Introduction
The seed cells of artificial nerves are Schwann
cells (SCs), however, the limited availability of these cells has
been a persistent challenge to clinical application. Stem cells can
be used as a source of seed cells as they lead to a reduced
probability of immunological rejection, and have been successfully
used in nerve tissue regeneration. A study by Heath (1) demonstrated that the use of stem cells
as seed cells is able to improve regeneration and increase the
compatibility of transplanted tissue with adjacent tissue. Due to
their low immunogenicity, stem cells may reduce or even eliminate
the requirement for immunosuppressive drugs.
Human bone marrow mesenchymal stem cells (MSCs)
differentiated into Schwann-like cells have been used as artificial
nerve seed cells, utilizing the sciatic nerve lesion model in rats
(2–4). However, another study indicated that
the content and ability of multidirectional differentiation of bone
marrow cells reduces with increasing age of the donor, posing
limitations on the use of this treatment in older patients
(5). In contrast, MSCs isolated
from human umbilical cord blood (hUCB) are not affected by donor
age. Furthermore, hUCB has been widely investigated due to its vast
source options, easy and atraumatic collection, low immunogenicity
(6) and high differentiation
capability. In the current study, the differentiation of hUCBMSCs
into Schwann-like cells in vitro was investigated under
specific induction conditions.
Materials and methods
Umbilical cord blood sample
Umbilical cord blood was provided by the Department
of Obstetrics of the First Affiliated Hospital of Bengbu Medical
College (Bengbu, China). The patients and their family members
provided signed informed consent documents prior to the study. This
study was performed with the approval of the ethical committee of
the First Affiliated Hospital of Bengbu Medical College (Bengbu,
China; no. 2012029).
Instruments and reagents
The following equipment was used in the proceeding
experiments: Refrigerated horizontal centrifuge 5810 R (Eppendorf,
Hamburg, Germany); CO2 incubator (Thermo Fisher
Scientific, Waltham, MA, USA); clean bench (Suzhou Purification
Engineering Installation Co., Ltd., Suzhou, China); inverted
microscope (CKX41SF; Olympus Corp., Tokyo, Japan); BD FACSCalibur
Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA); gene
amplification apparatus (Black King Kong EDC-810, Dongsheng
Chuangxin Biotechnology Co., Ltd, Beijing, China); nucleoprotein
detector (BioPhotometer plus; Eppendorf AG, Hamburg, Germany) and a
Tanon 3500 Gel Imaging System (Tanon Science and Technology Co.,
Ltd., Shanghai, China). The following reagents were used: MesenCult
Proliferation Kit (Human) (05411; Stemcell Technologies, Inc.,
Vancouver, Canada); hespan 6% hetastarch in 0.9% sodium chloride
injection (B Braun Medical, Inc., Bethlehem, PA, USA); fetal bovine
serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM)/F12
(Hyclone Laboratories, Logan UT, USA); lymphocyte separation media
(human) (ρ=1.077 g/ml) (Haoyang Biological Manufacture Co., Ltd,
Tianjin, China); pancreatin, β-sodium glycerophosphate and
3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich, St. Louis, MO,
USA); anti-CD34-fluorescein isothiocyanate (FITC),
CD44-phycoerythrin (PE) and CD73-PE (BioLegend, Inc., San Diego,
CA, USA); β-mercaptoethanol (β-ME; Amresco LLC, Solon, OH, USA);
retinoic acid (RA; #R2625; Sigma-Aldrich); recombinant human
fibroblast growth factor-basic (bFGF; #100-18B), recombinant human
platelet-derived growth factor-BB (PDGF-BB; #100-14B), recombinant
human heregulin β-1 (HRG; #100-03) and recombinant human β-nerve
growth factor (NGF; #450-01) (all from PeproTech, Inc., Rocky Hill,
NJ, USA); forskolin (FSK; #S1612; Beyotime Institute of
Biotechnology, Shanghai, China); anti-S100 (Maixin Biotech Co.,
Ltd, Fuzhou, China); rabbit anti-S100b (Beijing Biosynthesis
Biotechnology Co., Ltd., Beijing, China); mouse anti-GFAP (Maixin
Biotech Co., Ltd.); mouse anti-GFAP (#AG259; Beyotime Institute of
Biotechnology); rabbit anti-P75 (Beijing Biosynthesis Biotechnology
Co., Ltd.); E.Z.N.A. Total RNA Kit I (#R6834-00; Omega Bio-Tek,
Inc., Norcross, GA, USA); RevertAid reverse transcriptase (#EP0441;
Thermo Fisher Scientific, Pittsburgh, PA, USA), PCR Master Mix (2X)
(#K1621; Thermo Fisher Scientific); and non-biotin
immunohistochemical Elivision™plus kit (Maixin Biotech Co.,
Ltd).
