Introduction
Human bladder cancer is the fourth most common
malignancy in males, and the 10th most common malignancy in females
(1). There are currently numerous
therapeutic techniques available for the treatment of bladder
cancer, including intravesical chemotherapy, surgery, radiation
therapy and systemic chemotherapy, which are selected depending on
the extent of the disease (2,3).
Despite these various treatment options, patients with advanced
bladder cancer have a five-year survival rate of 20–40% (4). For patients with advanced bladder
cancer, cisplatin-based combination chemotherapy, for example
methotrexate, vinblastine, doxorubicin and cisplatin or gemcitabine
and cisplatin, is the current choice of treatment. However, one of
the most significant limitations in the efficacy of cisplatin-based
combination chemotherapy is that numerous tumors either demonstrate
an inherent resistance to the chemotherapy or acquire resistance
following an initial response (5).
The molecular mechanisms underlying this chemoresistance have yet
to be elucidated, although cisplatin resistance has been associated
with a reduction in the apoptotic response of certain types of
cancer cells (6). There is
therefore a requirement to develop novel drugs that are able to
reverse the resistance to chemotherapy and enhance sensitivity to
platinum-based chemotherapy drugs.
microRNAs (miRNAs) are endogenous, non-coding RNA
molecules of 19–25 nucleotides in length. The pairing of miRNAs
with target mRNAs carrying a partially complementary sequence in
the 3′ untranslated region (UTR) results in the translational
repression and/or degradation of the mRNA and thus the silencing of
associated genes (7–9). Alterations in the expression patterns
of miRNAs that regulate genes involved in cell proliferation,
differentiation or apoptosis have been found in various human
tumors (10–12). Therefore, miRNA expression profiles
may be utilized to determine the expected sensitivity of tumors to
chemotherapy prior to treatment, and changes in the expression of
miRNAs during treatment could act as markers for the response of
tumors to the chemotherapy. In a comparison of
doxorubicin-resistant and -sensitive MCF-7 breast cancer cell
lines, Kovalchuk et al (13) identified 137 deregulated miRNAs,
while 103 deregulated miRNAs were identified in a comparison of
cisplatin-resistant and -sensitive MCF-7 cells (13). In addition, Nordentoft et al
(14) found that the expression of
15 miRNAs correlated with chemotherapy response and that the
expression of five miRNAs correlated with survival time in patients
with bladder cancer. It was further found that a reduction in
miR-138 expression resulted in an increase in cisplatin
sensitivity, while an upregulation of miR-27a and miR-642
expression also increased cisplatin sensitivity (14).
Increasing evidence has indicated that there is an
association between chronic inflammation and tumorigenesis.
Cyclooxygenase-2 (COX-2) is a key regulator of
inflammation-producing prostaglandins and thus could be involved in
inflammation-associated cancer development (15). Increased COX-2 expression has been
found in a number of apoptosis-resistant cancer cells (16); therefore, COX-2 may be a potential
target in the reversal of this apoptosis resistance. The expression
of COX-2 has been shown to be modulated by several mechanisms,
including the action of miRNA (17). He et al (18) reported that miR-101 downregulation
in gastric cancer correlated with the overexpression of COX-2 and
tumor growth. miR-101 may therefore influence cisplatin sensitivity
in cancer cells through the inhibition of COX-2 expression.
To the best of our knowledge, the association
between miR-101 and cisplatin resistance in bladder cancer cells
has not been reported. In the present study, we explored the
association between cisplatin sensitivity and miR-101 in bladder
cancer.
Materials and methods
Cell culture
T24 human urinary bladder transitional cell
carcinoma cells were cultured in RPMI-1640 medium supplemented with
10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich, St.
Louis, MO, USA) and 1% penicillin/streptomycin sulfate (Beyotime
Institute of Biotechnology, Haimen, China) under an atmosphere of
95% air and 5% CO2 at 37°C. T24 human urinary bladder
transitional cell carcinoma cells were purchased from the Shanghai
Institute of Biochemistry and Cell Biology (Shanghai, China). The
cisplatin-resistant T24 cell line (T24/CDDP) was established as
described previously (6).
Transfection of miRNA mimics and
inhibitors
T24/CDDP and T24 cells were plated in six-well
plates (5×105 cells/well). A total of 100 nM miR-101
mimic or 100 nM miRNA mimic control was transfected in T24/CDDP
cells, and 100 nM miR-101 inhibitor or 100 nM miRNA inhibitor
control was transfected in T24 cells, using Lipofectamine™ 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The sequences were as follows: miR-101
mimic, 5′-UACAGUACUGUGAUAACUGAA-3′; miR-101 inhibitor,
5′-CUUCAGUUAUCACAGUACUGUA-3′; and negative control,
5′-GUGGAUAUUGUUGCCAUCA-3′.
Quantitative polymerase chain reaction
(qPCR) for miR-101
Total RNA from cells was extracted using TRIzol™
reagent (Invitrogen Life Technologies). Levels of the mature forms
of miR-101 in the cells were determined using a TaqMan®
miRNA assay kit (Applied Biosystems, Foster City, CA, USA), and
data were normalized to U6 expression (Applied Biosystems). The
qPCR procedure was performed according to the manufacturer’s
instructions. The fold change for miR-101 was calculated using the
2−ΔΔCt method (19).
