Introduction
Microglia, as immune effectors of the central
nervous system, respond to pathological conditions and participate
in the initiation and progression of neurological disorders such as
inflammation and brain tumor, by releasing potential neurotrophic
or cytotoxic molecules (1).
Increasing evidence indicates that chronic microglial activation
may also contribute to the development and progression of
neurodegenerative disorders, such as Alzheimer’s disease and
Parkinson’s disease (PD), neurotropic viral infections, stroke,
paraneoplastic disorders, traumatic brain injury, amyotrophic
lateral sclerosis and multiple sclerosis (2–5).
Thus, inhibition of activated microglia is an important therapeutic
route for neurodegenerative disorders.
Rifampicin is a macrocyclic antibiotic that is
extensively used against Mycobacterium tuberculosis and
other mycobacterial infections (6). The immunosuppressive properties of
rifampicin were first reported more than 30 years ago (7–9). Our
group previously demonstrated that rifampicin significantly
inhibits the lipopolysaccharide (LPS)-induced expression of
pro-inflammatory mediators, including inducible nitric oxide (NO),
NO synthase (iNOS), cyclooxygenase-2 (COX-2), tumor necrosis
factor-α (TNF-α), and interleukin-1β (IL-1β), as well as the
production of NO and prostaglandin E2 (PGE2).
Additionally, rifampicin inhibits nuclear factor-κB (NF-κB) via the
inhibitor of κB (IκB) pathway. Rifampicin also decreases the
phosphorylation of mitogen-activated protein kinases (MAPKs)
(10). However, the mechanism
through which rifampicin inhibits the production of LPS-induced
pro-inflammatory mediators and its neuroprotective effects are not
completely understood.
In this study, we investigated the effects of
rifampicin on morphological changes induced by LPS in BV2
microglia. Then, we investigated, in murine microglial BV2 cells,
the effects of rifampicin on two signaling pathway components
stimulated by LPS, the Toll-like receptor-4 (TLR-4) and NF-κB. Our
experiments, using the microglia-neuronal co-culture system,
demonstrated that rifampicin protects the neurons from
microglia-mediated LPS neurotoxicity, supporting that this
antibiotic may be effectively used in the prevention of
neurodegenerative diseases.
Materials and methods
Materials
Rifampicin (purity >98%), LPS, and
dimethylsulfoxide were purchased from Sigma-Aldrich (St. Louis, MO,
USA). The primary rabbit anti-human polyclonal antibody targeting
the NF-κB p65 subunit and the secondary goat anti-rabbit polyclonal
rhodamine-conjugated IgG antibody were obtained from Cell Signaling
Technology (Beverly, MA, USA). Dulbecco’s modified Eagle’s medium
(DMEM) containing L-arginine (200 mg/l), fetal bovine serum (FBS),
and other tissue culture reagents were from Gibco®
(Thermo Fisher Scientific, Waltham, MA, USA).
Cell cultures
BV2 immortalized murine microglia were provided by
the Cell Culture Center of the Chinese Academy of Medical Sciences
(Beijing, China). Cells were cultured in DMEM supplemented with 10%
FBS, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified
atmosphere of 5% CO2, at 37°C. In all experiments, the
BV2 microglial cells were pre-treated with 150 μM rifampicin for 2
h before the addition of LPS (1.0 mg/ml) in serum-free DMEM.
Primary cortical neurons were derived from the
cerebral cortices of one-day-old Sprague-Dawley rats that were
supplied by the Animal Experimental Center of the Southern Medical
University of China [License no. SCXK (yue) 2006–0015]. Animal
procedures were performed in accordance with the Guidelines for the
Care and Use of Laboratory Animals, which were determined by the
Ministry of Science and Technology of China. Primary cortical
neurons were derived from the cerebral cortices of 1-day-old
Sprague-Dawley rats using previously described procedures (11) with certain modifications: briefly,
the isolated tissues were incubated in 0.25% trypsin
(Sigma-Aldrich) in phosphate-buffered saline (PBS) for 15 min at
37°C. Following trypsinization, the tissues were rinsed in the
Gibco® NeurobasalTM medium containing 2% B27
supplement (both from Thermo Fisher Scientific) three times (5 min
each), and were mechanically dissociated using a fire-polished
pipette. Cells were seeded at a density of ~2×105
cells/ml on poly-L-lysine (0.1 mg/ml)-coated 24-well culture
polystyrene plates and incubated at 37°C with 5% CO2, at
saturated humidity. After 120 min, cells were gently rinsed three
times to remove detached tissues from the surface. Half of the
medium was replaced with fresh medium twice a week. Cultures were
monitored to ensure that neurons constituted ≥95% of the total
population. All experiments were performed using rat primary
cortical neurons cultured for 5 days. The primary cortical neurons
were verified using an immunofluorescence technique as described in
the following section.