Isolation and cultivation of
hUCBMSCs
A volume of 40–60 ml umbilical cord blood from
full-term normal delivery or cesarian-section fetuses were
collected under aseptic conditions and separated 6 h, subsequent to
anticoagulation, with heparin. Umbilical cord blood was mixed with
6% hetastarch at a volume ratio of 1:4 and incubated at room
temperature for 45 min. The supernatant was collected and added to
human lymphocyte separation media (volume ratio of 1:2). The
mononuclear cell suspension containing hUCBMSCs was obtained from
the cloudy white interphase following centrifugation for 20 min at
690 xg at room temperature. DMEM/F12 was added to the cells (volume
ratio of 1:5), and the cells were then washed twice with DMEM/F12
and counted with a hematocytometer. The hUCBMSCs were resuspended
in MesenCult complete medium (from the proliferation kit) at
1.0–1.5×107 cells/ml, seeded into a 25 cm2
plastic culture flask, and cultured at 37°C in a 5% CO2
humidified incubator. The cell culture media was changed daily for
the 5–7 days after seeding and then changed every 3 days. Cells of
90% confluence were digested by 0.25% trypsin (Sigma-Aldrich). The
morphological changes in the cultured cells were observed using an
inverted microscope.
hUCBMSC differentiation to osteocytes and
adipocytes
Osteogenic differentiation
Cells at passage 3 (P3) at a density of
5×104/cm2 were added to a six-well plate and
incubated in osteogenesis-inducing medium, consisting of DMEM/F12
containing 10 mM β-sodium glycerophosphate, 0.1 μM dexamethasone,
50 μg/ml vitamin-C (all Sigma-Aldrich) and 10% FBS. Cells in the
control group were not treated with this media. The cell culture
media was changed every three days. The cells were cultured for 21
days and then stained with 0.1% alizarin red (Sigma-Aldrich) for
calcium nodule staining.
Adipogenic differentiation
Cells at P3 were seeded into a 6-well plate in
adipogenesis-inducing medium, consisting of DMEM/F12 containing 1
μM dexamethasone, 10 μg/ml insulin, 0.5 mM IBMX, 0.1 μM
indomethacin (all Sigma-Aldrich), and 10% FBS. Control cells were
not treated with this media. The culture media was changed every 3
days. After 21 days, the cells were stained with 2% oil red O
staining solution (Sigma-Aldrich).
Directional differentiation of hUCBMSCs
to SCs
P3 cells were seeded onto 0.01% poly-L-lysine
(Sigma-Aldrich)-coated coverslips placed in a six-well plate. When
the adherent cells grew to 60–70% confluence, differentiation was
induced. First, cells were pre-induced with DMEM/F12 containing
0.25 mM β-ME and 10 ng/ml bFGF for 24 h. The culture media was then
changed to DMEM/F12 with 10% FBS. Following 24 h incubation, the
culture media was changed to DMEM/F12 supplemented with 35 ng/ml RA
for 24 h. The culture media was then changed to DMEM/F12 containing
10% FBS. Subsequent to another 24 h incubation, the cells were
incubated in DMEM/F12 supplemented with 5 μM FSK, 10 ng/ml bFGF, 5
ng/ml PDGF-BB, 100 ng/ml NGF and 200 ng/ml HRG for 4 days. The
control cells were cultured in DMEM/F12 containing 10% FBS. The
cell morphology was observed using an inverted microscope.