Cell viability assay
Cells were seeded into 96-well plates in RPMI-1640
medium containing 10% FBS. Following 24 h in culture, the cells
were treated with serial dilutions of cisplatin. Approximately 72 h
after cisplatin treatment, MTT was added to a final concentration
of 0.5 mg/ml and the cells were then incubated for 4 h at 37°C. The
optical density was read at 490 nm with a microplate
spectrophotometer (iMark; Bio-Rad, Hercules, CA, USA). Each
experiment was performed in triplicate and repeated three
times.
Western blot analysis
T24 or T24/CDDP cells were plated in six-well plates
(5×105 cells/well) and, 72 h after transfection with
miR-101 mimic or inhibitors, the cells were harvested and
homogenized with lysis buffer. Total protein was separated by
denaturing 10% SDS-PAGE. Western blot analysis was performed as
described previously (20). The
primary antibodies against COX-2 and GAPDH were purchased from Cell
Signaling Technology, Inc., (Danvers, MA, USA) and Santa Cruz
Biotechnology, Inc., (Santa Cruz, CA, USA), respectively. Protein
levels were normalized to GAPDH and the subsequent fold changes
were determined.
Apoptosis detection
Apoptosis was assessed by Annexin V/propidium iodide
(PI) staining 48 h after treatment using flow cytometry
(FACSCalibur™; BD Biosciences, Heidelberg, Germany), according to
the manufacturer’s instructions. The percentage of early apoptotic
cells was determined by the quantitative analysis of Annexin
V-fluorescein isothiocyanate-positive/PI-negative plots using the
CellQuest software (BD Biosciences).
Luciferase assay
The luciferase reporter plasmids were constructed as
described previously (21). A
fragment of the 3′UTR COX-2 mRNA, which carried a putative miR-101
complementary site (NM_000963; 3′-UTR, 1,735–1,741), and a mutant
variant were cloned into a pGL3 vector (pGL3-empty; Promega
Corporation, Madison, WI, USA) with a downstream firefly luciferase
gene (XbaI site) to obtain luciferase reporter constructs
(pGL3-COX2-wildtype and pGL3-COX2-mutant, respectively). The target
gene was predicted by TargetScan Human 6.2 (http://www.targetscan.org) as described previously
(22). Cells were seeded in
24-well plates and co-transfected with miR-101 mimics and either
pGL3-COX2-wildtype or pGL3-COX2-mutant.
siRNA transfection
siRNA against COX-2 (5′-AACTGCTCAACACCGGAATTT-3′)
and negative control was purchased from Cell Signaling Technology,