Immunofluorescence staining
For immunofluorescence staining, the cells were
fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1%
Triton X-100 for 10 min, and blocked with 5% bovine serum albumin
(BSA) for 30 min. The cells were then incubated with the primary
antibody targeting NF-κB p65 (1:100 dilution) overnight at 4°C.
After washing three times with PBS, the cells were incubated with
the secondary antibody for 1 h. Nuclei were stained with Hoechst
33258 (Sigma-Aldrich). Fluorescent images were captured on a laser
scanning confocal microscope (LSM 510 META; Carl Zeiss, Stuttgart,
Germany).
Total RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was isolated with the Invitrogen™ TRIzol
reagent (Thermo Fisher Scientific) according to the manufacturer’s
instructions. Total RNA (1.0 μg) was reverse transcribed using the
M-MLV reverse transcriptase (Promega Corp., Madison, WI, USA) to
synthesize complementary DNA. The primers used for qPCR were as
follows: TLR-4 forward (F), 5′-GCTTTCACCTCT GCCTTCAC-3′, and
reverse (R), 5′-CCAACGGCTCTG AATAAAGTG-3′; 3-phosphate
dehydrogenase (GAPDH) F, 5′-TCACCACCATGGAGAAGGC-3′, and R,
5′-GCTAAG CAGTTGGTGGTGCA-3′. qPCR was performed with the following
cycling parameters: 40 cycles of denaturation at 94°C for 20 sec,
annealing at 62°C for 30 sec, and extension at 72°C for 30 sec. The
SYBR-Green qPCR Master Mix 2 kit (Takara Bio Inc., Otsu, Japan) was
used in all samples, and the reactions were carried out in a 20 μl
final reaction volume, using an LC480 qPCR machine (Roche, Basel,
Switzerland). The mRNA expression levels of target genes were
calculated based on standard curve analysis with the absolute
quantification method, and were expressed relative to the level of
GAPDH, a housekeeping gene used as an endogenous control
(12).
Cytotoxicity assay in a co-culture of
microglia and neurons
The BV2 microglial cells were co-cultured with
primary cortical neurons to study the regulation of neuronal
survival by the LPS-stimulated microglia. The BV2 microglial cells
were grown in Transwell inserts (pore size, 0.4 μm; Corning Life
Sciences, Tewksbury, MA, USA), and LPS (1.0 μg/ml) was added. The
neurons were then transferred onto the inserts containing BV2
cells. In the Transwell co-culture system, microglial cells
communicate with neurons through the semi-permeable membrane
without direct cell contact (13).
Cell viability was assessed by measurement of released lactate
dehydrogenase (LDH), using the CytoTox-96 kit from Promega Corp.,
according to the manufacturer’s instructions.
Detection of apoptosis in a co-culture of
microglia and neurons
In the co-culture system described above, apoptotic
neuronal cells were detected by the terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay (Roche,
Basel, Switzerland). After each treatment, the TUNEL assay was
performed according to manufacturer’s instructions, and all nuclei
were counterstained with 5 mg/ml of Hoechst 33342 for 10 min at
37°C. The labeled neuronal cells were examined under an LSM 510
META laser scanning confocal microscope. Neuronal cells were
considered to be apoptotic when their nuclei were co-stained with
Hoechst 33342 and TUNEL. We counted the number of apoptotic cells
in 100 randomly chosen neurons, observed at different
magnifications.
Statistical analysis
Quantitative data were expressed as the mean ±
standard error of the mean of at least three independent
experiments. Comparisons between two groups were analyzed using
Student’s t-tests. A value of p<0.05 was considered to indicate
statistically significant differences.
