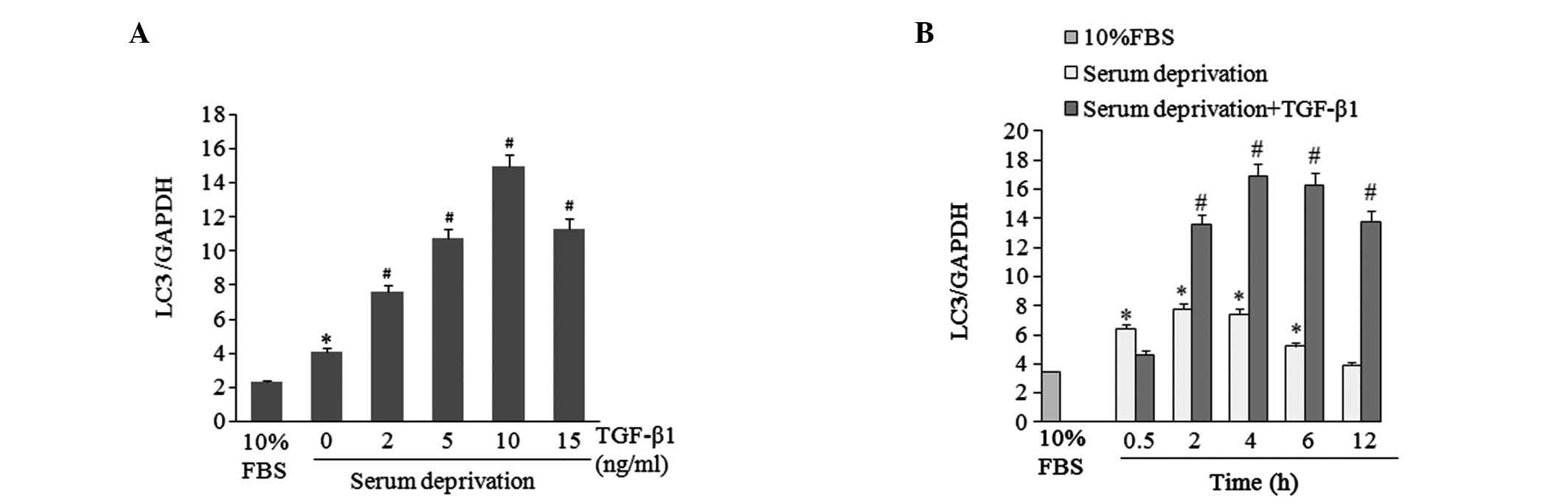
TGF-β1 induces mRNA expression of the
autophagic marker Map1LC3
The levels of LC3II, a proteolytic product of the
Map1LC3 protein, is closely associated with the number of
autophagic lysosomes present, and is widely used as a molecular
marker for autophagy (21). In the
present study, mRNA expression levels of Map1LC3 in HSC-T6 cells
were analyzed under various treatment conditions. HSC-T6 cells were
incubated in 10% FBS or serum-free medium with different
concentrations of TGF-β1 (0, 2, 5, 10, 15 ng/ml) for 4 h. qPCR
results indicated that serum deprivation induced Map1LC3 mRNA
expression in HSC-T6 cells, and TGF-β1 treatment further elevated
the levels in a dose-dependent manner (Fig. 2A). LC3 mRNA expression levels were
then determined in HSC-T6 cells treated with 10 ng/ml TGF-β1 for
various time periods. From 2 to 12 h, TGF-β1 stimulated higher
levels of LC3 mRNA expression than those stimulated by serum
deprivation alone (Fig. 2B).