Inc., and the transfection was performed according to the
manufacturer’s instructions. The transfection efficiency was
evaluated using fluorescence microscopy by calculating the
percentage of fluorescein-labeled cells. Cells were lysed in lysis
buffer 48 h after transfection for western blot analyses.
Nude mouse xenografts
Nude mouse xenografts were performed as previously
described (23). Nude mice were
purchased from Vital River Laboratories (Beijing, China) and
maintained at the Experimental Animal Center of Jiangsu University
(Jiangsu, China). Each mouse was subcutaneously inoculated with
2×106 T24/CDDP cells, which had been transfected with
miR-101 or miR-control, in 0.2 ml medium in the forelimb. Tumor
sizes were measured every three days in two dimensions using a
caliper and the volume (V) (mm3) was calculated using
the following formula: V = 0.5 × larger diameter × (smaller
diameter)2. The mice were sacrificed and the tumors were
excised and weighed 21 days after the initial inoculation. All
procedures involving animals were approved by the Animal Care and
Use Committee of Jiangsu University (Zhenjiang, China).
Statistical analysis
Quantitative data are presented as the mean ±
standard deviation. The Student’s t-test was used to determine
statistical significance. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-101 is downregulated in T24/CDDP
cells as compared with T24 cells
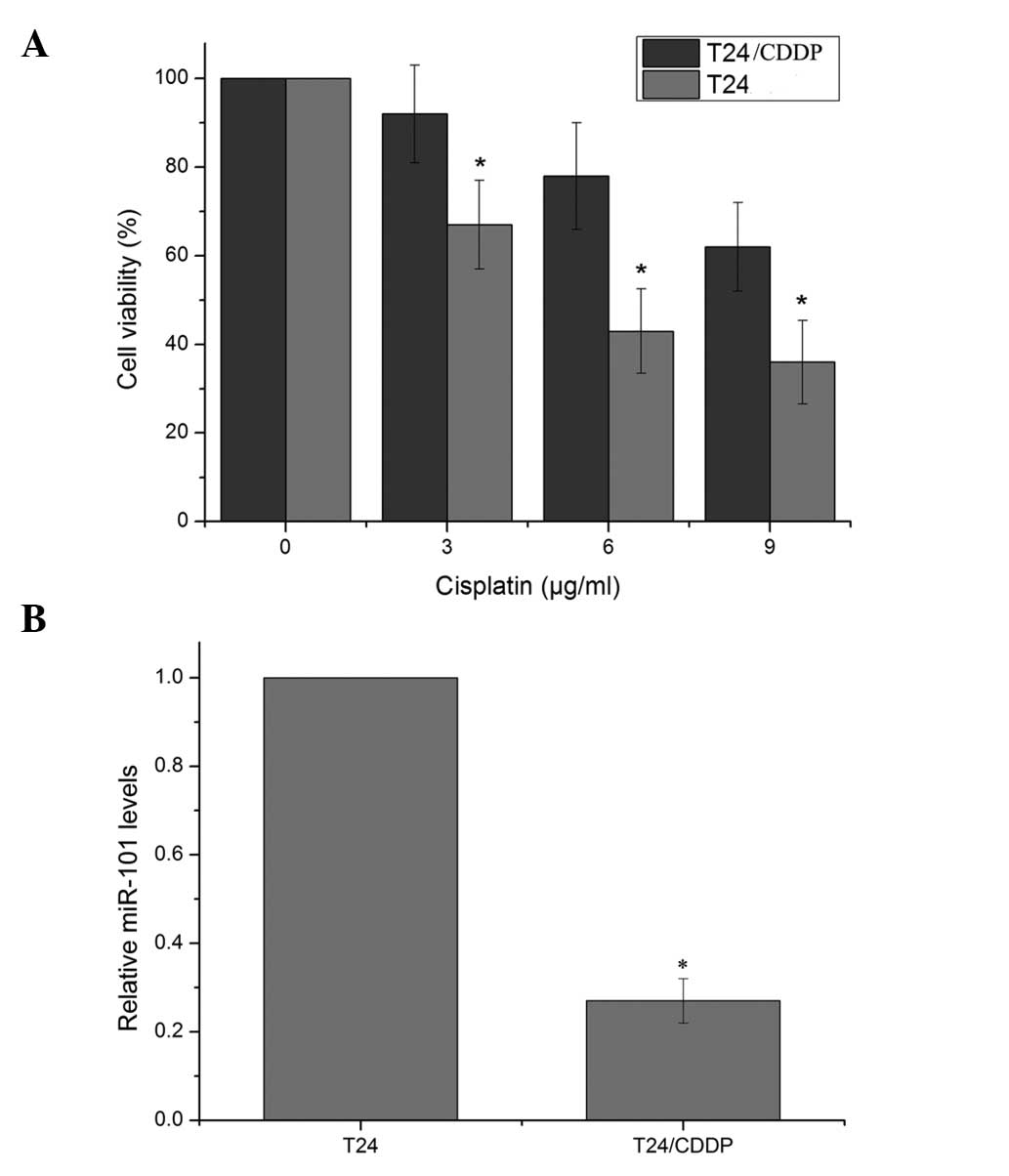
The doses of cisplatin required for a 50% inhibition
of T24 and T24/CDDP cells were 5.2 and 14.7 μg/ml, respectively
(Fig. 1). The resistance to
cisplatin in the T24/CDDP cells was 2.83-fold greater than that in
the parent T24 cells. qPCR for miR-101 expression further verified
that miR-101 was significantly downregulated in T24/CDDP cells,
with an expression level of 0.27±0.05 relative to that in the
parental cells.
miR-101 modulates the sensitivity of
cisplatin in T24 cells
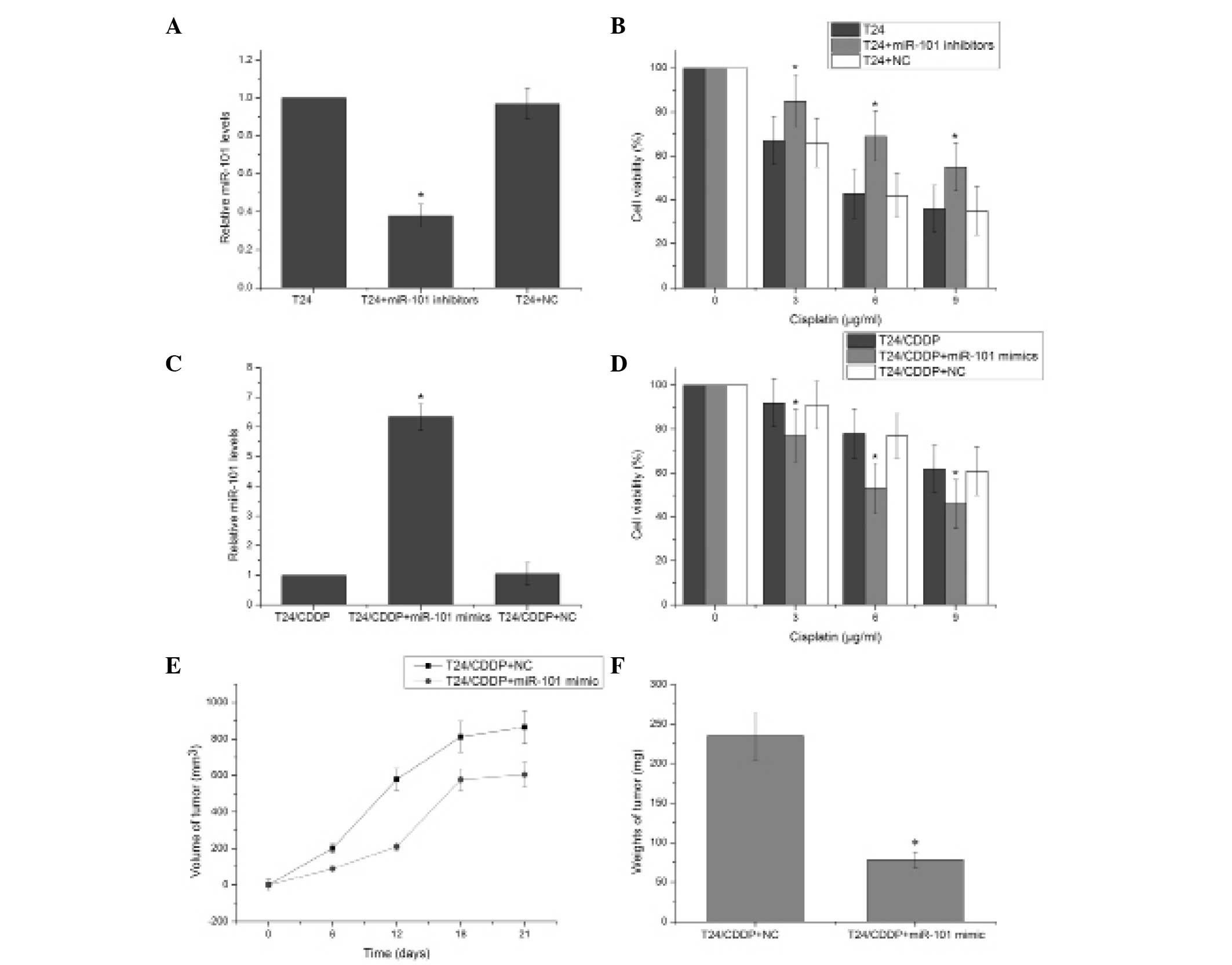
To directly assess the association between miR-101
and cisplatin resistance in the T24 cells, the expression of
miR-101 was further downregulated in the T24 cell line. The miR-101
inhibitor effectively reduced the expression of miR-101 (P<0.05)
(Fig 2A). T24 cells transfected
with the miR-101 inhibitor exhibited significantly enhanced
resistance to cisplatin compared with the negative control
transfected cells (P<0.05) (Fig.