Results
Effect of rifampicin on morphological
changes induced by LPS in BV2 microglia
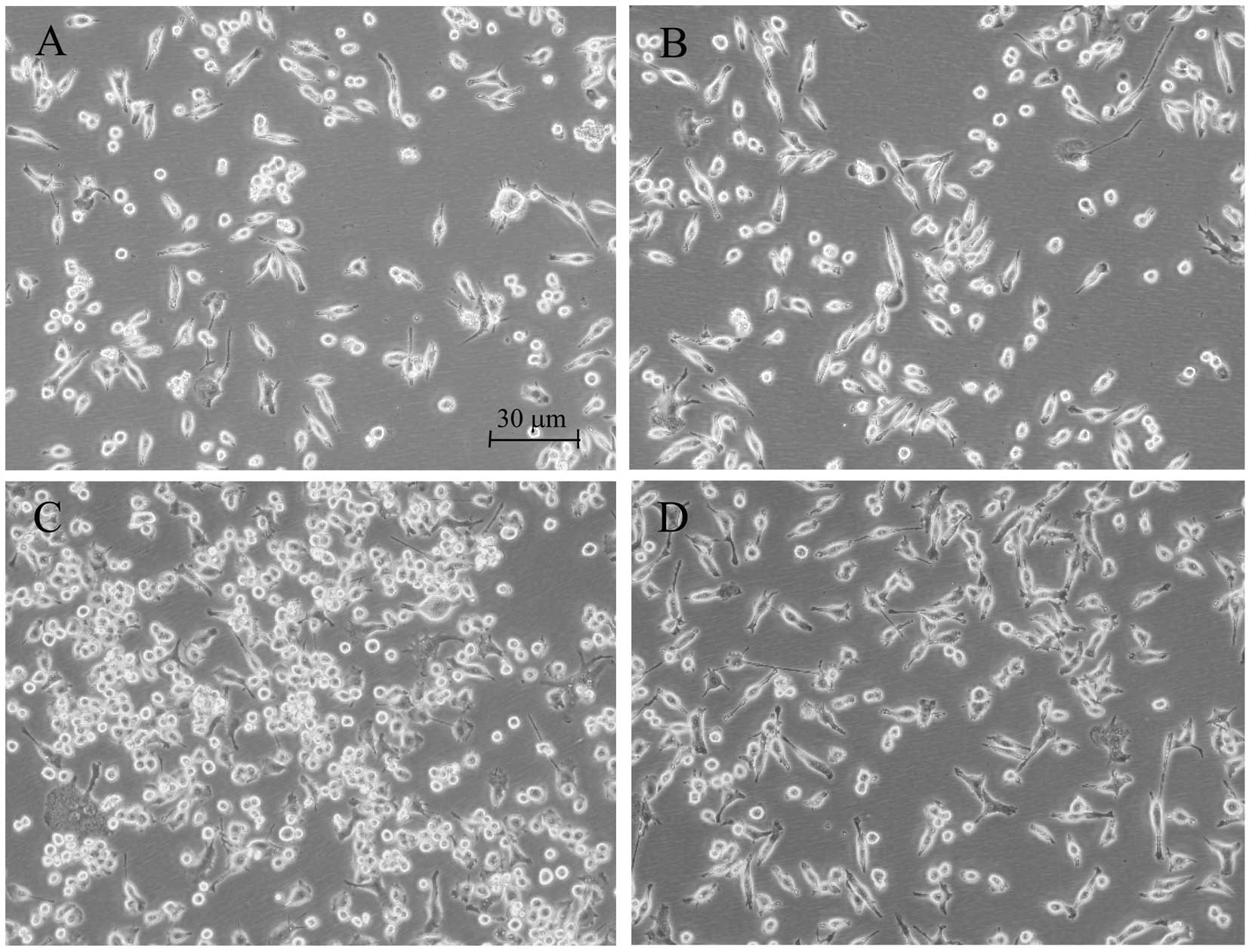
Morphological alterations of the microglia are
suitable indicators of the effects of different agents. Enlargement
of the microglial cell body and loss of ramifications, along with
development of an amoeboid shape, are commonly caused by LPS
(14). While those changes were
clearly observed in the LPS-treated BV2 microglia, rifampicin
markedly improved morphological changes that were caused by LPS,
and branch-like morphology was observed in these rifampicin-treated
cells (Fig. 1).
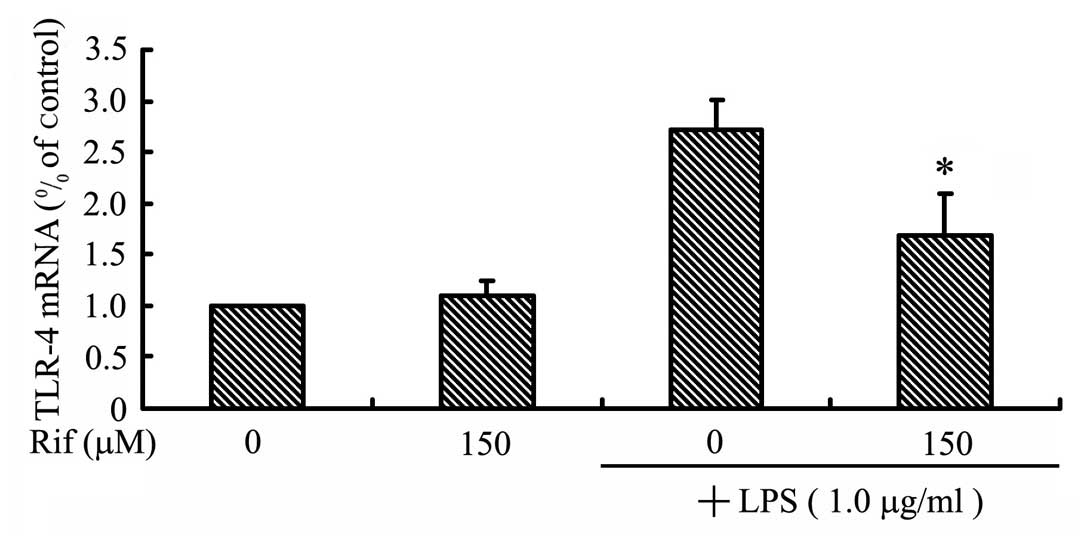
Rifampicin inhibits TLR-4 expression in
LPS-stimulated BV2 microglia
To examine the effect of rifampicin on TLR-4
expression, we measured the levels of the TLR-4 mRNA in
LPS-stimulated BV2 microglia. The BV2 microglia were pre-treated
with rifampicin for 2 h, and then stimulated with LPS for 2 h prior
to RT-qPCR analysis. As shown in Fig.