Immunocytochemistry
Cells grown on glass coverslips were fixed with 4%
paraformaldehyde for 30 min and washed with PBS (pH 7.4) 3 times,
for 3 min each. The fixed cells were incubated in 3% hydrogen
peroxide solution for 10 min to block endogenous catalase, washed
with PBS, and then incubated with the anti-S100, GFAP and P75
primary antibodies for 60 min at room temperature. The cells were
washed 3 times with PBS, incubated for 20 min in polymer
reinforcing agent (agent A from the Elivision kit), washed with
PBS, and then incubated for 30 min in enzyme-labeled resistant
rat/rabbit polymer (agent B). The slides were developed with
diaminobenzidine (Maixin Biotech Co., Ltd, Fuzhou, China) and
counterstained with hematoxylin. The samples were examined using an
inverted microscope.
Reverse transcription (RT) and polymerase
chain reaction (PCR)
Total RNA was extracted with the E.Z.N.A. Total RNA
Kit, cDNA was synthesized with the RevertAid reverse transcriptase
and the product was amplified using PCR Master Mix (2X). The
primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai,
China) (Table I). The PCR cycling
parameters were as follows: Initial denaturation step (95°C, 3 min)
followed by 35 cycles of denaturation (95°C, 30 sec), annealing (30
sec; S100b, 54.4°C; GFAP, 59.9°C; P75, 59.9°C; β-actin, 55.6°C) and
primer extension (72°C, 45 sec) followed by final extension
incubation (72°C, 10 min). The products were analyzed using a gel
imaging system following 1.5% agarose gel electrophoresis (100V, 25
min).
 | Table IPrimer sequences and expected product
sizes following polymerase chain reaction. |
Table I
Primer sequences and expected product
sizes following polymerase chain reaction.
| Gene | Direction | Sequence | Size (bp) |
|---|
| S100b | Sense |
5′-GGAAATCAAAGAGCAGGAGGT-3′ | 254 |
| Antisense |
5′-ATTAGCTACAACACGGCTGGA-3′ | |
| GFAP | Sense |
5′-GTCCATGTGGAGCTTGACG-3′ | 406 |
| Antisense |
5′-CATTGAGCAGGTCCTGGTAC-3′ | |
| P75 | Sense |
5′-TGGACAGCGTGACGTTCTCC-3′ | 371 |
| Antisense |
5′-GATCTCCTCGCACTCGGCGT-3′ | |
| β-actin | Sense |
5′-GGGACCTGACTGACTACCTC-3′ | 546 |
| Antisense |
5′-ACTCGTCATACTCCTGCTTGCTG-3′ | |
Western blotting
The cultured cells were washed with PBS and lysed
with a lysis buffer (50 mM Tris-HCl, 150 mM NaCl buffer, 1% NP-40,
0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM sodium
orthovanadate, 10 mM sodium fluoride, 4 μg/ml leupeptin, 1 μg/ml
aprotinin and 100 μg/ml PMSF; all from Sigma-Aldrich), and the
total protein was extracted. The concentration of the total protein
was measured using an enhanced bicinchoninic acid protein assay kit
(Beyotime Biotech, Shanghai, China). The proteins were denatured by
boiling for 5 min. A total of 45 μg protein was separated by 12%
SDS-PAGE (Sigma-Aldrich) and transferred to a nitrocellulose (NC)
membrane (EMD Millipore, Billerica, MA, USA). The NC membranes were
blocked with 5% skimmed milk prior to incubation with the primary
antibodies at 37°C for 1 h followed by overnight incubation at 4°C.
The primary antibodies used were those against S100b (1:200), GFAP
(1:300) and P75 (1:200). The membranes were incubated with
secondary antibodies goat anti-rabbit IgG (1:6,000) and goat
anti-mouse IgG (1:6,000) and then exposed using Luminata Crescendo
premixed horseradish peroxidase chemiluminescence substrate (EMD
Millipore).