2B). This suggests that decreasing miR-101 expression
contributes to cisplatin resistance in T24 cells. The miR-101 mimic
significantly induced the expression of miR-101 in T24/CDDP cells
(P<0.05) (Fig. 2C), and MTT
assay revealed that T24/CDDP cells transfected with the miR-101
mimics exhibited significantly decreased resistance to cisplatin
compared with the negative control-transfected cells (Fig. 2D). Tumor growth curves were
generated by measuring the size of tumors in nude mice over a
period of 21 days. Following T24/CDDP cell injection, the volume of
the tumors in the miR-101 mimic and negative control groups showed
differential progression (Fig.
2E). Additionally, the tumors in the T24/CDDP plus miR-101
mimic group were significantly lighter than those in the negative
control group (P<0.05) (Fig.
2F). These results suggest that miR-101 regulates cisplatin
resistance in the T24 human bladder cancer cell line.
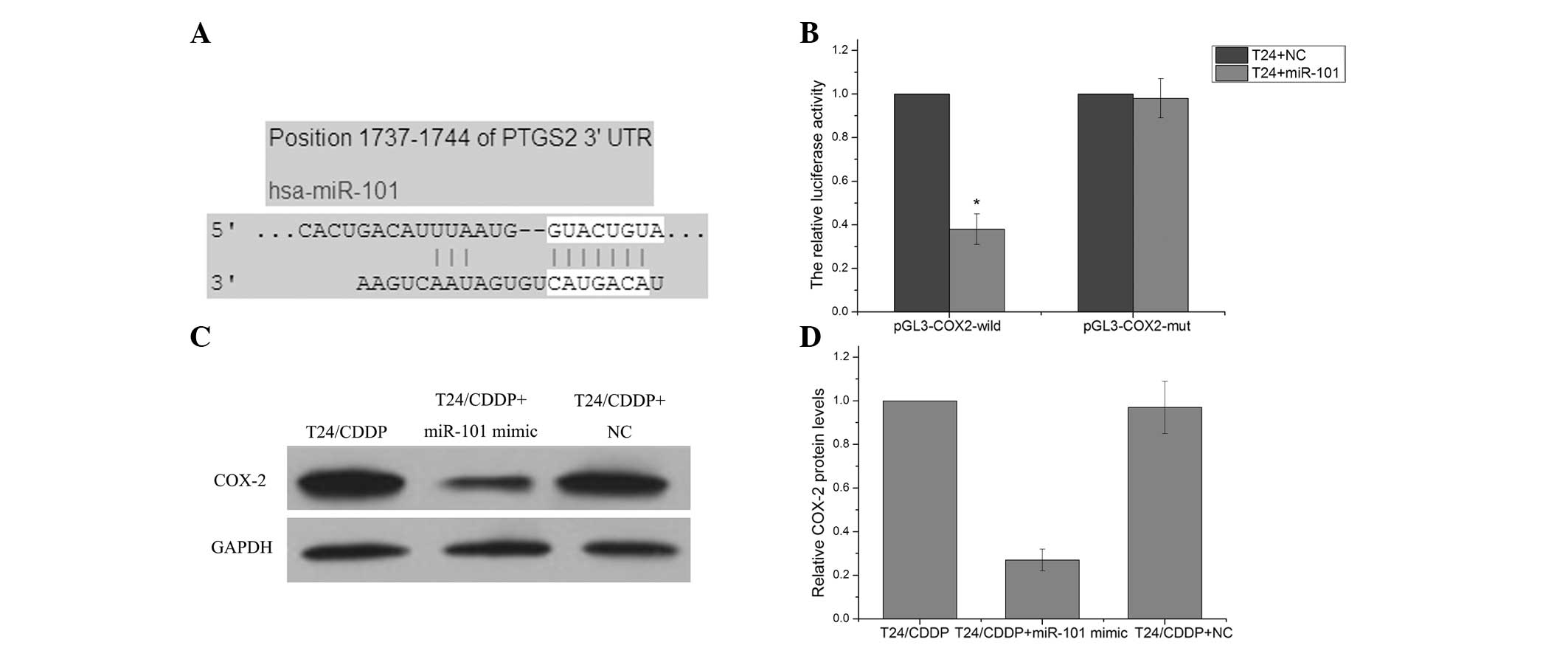
COX-2 is a direct target gene of
miR-101
TargetScan Human 6.2 (http://www.targetscan.org) predicted that COX-2 was a
target gene of the miR-101 (Fig.