2, the TLR-4 mRNA level increased in LPS-stimulated BV2
microglia, but was significantly reduced by treatment with
rifampicin. This result indicates that rifampicin inhibits
TLR-4 expression, which we hypothesized may lead to
inhibited activation of the NF-κB, MAPK and Akt pathways.
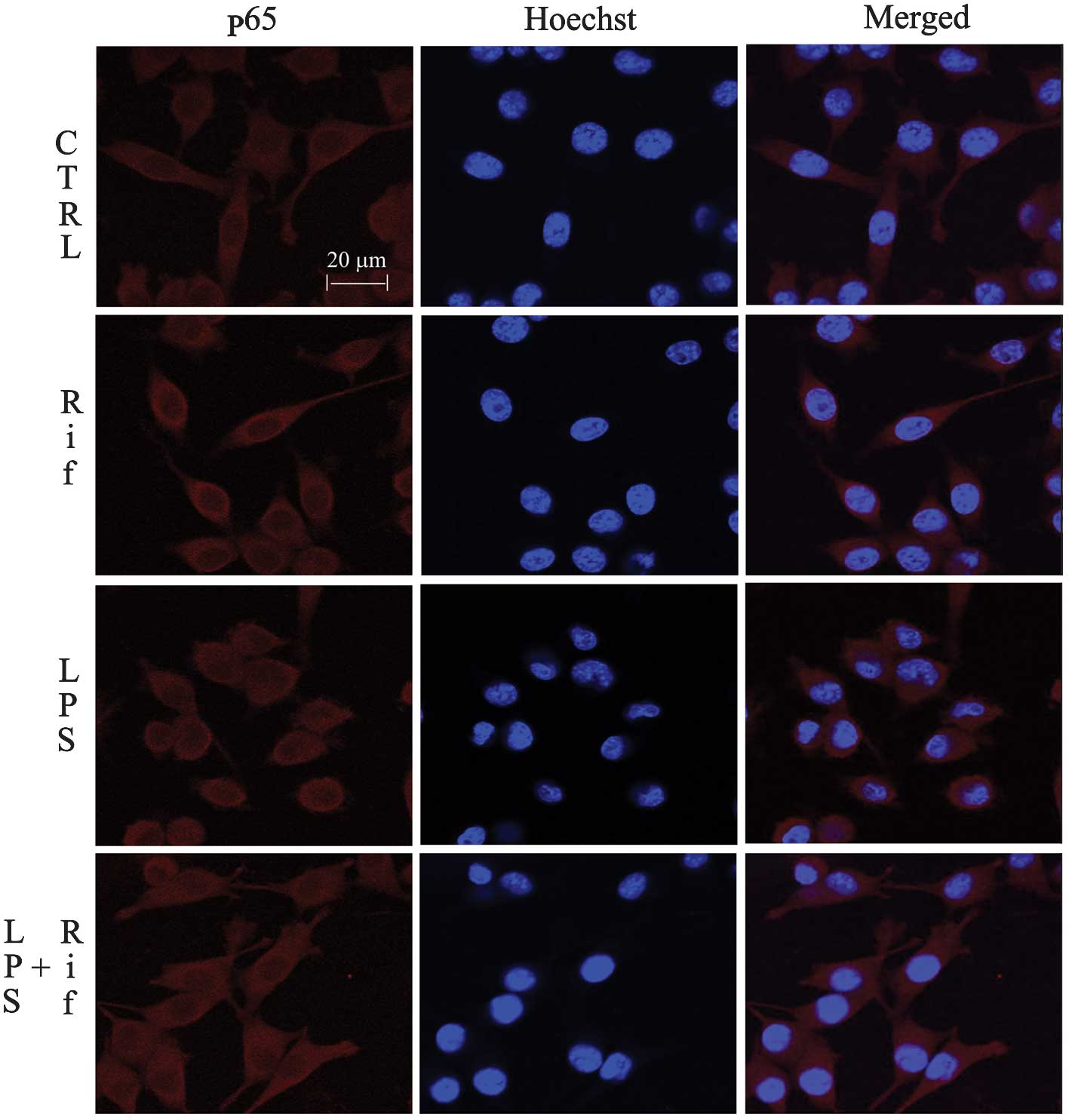
Effects of rifampicin on the NF-κB
signaling pathway
Activation of NF-κB leads to its translocation to
the nucleus, where it mediates the transcriptional regulation of
pro-inflammatory genes. The activation and nuclear translocation of
NF-κB is a key step in LPS-stimulated microglial activation. We
investigated the regulation of NF-κB by rifampicin using
immunofluorescence staining. As shown in Fig. 3, the NF-κB p65 subunit was
primarily retained in the cytoplasm in unstimulated cells; however,
following stimulation with LPS, the cytoplasmic NF-κB p65 level was
reduced, accompanied by an increase in the nuclear NF-κB p65 level.