Results
Isolation and culture of hUCBMSCs in
vitro
The mononuclear cells of the primary passage were
seeded in a plastic culture flask supplemented with MesenCult
complete medium. After 72 h, a small number of cells adhered to the
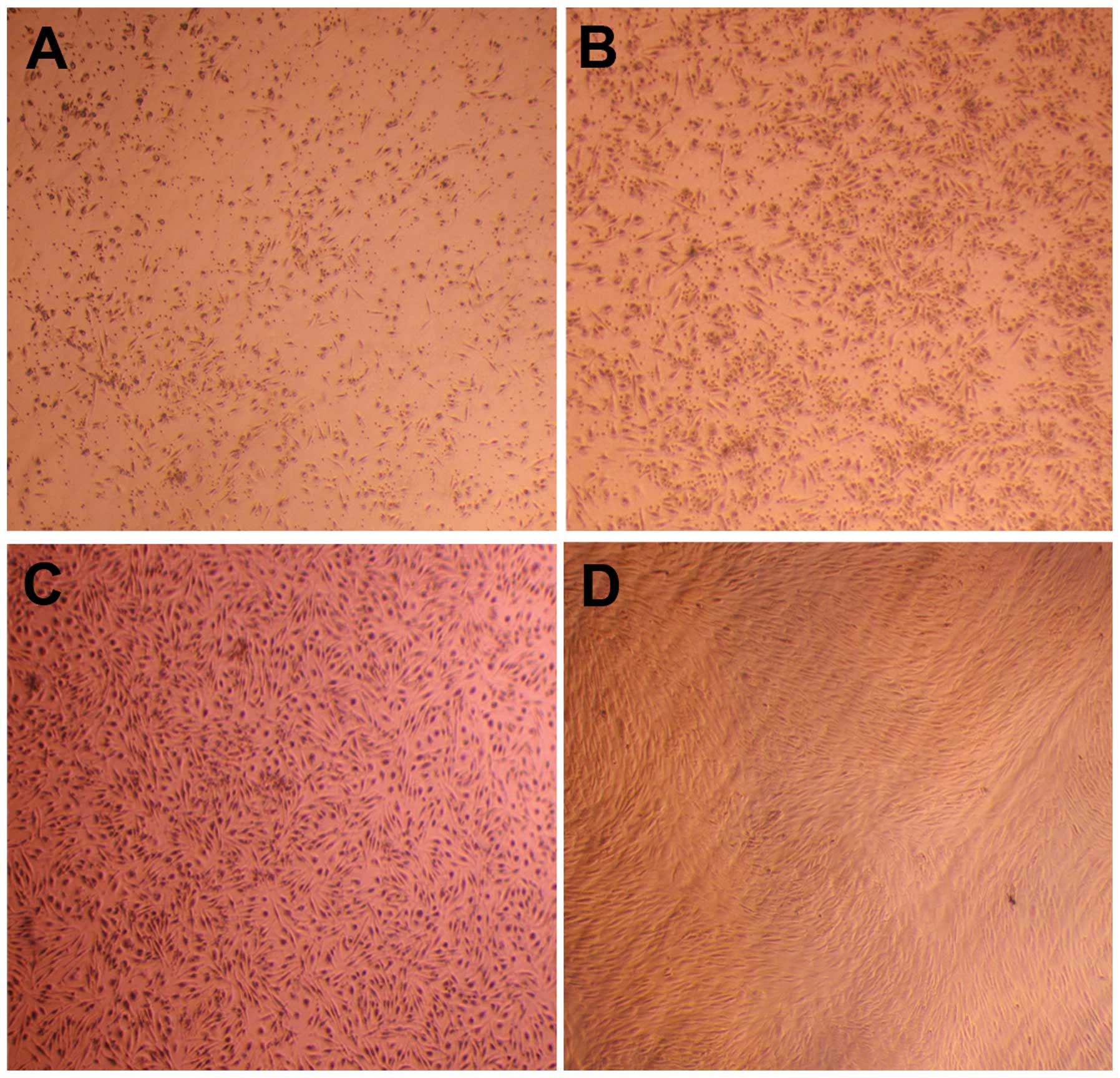
plastic culture flask (Fig. 1A).
After five days, numerous adherent cells were observed, the
majority of which were a short-fusiform shape and evenly
distributed (Fig. 1B). A number of
round-shaped osteoclast-like cells were also observed. With
increased culture time, the cell number increased and colony
formation occurred. Following two weeks of culturing, the number of
mixed round-shaped cells was substantially reduced, while that of
the short fusiform-shaped cells increased, and the morphology
changed into a long-fusiform shape. Following 3 weeks, when the
cell confluence reached 90%, the cells were similar to fibroblast
cells, with a uniformly long-fusiform shape and smaller size
(Fig. 1C). Nearly all other types
of cells were absent and only the uniformly fibroblast-like
hUCBMSCs remained upon subculturing at the third passage (Fig. 1D). The cells were broad and flat
and appeared similar to bone marrow MSCs. A number of the cells
began to present signs of aging and the rate of amplification
reduced when subcultured to the seventh passage.