3A). A luciferase reporter vector with the putative COX-2 3′
UTR target site for the miR-101 downstream of the luciferase gene
(pGL3-COX2-wildtype) was therefore constructed in order to test
this in vitro. The luciferase reporter vector, together with
the miR-101 mimic or the negative control, was transfected into
T24/CDDP cells. A significant decrease in relative luciferase
activity was noted when pGL3-COX2-wildtype was co-transfected with
the miR-101 mimics into T24/CDDP cells. However, the
co-transfection of the pGL3-COX2-mutant with the miR-101 mimic did
not affect reporter activity (P>0.05) (Fig. 3B). Western blot assay revealed that
T24/CDDP cells transfected with miR-101 mimic exhibited
significantly decreased protein levels of COX-2 as compared with
the cells transfected with the negative control (Fig. 3C and D). These results suggest that
COX-2 is a target gene of miR-101.
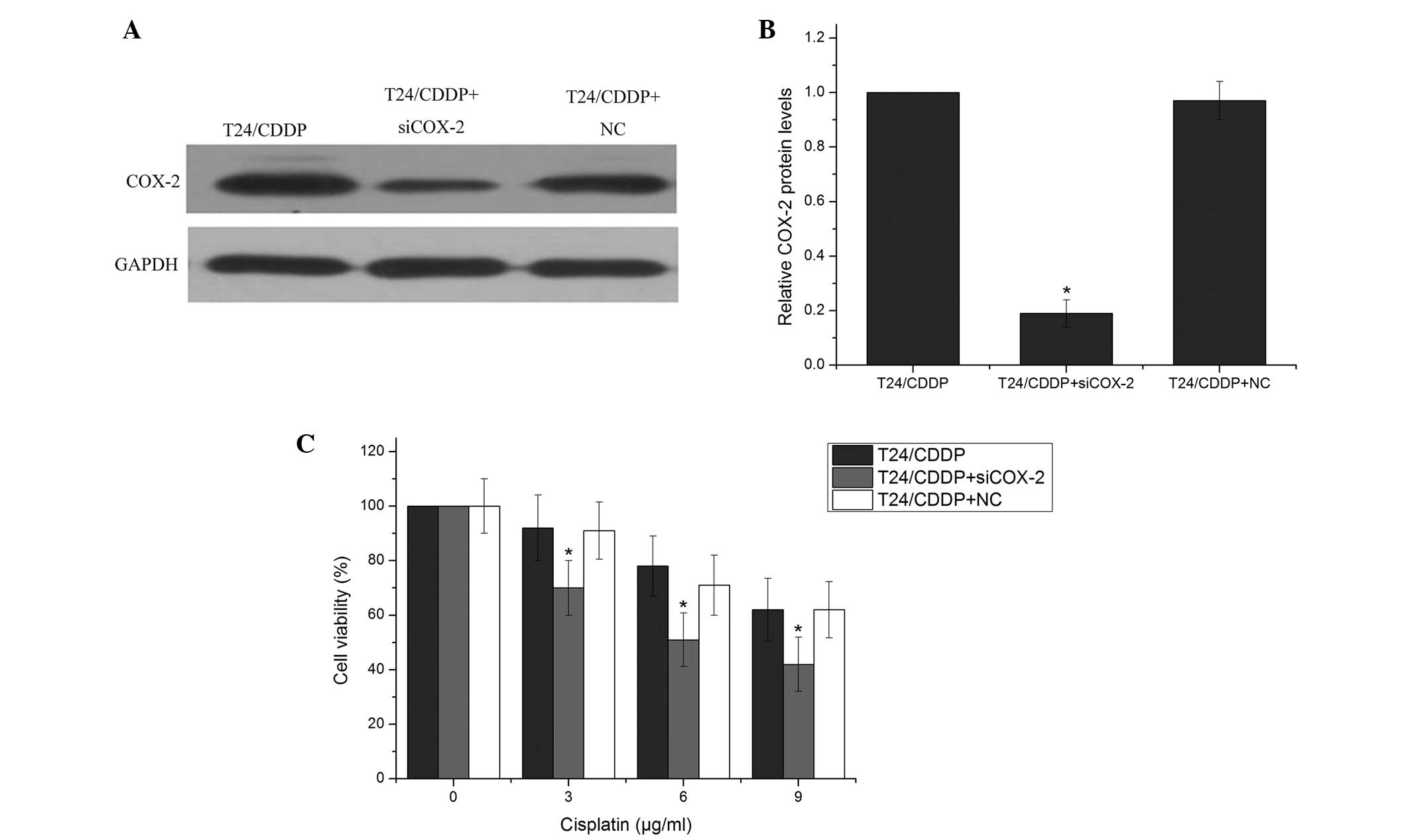
COX-2 is involved in cisplatin resistance
in T24/CDDP cells
Previous studies have shown that COX-2 is involved
in drug resistance in various types of cancer (24,25);
however, its role in cisplatin sensitivity in the T24/CDDP cell
line remains unclear. To explore the association between COX-2 and
cisplatin-induced cytotoxicity, COX-2 siRNA or a negative control
was transfected into T24/CDDP cells, followed by treatment with
various doses of cisplatin. COX-2 siRNA effectively reduced the
COX-2 protein level (Fig. 4A and
B). Furthermore, T24/CDDP cells that were treated with COX-2
siRNA had decreased survival compared with the control group
(Fig. 4C). Of note, the reduced
survival rates exhibited by the COX-2 siRNA-treated T24/CDDP cells
were similar to those exhibited by the cells with miR-101
overexpression. This suggests that miR-101 reversed cisplatin
resistance via the regulation of COX-2 in the T24/CDDP cells.