Treatment with 150 μM rifampicin blocked NF-κB p65 nuclear
translocation in LPS-stimulated BV-2 cells. This result suggests
that rifampicin may suppress pro-inflammatory enzymes and
pro-inflammatory cytokines by inhibiting NF-κB activation.
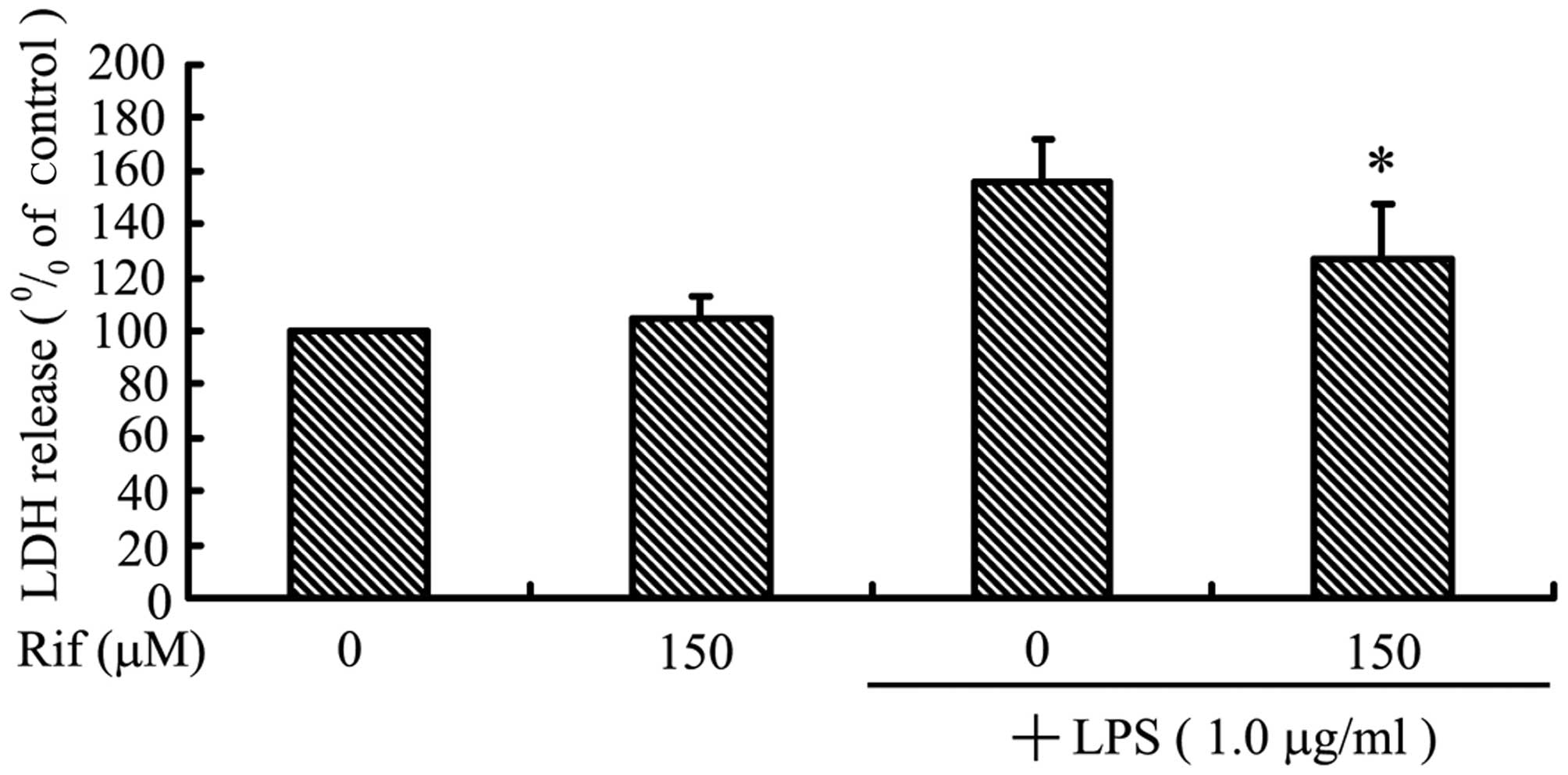
Rifampicin decreases microglial-induced
cortical neuron death in a co-culture system
In order to investigate whether rifampicin can
protect against neuronal death induced by microglial activation, we
used a co-culture system with cortical neurons and BV2 microglia.