Identification of surface markers by flow
cytometry
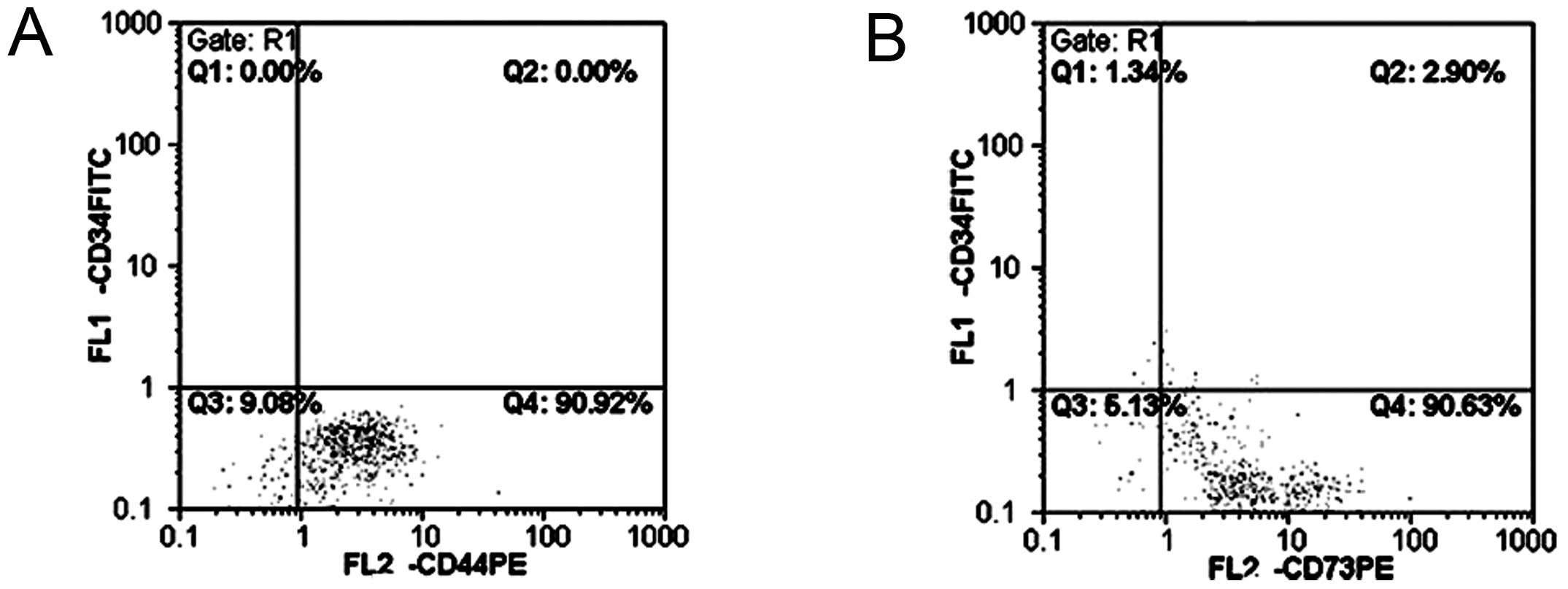
hUCBMSCs exhibited a lack or reduced expression of
hematopoietic stem/progenitor cell antigen CD34, and highly
expressed the MSC surface antigens CD44 and CD73 (Fig. 2).
Differentiation of hUCBMSCs to osteocytes
and adipocytes and their identification
Osteogenic differentiation
One week after induction, the cells became 100%
confluent and aggregated during culture. Clear thickening and
nodosity in regions were observed following 2 weeks, and major red
cell nodules were observed after three weeks following staining
with alizarin red (Fig. 3A).
Adipogenic differentiation
Enlarged cells and tiny lipid droplets were observed
in the hyaloplasm 1 week subsequent to induction. Highly refractive
lipid droplets were formed in the hyaloplasm after 2 weeks.
Following cultivation for 3 weeks, tiny lipid droplets increased in
number and merged to form larger droplets filling the entire cell.