miR-101 regulates cisplatin sensitivity
in T24 cells by targeting COX-2
It was hypothesized in this study that miR-101
modulates the cisplatin resistance of bladder cancer cells by
repressing COX-2 protein expression. To ascertain this, miR-101
inhibitor or negative control was transfected into T24 cells for
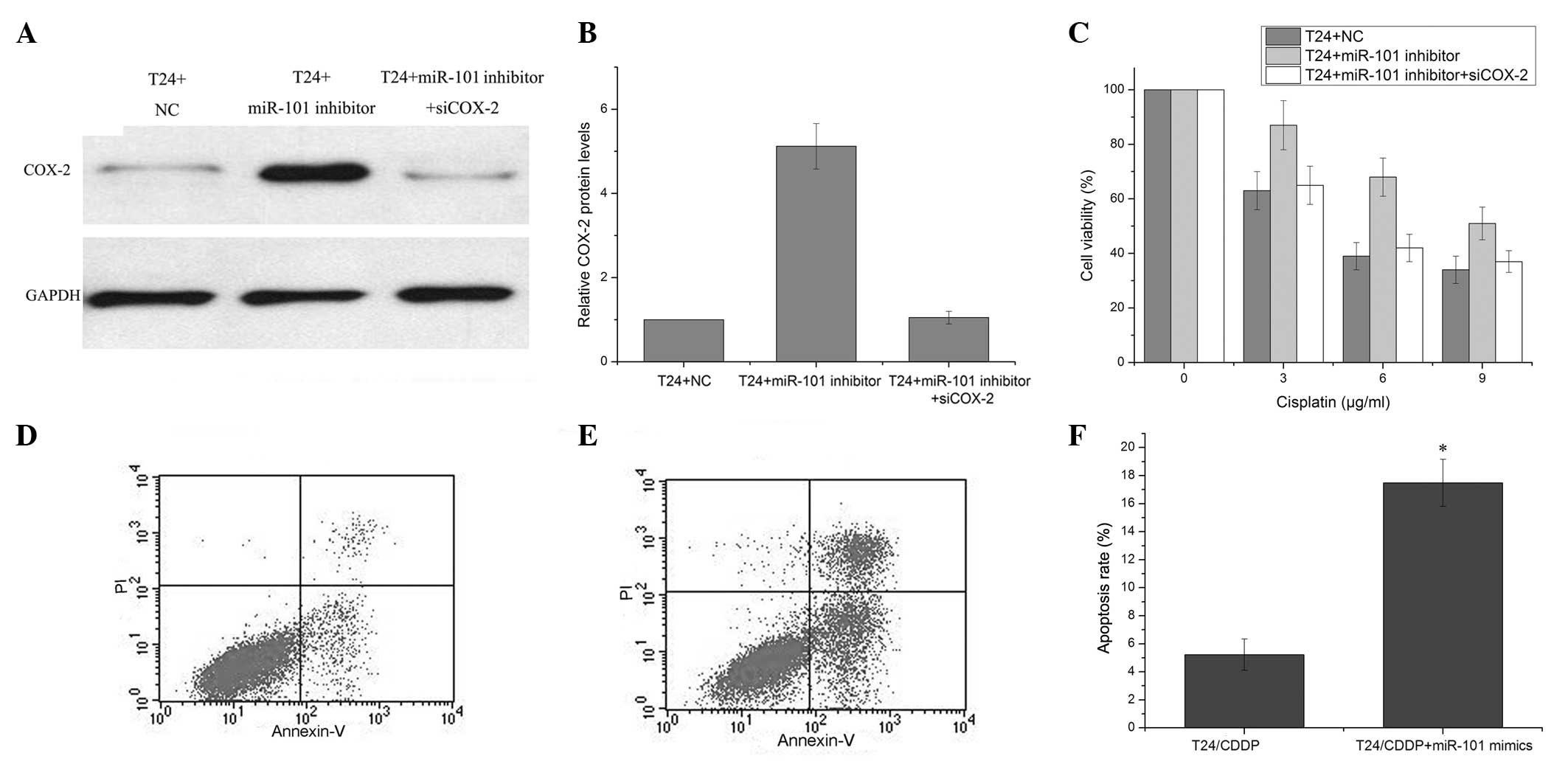
the detection of changes in COX-2 expression levels. Western blot
analysis demonstrated significantly increased COX-2 protein levels
in the miR-101 mimic-transfected T24 cells as compared with the
negative control-transfected T24 cells 72 h after transfection,.
This was observed to be in part alleviated by transfection with
COX-2 siRNA (Fig. 5A and B).