We examined cortical neuron viability following co-culture with
LPS-activated BV2 microglia using the LDH assay. As shown in
Fig. 4A, cortical neurons in
control inserts without LPS-stimulated BV2 microglia did not
undergo cell death. By contrast, LPS treatment alone led to a high
level of cortical neuron death in co-culture, suggesting that
LPS-activated microglia secrete pro-inflammatory cytokines that can
migrate through the insert, inducing death of the neuronal cells.
Treatment with rifampicin markedly reduced the death of cortical
neurons: cell viability was increased by ~28.6% when the
LPS-stimulated BV2 microglia were pre-treated with rifampicin.
Apoptosis was determined by the TUNEL assay. As
shown in Fig. 4B, cortical neurons
were stained with Hoechst 33342 (blue), and apoptotic neurons were
stained green using the TUNEL method (Fig. 4B). Co-culture with BV2 microglia
exposed to LPS alone resulted in a significant increase in the
number of apoptotic cortical neurons compared to control cells. As
expected, administration of rifampicin reduced the number of
apoptotic cortical neurons (Fig.
4C).
Discussion
Rifampicin has been reported to exert
neuroprotective effects in various disease models (15–19).
Rifampicin-induced cytoprotection and suppression of β-amyloid
aggregation indicate its potential application in the treatment of
PD (20,21). An in vivo study showed that
rifampicin attenuates MPTP-induced neurodegeneration in
nigrostriatal dopamine neurons of mouse brains (22). We previously showed that rifampicin
pre-treatment causes a dose-dependant increase in cell viability
and a reduction in α-synuclein expression (23). Rifampicin-induced neuroprotection
was previously attributed to its free radical-scavenging ability
(18). We found that rifampicin
pre-treatment protects PC12 cells against rotenone-induced cell
death. Qualitative and quantitative analysis revealed that
rifampicin significantly suppresses rotenone-induced apoptosis by
ameliorating mitochondrial oxidative stress (24). We also demonstrated that rifampicin
reduces microglial activation and improves neuronal survival during
inflammation (10). However, the
mechanism through which rifampicin inhibits microglial inflammation
and its neuroprotective effects are not completely understood.
Our previous in vivo study showed that
rifampicin significantly inhibits the LPS-induced expression of
pro-inflammatory mediators, including inducible iNOS, COX-2, TNF-α,
and IL-1β, as well as the production of NO and PGE2
(10). The morphology of microglia
cells (Fig. 1), along with data on
the expression and synthesis of iNOS, COX-2, TNF-α and IL-1β
indicate that rifampicin may induce pro-inflammatory changes in the
microglia.
The principal cell surface receptor for the LPS
component of antitoxin is TLR-4, member of a highly conserved
family of receptors specific to highly conserved bacterial and
viral components; these receptors play key roles in activating a
cascade of pro-inflammatory events in response to pathogens
(25,26). Therefore, treatments that attenuate
TLR-4-associated inflammatory cascades may prove beneficial to
microglial activation and prevent neurodegenerative processes. Our
results indicate that rifampicin pre-treatment inhibits the
LPS-induced TLR-4 expression (Fig. 2). We therefore hypothesized that
the underlying molecular mechanism may involve interference with
the LPS-triggered increase in TLR-4 expression. Our previous
study showed that rifampicin decreases the phosphorylation of MAPKs
(10). Collectively, these results
indicate that rifampicin may inhibit NF-κB, p38, JNK, and MAPK
activation through downregulation of TLR-4 expression.
The activation and nuclear translocation of NF-κB is
a key step in LPS-stimulated microglial activation, and mediates
the transcriptional regulation of pro-inflammatory genes (27,28).