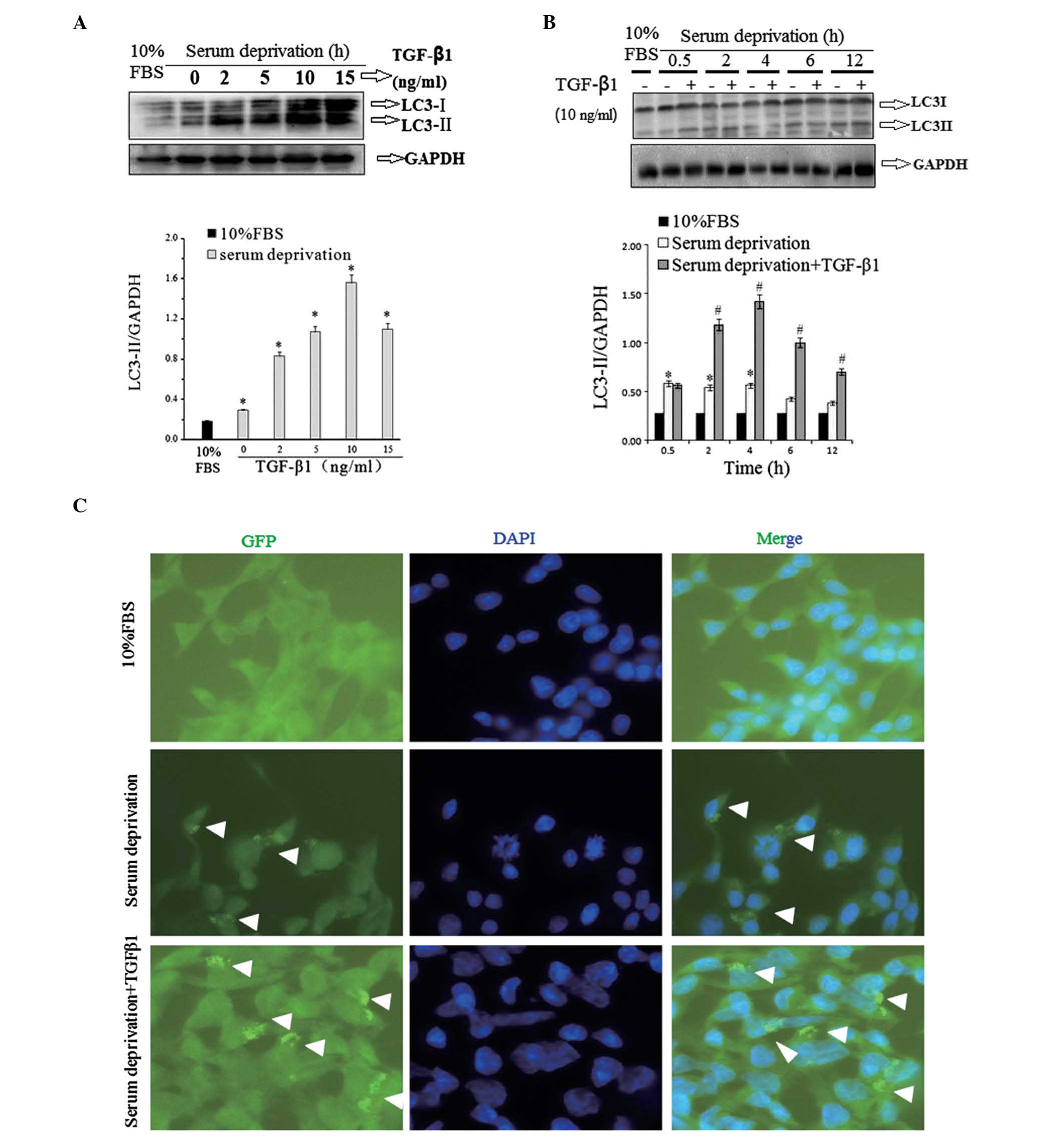
TGF-β1 increases LC3II protein
levels
Levels of LC3II protein were also measured by
western blot analysis to enable further consideration of the effect
of TGF-β1 on autophagy in HSC-T6 cells. The cells subjected to
western blotting were treated under similar conditions to those
used in the Map1LC3 mRNA expression analysis. 4-h serum deprivation
significantly increased LC3II protein levels in HSC-T6 cells
compared with levels in FBS-cultured cells, and TGF-β1 treatment
further elevated the LC3II protein levels in a dose-dependent
manner (Fig. 3A). In the
time-course study, it was observed that from 2 to 12 h, TGF-β1 (10
ng/ml) treatment increased LC3II protein levels to a greater extent
than serum deprivation alone (Fig.