Enhanced cell viability was observed in the T24 cells transfected
with miR-101 inhibitor as compared with negative
control-transfected cells, which could be partly alleviated by the
COX-2 siRNA (Fig. 5C). miR-101 may
therefore play a role in the development of cisplatin resistance,
in part through the modulation of apoptosis. To confirm this
hypothesis, cisplatin-induced apoptosis was evaluated in T24/CDDP
cells following transfection with the miR-101 mimic or the negative
control. A marked increase in apoptosis, as assessed by flow
cytometry, was observed in the T24/CDDP cells transfected with the
miR-101 mimic following cisplatin treatment, as compared with the
negative control-transfected cells (Fig. 5D–F). These results showed that
miR-101 modulates the cisplatin resistance of T24/CDDP cells, and
this may be through the inhibition of COX-2 protein expression.
Discussion
Resistance to chemotherapeutic agents is a critical
issue in cancer treatment, including the treatment of bladder
cancer. This resistance may result from the failure of the
apoptosis that is activated in response to drug treatment. Combined
chemotherapy using cisplatin resulted in a response rate of 60–80%
in patients with advanced bladder cancer, but the subsequent
emergence of chemoresistance meant that complete remission was
sustained in only 15% of affected patients (26). An enhanced understanding of the
mechanisms contributing to cisplatin resistance may facilitate
predictions of the clinical response to therapy. The aim of this
study was to gain insight into the pathway leading to
cisplatin-induced apoptosis in human bladder cancer cells. This
study showed for the first time that cisplatin-induced drug
resistance was associated with the downregulation of miR-101 in the
T24 bladder cancer cell line. miR-101 may regulate the survival of
T24 cells by modulating COX-2 expression.
Overexpression of miR-101 has been found to suppress
cell proliferation and impair invasive potential in prostate
cancer, bladder carcinoma and colon cancer (27–29).
Additionally, Zhang et al (30) demonstrated that the downregulation
of miR-101 in non-small cell lung cancer acted as a tumor promoter
by stimulating cell proliferation and invasion and inhibiting
paclitaxel-induced apoptosis. In an in vitro study of
gastric cancer, Wang et al (31) showed that gastric cancer cell
proliferation, migration and invasion could be inhibited by the
ectopic expression of miR-101. Furthermore, Chen et al found
that ectopic miR-101 acted to sensitize human tumor cells to
radiation via the inhibition of DNA repair by targeting ataxia
telangiectasia mutated and DNA-dependent protein kinase catalytic
subunits (32). The present study
found that miR-101 expression was significantly downregulated in
cisplatin-resistant bladder cancer cells as compared with that in
parent cells. This observed downregulation of miR-101 may be
involved in the occurrence of cisplatin resistance. The biological
functions of miR-101 in bladder cancer were further explored.
Expression of miR-101 in cisplatin-resistant bladder cancer cells
inhibited cell proliferation and promoted apoptosis. The identified
role of miR-101 in bladder cancer cells from this study was
consistent with that observed in other cancers (33). Taken together, these results
further confirm that miR-101 functions as a tumor suppressor.
Following TargetScan software analysis, it was
predicted that COX-2 could be a direct target of miR-101 due to a
seed region within miR-101, which is able to bind to the COX-2 mRNA
3′-UTR. A luciferase reporter vector (pGL3-COX2-wildtype)
containing the 3′-UTR of COX-2 with a putative miR-101
complementary region was constructed. A significantly lower level
of luciferase activity was detected when T24/CDDP cells were
co-transfected with the miR-101 mimic and pGL3-COX2-wildtype,
indicating a direct interaction between miR-101 and COX-2 mRNA. It
has been suggested that COX-2 inhibitors can sensitize
drug-resistant tumor cells to chemotherapy (34). Nonsteroidal anti-inflammatory drugs
are often used for the treatment of colon cancer, either alone or
in combination with other chemotherapeutic agents. Combining
flurbiprofen, sulindac or indomethacin with 5-fluorouracil resulted
in an enhancement of cytotoxicity (35). In this study, the re-expression of
miR-101 in cisplatin-resistant bladder cancer cells decreased COX-2
expression, inhibited cell proliferation and promoted apoptosis.
The results of the siRNA assays confirmed that COX-2 could modulate
cisplatin sensitivity in bladder cancer cells. Knockdown of COX-2
significantly decreased cell survival with an overall effect that
was similar to that of miR-101 overexpression.
In conclusion, this study has presented the first
evidence, to the best of our knowledge, that miR-101 may be
involved in the development of cisplatin resistance in human
bladder cancer cell lines. miR-101 was shown to modulate the
resistance of bladder cell lines to cisplatin, at least in part,
through targeting COX-2 expression. Therapeutic strategies
targeting drug resistance associated with miRNAs, such as
hsa-miR-101, may be another promising way to enhance therapeutic
effects.
Acknowledgements
This study was supported by funding from the Science
and Technology Support Plan of Zhenjiang City (FZ2012003).
References
|
1
|
Bo J, Yang G, Huo K, et al: microRNA-203
suppresses bladder cancer development by repressing bcl-w
expression. FEBS J. 278:786–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Donat SM: Integrating perioperative
chemotherapy into the treatment of muscle-invasive bladder cancer:
strategy versus reality. J Natl Compr Canc Netw. 7:40–47. 2009.
|
|
3
|
Kakizoe T, Mucci LA, Albertsen PC and
Droller MJ: Screening for bladder cancer: theoretical and practical
issues in considering the treated and untreated natural history of
the various forms of the disease. Scand J Urol Nephrol Suppl.