In the present study, treatment with 150 μM rifampicin blocked
NF-κB p65 nuclear translocation in LPS-stimulated BV-2 cells
(Fig. 3). A recent study used a
luciferase reporter assay to investigate the possibility that
rifampicin inhibits NF-κB transcriptional activity. Further
investigation demonstrated that rifampicin blocks the
phosphorylation and subsequent degradation of IκB in LPS-induced
BV2 cells (10). It is
hypothesized that rifampicin markedly inhibits the nuclear
translocation of NF-κB p65 (10).
These results, also confirmed by the current study, suggest that
rifampicin may suppress pro-inflammatory enzymes and
pro-inflammatory cytokines through inhibiting the activation of
NF-κB.
Microglial activation has been considered harmful
for neurons, and can lead to neuronal apoptosis (29). Microglial involvement in
neurodegenerative diseases is well-established, microglial
activation and neuroinflammation being common features of these
neuropathologies (30). Neurotoxic
microglial-neuronal interactions have been implicated in the
pathogenesis of various neurodegenerative diseases, and have been
recognized as critical for the understanding of the underlying
mechanism of neuron diseases (31,32).
In order to investigate whether rifampicin can rescue neuronal
death induced by microglial activation, we used cortical neurons
and BV2 microglia in a co-culture system. Our results clearly
indicate that when cortical neurons are co-cultured with
LPS-stimulated BV2 microglia, neuronal cell death is increased by
56.0%, and the number of apoptotic neurons is increased by 49.0%.
However, treatment with rifampicin in our LPS-induced co-culture
system increased cell viability by 28.6% and reduced the apoptotic
cell number by 28.0%. (Fig. 4).
These data suggest that rifampicin decreases cortical neuron
apoptosis through inhibition of microglial activation in the
microglial-neuronal co-culture system. Together, these results
provide strong evidence that rifampicin can protect neurons from
microglial-mediated LPS neurotoxicity.
In conclusion, the present study demonstrated that
rifampicin inhibits the LPS-stimulated expression of TLR-4.
When cortical neurons were co-cultured with LPS-stimulated BV2
microglia, pre-treatment with rifampicin increased neuronal
viability and reduced the number of apoptotic cells. Our
observations suggest that rifampicin may be used as a therapeutic
agent for the treatment of neurodegenerative diseases.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of China (nos. 81200930 and 81371442), the
Natural Science Foundation of Guangdong Province (nos.
S2012040007768 and S2011040003038), the Medical Research Fund of
Guangdong Province (no. B2012190), the Ph.D. Program Foundation of
Ministry of Education of China (no. 20124401120016), the
Cultivation and Innovation Fund of The First Affiliated Hospital of
Jinan University (no. 201222), and the Fundamental Research Funds
for the Central Universities (no. 21612307).
References
|
1
|
Zhang H, Wang FW, Yao LL and Hao AJ:
Microglia - friend or foe. Front Biosci (Schol Ed). 3:869–883.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith JA, Das A, Ray SK and Banik NL: Role
of pro-inflammatory cytokines released from microglia in
neurodegenerative diseases. Brain Res Bull. 87:10–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bernardi A, Frozza RL, Meneghetti A, et
al: Indomethacin-loaded lipid-core nanocapsules reduce the damage
triggered by Aβ1–42 in Alzheimer’s disease models. Int J
Nanomedicine. 7:4927–4942. 2012.PubMed/NCBI
|
|
4
|
Sung YH, Kim SC, Hong HP, et al: Treadmill
exercise ameliorates dopaminergic neuronal loss through suppressing
microglial activation in Parkinson’s disease mice. Life Sci.
91:1309–1316. 2012.
|
|
5
|
Dibaj P, Zschüntzsch J, Steffens H, et al:
Influence of methylene blue on microglia-induced inflammation and
motor neuron degeneration in the SOD1(G93A) model for ALS. PLoS
One. 7:e439632012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yulug B, Kilic U, Kilic E and Bähr M:
Rifampicin attenuates brain damage in focal ischemia. Brain Res.
996:76–80. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paunescu E: In vivo and in vitro
suppression of humoral and cellular response by rifampicin. Nature.