3B). The results indicated that the LC3II protein levels were
associated with Map1LC3 mRNA expression.
Detection of GFP-LC3 fusion protein
aggregation in HSC-T6 cells by immunofluorescence staining
Immunofluorescence staining exhibited a clear
increase in GFP-LC3 autophagic punctate in the TGF-β1-treated group
compared with the group with serum deprivation without TGF-β1
(Fig. 3C). Autophagic punctate was
calculated using image-pro-plus 5.0 software analysis (Media
Cybernetics, Bethesda, MD, USA). GFP-LC3 aggregation in cells with
serum deprivation (11.70±1.31, P>0.05 vs. 10% FBS group) and
cells with serum deprivation combined with TGF-β1 treatment
(19.33±2.76, P<0.05 vs. 10% FBS group) was greater than the
level of aggregation in the control group (7.32±0.85).
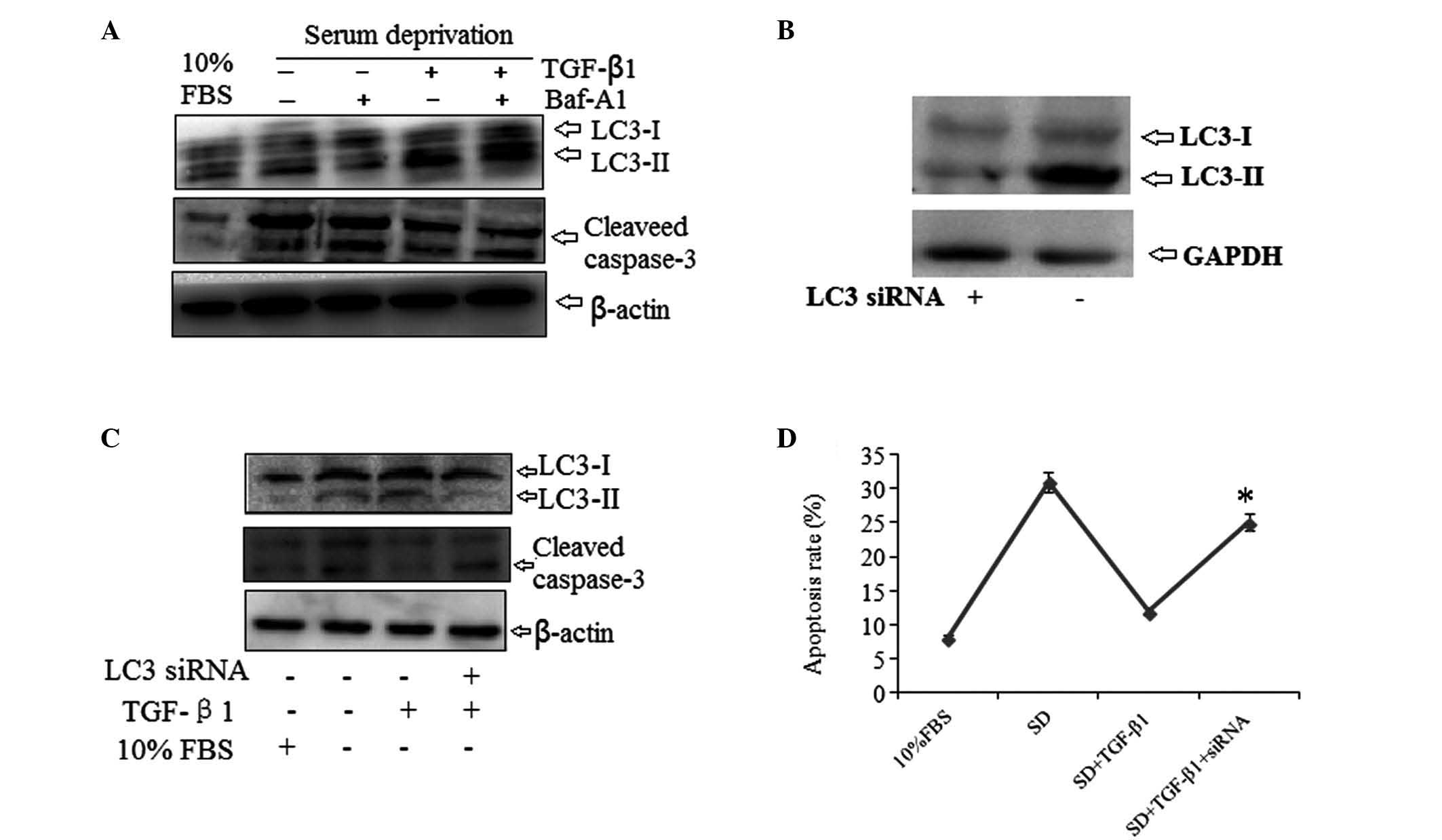
TGF-β1 inhibits HSC-T6 cell apoptosis
through induction of autophagy
To evaluate the role of autophagy in the
anti-apoptotic effect of TGF-β1 in serum-deprived HSC-T6 cells, the
effect of the specific autophagy inhibitor bafilomycin A1 and the
effect of LC3 gene knockdown were assessed. Bafilomycin A1
treatment significantly reduced the inhibitory effect of TGF-β1 on
cleaved caspase-3 expression (Fig.
4A). In addition, HSC-T6 cells were transfected with a specific
LC3 small interfering (si)RNA for 24–48 h in order to knockdown the
LC3 gene (Fig. 4B). When
serum-deprived cells were treated with TGF-β1, cells transfected
with LC3 siRNA displayed visibly increased cleaved caspase-3 levels
compared with cells transfected with the vector without TGF-β1
treatment (Fig. 4C). The effect of
LC3 knockdown on cell apoptosis was also evaluated using flow
cytometry. The anti-apoptotic effect of TGF-β1 in serum-deprived
HSC-T6 cells was significantly reduced in cells transfected with
LC3 siRNA (Fig. 4D). These results
implied that TGF-β1 acted through autophagy activation to
ameliorate serum deprivation-induced apoptosis in HSC-T6 cells.
Discussion