191–212. 2008. View Article : Google Scholar
|
|
4
|
Noguchi S, Mori T, Hoshino Y, et al:
MicroRNA-143 functions as a tumor suppressor in human bladder
cancer T24 cells. Cancer Lett. 307:211–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andrews PA and Howell SB: Cellular
pharmacology of cisplatin: perspectives on mechanisms of acquired
resistance. Cancer Cells. 2:35–43. 1990.PubMed/NCBI
|
|
6
|
Cho HJ, Kim JK, Kim KD, et al:
Upregulation of Bcl-2 is associated with cisplatin-resistance via
inhibition of Bax translocation in human bladder cancer cells.
Cancer Lett. 237:56–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim VN and Nam JW: Genomics of microRNA.
Trends Genet. 22:165–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shyu AB, Wilkinson MF and van Hoof A:
Messenger RNA regulation: to translate or to degrade. EMBO J.
27:471–481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iorio MV, Ferracin M, Liu CG, et al:
MicroRNA gene expression deregulation in human breast cancer.
Cancer Res. 65:7065–7070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Volinia S, Calin GA, Liu CG, et al: A
microRNA expression signature of human solid tumors defines cancer
gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nordentoft I, Birkenkamp-Demtroder K,
Agerbæk M, et al: miRNAs associated with chemo-sensitivity in cell
lines and in advanced bladder cancer. BMC Med Genomics. 5:402012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Greenhough A, Smartt HJ, Moore AE, et al:
The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and
adaptation to the tumour microenvironment. Carcinogenesis.
30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hasegawa K, Ishikawa K, Kawai S, et al:
Overcoming paclitaxel resistance in uterine endometrial cancer
using a COX-2 inhibitor. Oncol Rep. 30:2937–2944. 2013.PubMed/NCBI
|
|
17
|
Wu XL, Cheng B, Li PY, et al: MicroRNA-143
suppresses gastric cancer cell growth and induces apoptosis by
targeting COX-2. World J Gastroenterol. 19:7758–7765. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He XP, Shao Y, Li XL, et al:
Downregulation of miR-101 in gastric cancer correlates with
cyclooxygenase-2 overexpression and tumor growth. FEBS J.
279:4201–4212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
20
|
Xia L, Zhang D, Du R, et al: miR-15b and
miR-16 modulate multidrug resistance by targeting BCL2 in human
gastric cancer cells. Int J Cancer. 123:372–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Srivastava SK, Tetsuka T, Daphna-Iken D
and Morrison AR: IL-1 beta stabilizes COX II mRNA in renal
mesangial cells: role of 3′-untranslated region. Am J Physiol.
267:F504–F508. 1994.PubMed/NCBI
|
|
22
|
Xiang KM and Li XR: MiR-133b acts as a
tumor suppressor and negatively regulates TBPL1 in colorectal
cancer cells. Asian Pac J Cancer Prev. 15:3767–3772. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu T, Zhou Q, Zhou J, et al: Carboxyl
terminus of Hsp70-interacting protein (CHIP) contributes to human
glioma oncogenesis. Cancer Sci. 102:959–966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang JH, Song KH, Jeong KC, et al:
Involvement of Cox-2 in the metastatic potential of
chemotherapy-resistant breast cancer cells. BMC Cancer. 11:3342011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akutsu Y, Hanari N, Yusup G, et al: COX2
expression predicts resistance to chemoradiotherapy in esophageal
squamous cell carcinoma. Ann Surg Oncol. 18:2946–2951. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hong JH, Lee E, Hong J, Shin YJ and Ahn H:
Antisense Bcl2 oligonucleotide in cisplatin-resistant bladder
cancer cell lines. BJU Int. 90:113–117. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Friedman JM, Liang G, Liu CC, et al: The
putative tumor suppressor microRNA-101 modulates the cancer
epigenome by repressing the polycomb group protein EZH2. Cancer
Res. 69:2623–2629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strillacci A, Griffoni C, Sansone P, et
al: MiR-101 downregulation is involved in cyclooxygenase-2
overexpression in human colon cancer cells. Exp Cell Res.
315:1439–1447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Varambally S, Cao Q, Mani RS, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang JG, Guo JF, Liu DL, Liu Q and Wang
JJ: MicroRNA-101 exerts tumor-suppressive functions in non-small
cell lung cancer through directly targeting enhancer of zeste
homolog 2. J Thorac Oncol. 6:671–678. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang HJ, Ruan HJ, He XJ, et al:
MicroRNA-101 is down-regulated in gastric cancer and involved in
cell migration and invasion. Eur J Cancer. 46:2295–2303. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen S, Wang H, Ng WL, Curran WJ and Wang
Y: Radiosensitizing effects of ectopic miR-101 on non-small-cell
lung cancer cells depend on the endogenous miR-101 level. Int J
Radiat Oncol Biol Phys. 81:1524–1529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Y, An Y, Wang Y, et al: miR-101
inhibits autophagy and enhances cisplatin-induced apoptosis in
hepatocellular carcinoma cells. Oncol Rep. 29:2019–2024.
2013.PubMed/NCBI
|
|
34
|
Hwang MK, Kang NJ, Heo YS, Lee KW and Lee
HJ: Fyn kinase is a direct molecular target of delphinidin for the
inhibition of cyclooxygenase-2 expression induced by tumor necrosis
factor-alpha. Biochem Pharmacol. 77:1213–1222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Duffy CP, Elliott CJ, O’Connor RA, et al:
Enhancement of chemotherapeutic drug toxicity to human tumour cells
in vitro by a subset of non-steroidal anti-inflammatory drugs
(NSAIDs). Eur J Cancer. 34:1250–1259. 1998. View Article : Google Scholar : PubMed/NCBI
|