228:1188–1189. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nilsson BS: Rifampicin: an
immunosuppressant? Lancet. 2:3741971. View Article : Google Scholar
|
|
9
|
Dajani BM, Canadi MS, Thompson JS and
Kasik JE: Rifampicin: an immunosuppressant? Lancet. 2:19041972.
|
|
10
|
Bi W, Zhu L, Wang C, et al: Rifampicin
inhibits microglial inflammation and improves neuron survival
against inflammation. Brain Res. 1395:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singer CA, Figueroa-Masot XA, Batchelor
RH, et al: The mitogen-activated protein kinase pathway mediates
estrogen neuroprotection after glutamate toxicity in primary
cortical neurons. J Neurosci. 19:2455–2463. 1999.
|
|
12
|
Kiefer T, Hirt C, Schüler F, et al:
Statistical analysis of results obtained by real-time PCR for
improvement of absolute quantification of target sequences. Clin
Lab. 58:465–470. 2012.PubMed/NCBI
|
|
13
|
Bureau G, Longpré F and Martinoli MG:
Resveratrol and quercetin, two natural polyphenols, reduce
apoptotic neuronal cell death induced by neuroinflammation. J
Neurosci Res. 86:403–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kloss CU, Bohatschek M, Kreutzberg GW and
Raivich G: Effect of lipopolysaccharide on the morphology and
integrin immunoreactivity of ramified microglia in the mouse brain
and in cell culture. Exp Neurol. 168:32–46. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Namba Y, Kawatsu K, Izumi S, Ueki A and
Ikeda K: Neurofibrillary tangles and senile plaques in brain of
elderly leprosy patients. Lancet. 340:9781992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chui DH, Tabira T, Izumi S, Koya G and
Ogata J: Decreased beta-amyloid and increased abnormal Tau
deposition in the brain of aged patients with leprosy. Am J Pathol.
145:771–775. 1994.PubMed/NCBI
|
|
17
|
Tomiyama T, Kaneko H, Kataoka K, et al:
Rifampicin inhibits the toxicity of pre-aggregated amyloid peptides
by binding to peptide fibrils and preventing amyloid-cell
interaction. Biochem J. 322:859–865. 1997.
|
|
18
|
Tomiyama T, Shoji A, Kataoka K, et al:
Inhibition of amyloid beta protein aggregation and neurotoxicity by
rifampicin. Its possible function as a hydroxyl radical scavenger.
J Biol Chem. 271:6839–6844. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tomiyama T, Asano S, Suwa Y, et al:
Rifampicin prevents the aggregation and neurotoxicity of amyloid
beta protein in vitro. Biochem Biophys Res Commun. 204:76–83. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kapurniotu A: Targeting alpha-synuclein in
Parkinson’s disease. Chem Biol. 11:1476–1478. 2004.
|
|
21
|
Bradbury J: New hope for mechanism-based
treatment of Parkinson’s disease. Drug Discov Today. 10:80–81.
2005.PubMed/NCBI
|
|
22
|
Oida Y, Kitaichi K, Nakayama H, et al:
Rifampicin attenuates the MPTP-induced neurotoxicity in mouse
brain. Brain Res. 1082:196–204. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu J, Wei C, Xu C, Bennett MC, Zhang G, Li
F, et al: Rifampicin protects PC12 cells against
MPP+-induced apoptosis and inhibits the expression of an
alpha-synuclein multimer. Brain Res. 1139:220–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Sun Y, Zeng Z and Tao E:
Rifampicin inhibits apoptosis in rotenone-induced differentiated
PC12 cells by ameliorating mitochondrial oxidative stress. Neural
Regen Res. 5:251–256. 2010.
|
|
25
|
Ozato K, Tsujimura H and Tamura T:
Toll-like receptor signaling and regulation of cytokine gene
expression in the immune system. Biotechniques. (Suppl): 66–68.
7072 passim. 2002.PubMed/NCBI
|
|
26
|
Bi W, Jing X, Zhu L, Liang Y, Liu J, Yang
L, Xiao S, et al: Inhibition of 26S protease regulatory subunit 7
(MSS1) suppresses neuroinflammation. Plos One. 7:e361422012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Broad A, Jones DE and Kirby JA: Toll-like
receptor (TLR) response tolerance: a key physiological ‘damage
limitation’ effect and an important potential opportunity for
therapy. Curr Med Chem. 13:2487–2502. 2006.
|
|
28
|
Medzhitov R: Toll-like receptors and
innate immunity. Nat Rev Immunol. 1:135–145. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brown GC and Neher JJ: Inflammatory
neurodegeneration and mechanisms of microglial killing of neurons.
Mol Neurobiol. 41:242–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Polazzi E and Monti B: Microglia and
neuroprotection: from in vitro studies to therapeutic applications.
Prog Neurobiol. 92:293–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shie FS, Chen YH, Chen CH and Ho IK:
Neuroimmune pharmacology of neurodegenerative and mental diseases.
J Neuroimmune Pharmacol. 6:28–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Liu L, Barger SW, Mrak RE and
Griffin WS: Vitamin E suppression of microglial activation is
neuroprotective. J Neurosci Res. 66:163–170. 2001. View Article : Google Scholar : PubMed/NCBI
|