In the present study, serum deprivation was noted to
significantly increase levels of apoptosis in HSC-T6 cells,
accompanied by autophagy activation (measured by LC3II levels and
protein aggregation). It was also demonstrated that TGF-β1
ameliorated serum deprivation-induced apoptosis via activation of
autophagy.
TGF-β1, which is one of the strongest inducers of
extracellular matrix (ECM) production, serves a central purpose in
the process of fibrogenesis in fibrotic diseases (22,23).
TGF-β1 exhibits pro-apoptotic or anti-apoptotic effects depending
on the cell type, in addition to other factors (24,25).
The pro-apoptotic effects of TGF-β1 are well documented in various
cell types, including immune cells (26) and epithelial cells (27). However, TGF-β1 has also been
demonstrated to inhibit apoptosis in certain cell types, including
murine macrophages (28),
mesenchymal cells (29),
fibroblasts and myofibroblasts (30). In the present study, TGF-β1 was
demonstrated to protect HSC-T6 cells against the apoptosis and
total cell death caused by serum deprivation.
Multiple previous studies have demonstrated the
central role of TGF-β1 in the initiation and development of liver
fibrosis: One study indicated that TGF-β1 simultaneously induces
autophagy and apoptosis in mammary epithelial cells (19). However, in mesangial cells,
TGF-β1-induced autophagy has been demonstrated to produce
anti-apoptotic effects (20). Upon
activation of autophagy, the cytoplasmic protein form of LC3 (LC3I)
is processed and recruited to autophagosomes, where LC3II is
generated by site-specific proteolysis and lipidation near the
C-terminus. Thus, the formation of cellular autophagosome punctate
containing LC3II is a marker of autophagic activation. The results
of the current study indicated that TGF-β1 treatment increased LC3
mRNA expression in serum-deprived HSC-T6 cells in a concentration-
and time-dependent manner. In addition, TGF-β1 increased LC3II
protein levels in a similar manner.
Autophagy can be monitored by GFP-LC3 labeling, in
which formation of autophagosome punctate containing GFP-LC3 is
detected by indirect immunofluorescence staining or direct
fluorescence microscopy (21). In
the present study, this method was used to observe that in HSC-T6
cells transiently transfected with GFP-LC3, LC3 punctate aggregated
around the nuclear membrane. TGF-β1 treatment led to the relocation
of LC3 from the cytoplasm to the caryotheca, indicating that TGF-β1
induces autophagy in HSC-T6 cells.
Autophagy is closely associated with apoptosis and
shares similar signaling pathways and stimulating factors,
differing only in the threshold values (3). In general, the relationship between
autophagy and apoptosis is mutual inhibition. Autophagy activation
may inhibit cell apoptosis, but excessive autophagy may cause
autophagic cell death (31).
TGF-β1 is an important regulator of autophagy and apoptosis
(32,33), and in the present study, it was
demonstrated that in serum-deprived cells, incubation with TGF-β1
significantly suppressed cleaved caspase-3 expression levels.
Blocking autophagy activation with bafilomycin A1 or LC3 siRNA
significantly reduced the inhibitory effect of TGF-β1 on cleaved
caspase-3 expression. TGF-β1 regulates autophagy through both SMAD
and non-SMAD signaling pathways (33); however, the precise mechanism by
which TGF-β1 regulates autophagy in HSC-T6 cells requires further
research.
In conclusion, the present study demonstrated that
TGF-β1 ameliorated apoptosis in serum-deprived HSC-T6 cells via a
mechanism mediated by autophagy activation. Further studies are
required to elucidate the mechanism by which TGF-β1 activates
autophagy in HSC-T6 cells in vitro and in vivo.
Acknowledgements
The present study was funded by the Wang Bao-en
Foundation of Hepatic Fibrosis and the Chinese Foundation for
Hepatitis Prevention and Control (no. CFHPC20131014).
References
|
1
|
Ni HM, Williams JA, Yang H, Shi YH, Fan J
and Ding WX: Targeting autophagy for the treatment of liver
diseases. Pharmacol Res. 66:463–474. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klionsky DJ: The autophagy connection. Dev
Cell. 19:11–12. 2010. View Article : Google Scholar
|
|
3
|
González-Polo RA, Boya P, Pauleau AL, et
al: The apoptosis/autophagy paradox: autophagic vacuolization
before apoptotic death. J Cell Sci. 118:3091–3102. 2005.PubMed/NCBI
|
|
4
|
Komatsu M: Liver autophagy: physiology and
pathology. J Bio Chem. 152:5–15. 2012.PubMed/NCBI
|
|
5
|
Sasaki M, Miyakoshi M, Sato Y and Nakanuma
Y: Autophagy may precede cellular senescence of bile ductular cells
in ductular reaction in primary biliary cirrhosis. Dig Dis Sci.
57:660–666. 2012. View Article : Google Scholar
|
|
6
|
Bhogal RH and Afford SC: Autophagy and the
Liver. Autophagy - A Double-Edged Sword - Cell Survival or Death?
Bailly Yannick: InTech; Rijeka, Croatia: pp. 165–185. 2013
|
|
7
|
Rautou PE, Mansouri A, Lebrec D, Durand F,
Valla D and Moreau R: Autophagy in liver diseases. J Hepatol.
53:1123–1134. 2010. View Article : Google Scholar
|
|
8
|
Hilscher M, Hernandez-Gea V and Friedman
SL: Autophagy and mesenchymal cell fibrogenesis. Biochim Biophys
Acta. 1831.972–978. 2012.PubMed/NCBI
|
|
9
|
Hidvegi T, Ewing M, Hale P, et al: An
autophagy-enhancing drug promotes degradation of mutant
alpha1-antitrypsin Z and reduces hepatic fibrosis. Science.
329:229–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ and
Choi ME: Autophagy promotes intracellular degradation of type I
collagen induced by transforming growth factor (TGF)-β1. J Biol
Chem. 287:11677–11688. 2012.PubMed/NCBI
|
|
11
|
Hernández-Gea V, Ghiassi-Nejad Z,
Rozenfeld R, et al: Autophagy releases lipid that promotes
fibrogenesis by activated hepatic stellate cells in mice and in
human tissues. Gastroenterology. 142:938–946. 2012.PubMed/NCBI
|
|
12
|
Thoen LF, Guimarães EL, Dollé L, et al: A
role for autophagy during hepatic stellate cell activation. J
Hepatol. 55:1353–1360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yin XM, Ding WX and Gao W: Autophagy in
the liver. Hepatology. 47:1773–1785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deretic V and Levine B: Autophagy,
immunity and microbial adaptations. Cell Host Microbe. 5:527–549.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Friedman SL: Hepatic fibrosis - overview.
Toxicology. 254:120–129. 2008. View Article : Google Scholar
|
|
17
|
Brenner DA: Molecular pathogenesis of
liver fibrosis. Trans Am Clin Climatol Assoc. 120:361–368.
2009.PubMed/NCBI
|
|
18
|
Iimuro Y and Brenner DA: Matrix
metalloproteinase gene delivery for liver fibrosis. Pharm Res.
25:249–258. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gajewska M, Gajkowska B and Motyl T:
Apoptosis and autophagy induced by TGF-β1 in bovine mammary
epithelial BME-UV1 cells. J Physiol Pharmacol. 56(Suppl 3):
143–157. 2005.
|
|
20
|
Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee
SJ and Choi ME: TGF-{beta}1 protects against mesangial cell
apoptosis via induction of autophagy. J Biol Chem. 285:37909–37919.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar
|
|
22
|
Leask A and Abraham DJ: TGF beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakerakanti S and Trojanowska M: The role
of TGF-β receptors in fibrosis. Open Rheumatol J. 6:156–162.
2012.
|
|
24
|
Roberts AB: Molecular and cell biology of
TGF-beta. Miner Electrolyte Metab. 24:111–119. 1998. View Article : Google Scholar
|
|
25
|
Roberts AB and Sporn MB: Physiological
actions and clinical applications of transforming growth
factor-beta (TGF-beta). Growth Factors. 8:1–9. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brown TL, Patil S, Cianci CD, Morrow JS
and Howe PH: Transforming growth factor-beta induces caspase
3-independent cleavage of alphaII-spectrin (alpha-fodrin)
coincident with apoptosis. J Biol Chem. 274:23256–23262. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dai C, Yang J and Liu Y: Transforming
growth factor-beta1 potentiates renal tubular epithelial cell death
by a mechanism independent of Smad signaling. J Biol Chem.
278:12537–12545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chin BY, Petrache I, Choi AM and Choi ME:
Transforming growth factor beta1 rescues serum deprivation-induced
apoptosis via the mitogen-activated protein kinase (MAPK) pathway
in macrophages. J Biol Chem. 274:11362–11368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hara T, Kamura T and Nakayama K, Oshikawa
K, Hatakeyama S and Nakayama K: Degradation of p27(Kip1) at the
G(0)–G(1) transition mediated by a Skp2-independent ubiquitination
pathway. J Biol Chem. 276:48937–48943. 2001.PubMed/NCBI
|
|
30
|
Chen HH, Zhao S and Song JG: TGF-beta1
suppresses apoptosis via differential regulation of MAP kinases and
ceramide production. Cell Death Differ. 10:516–527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang RC and Levine B: Autophagy in
cellular growth control. FEBS Lett. 584:1417–1426. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Suzuki HI, Kiyono K and Miyazono K:
Regulation of autophagy by transforming growth factor-β (TGF-β)
signaling. Autophagy. 6:645–647. 2010.
|