The lipid droplets in the hyaloplasm turned red when stained with
oil red O (Fig. 3B).
Morphological changes following
directionally-induced differentiation from hUCBMSCs to Schwann-like
cells
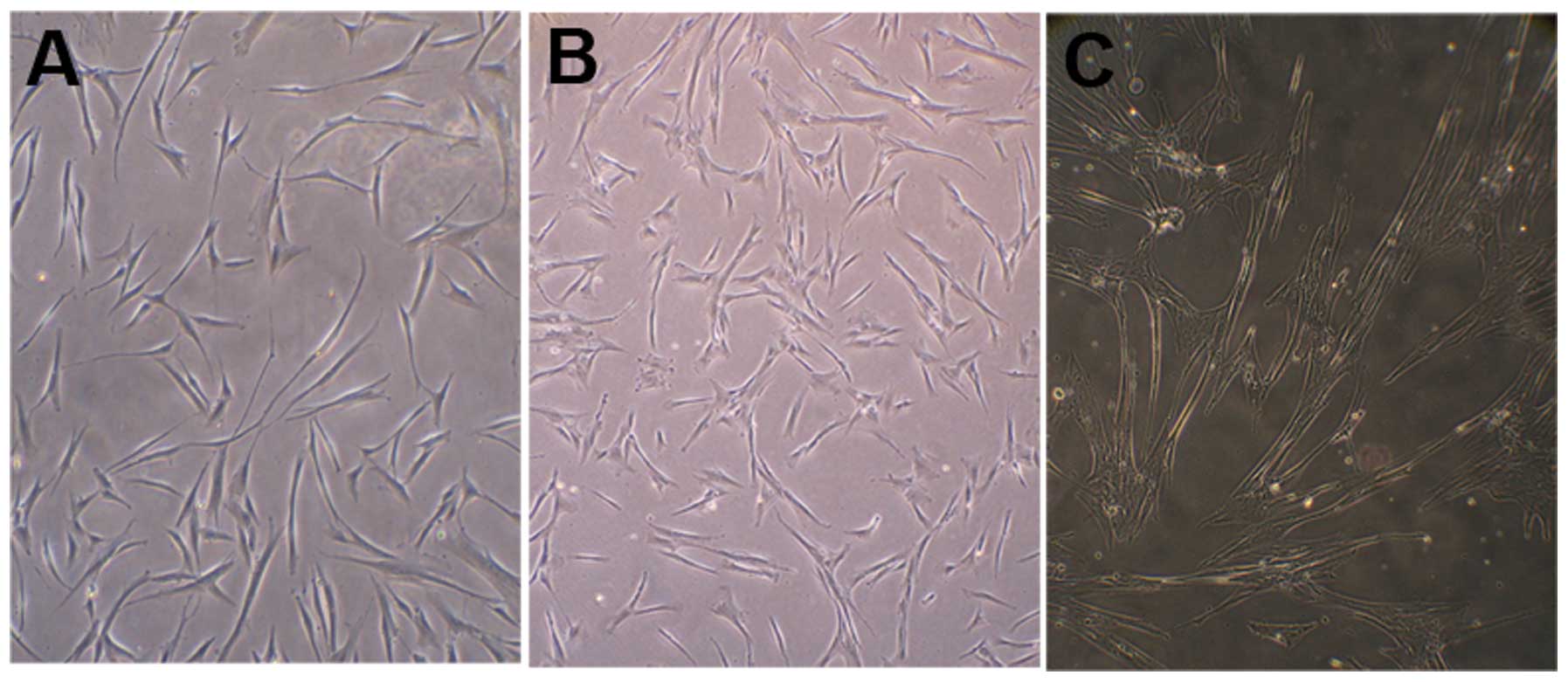
The cell body of the long-fusiform MSCs began
retracting and the edge appearance became irregular following
pre-induction with β-ME and bFGF (Fig.
4A). The edges retracted further after induction with RA and
changed to an irregular conical or triangular shape (Fig. 4B). Following induction with medium
containing FSK, bFGF, PDGF-BB, NGF and HRG, the frequency of the
cell body contractions and the spaces between the cells increased.
Subsequent to induction, the cell morphology altered to a slender
spindle or triangular shape similar to Schwann cells (Fig. 4C).
Schwann-like cells express glial cell
markers
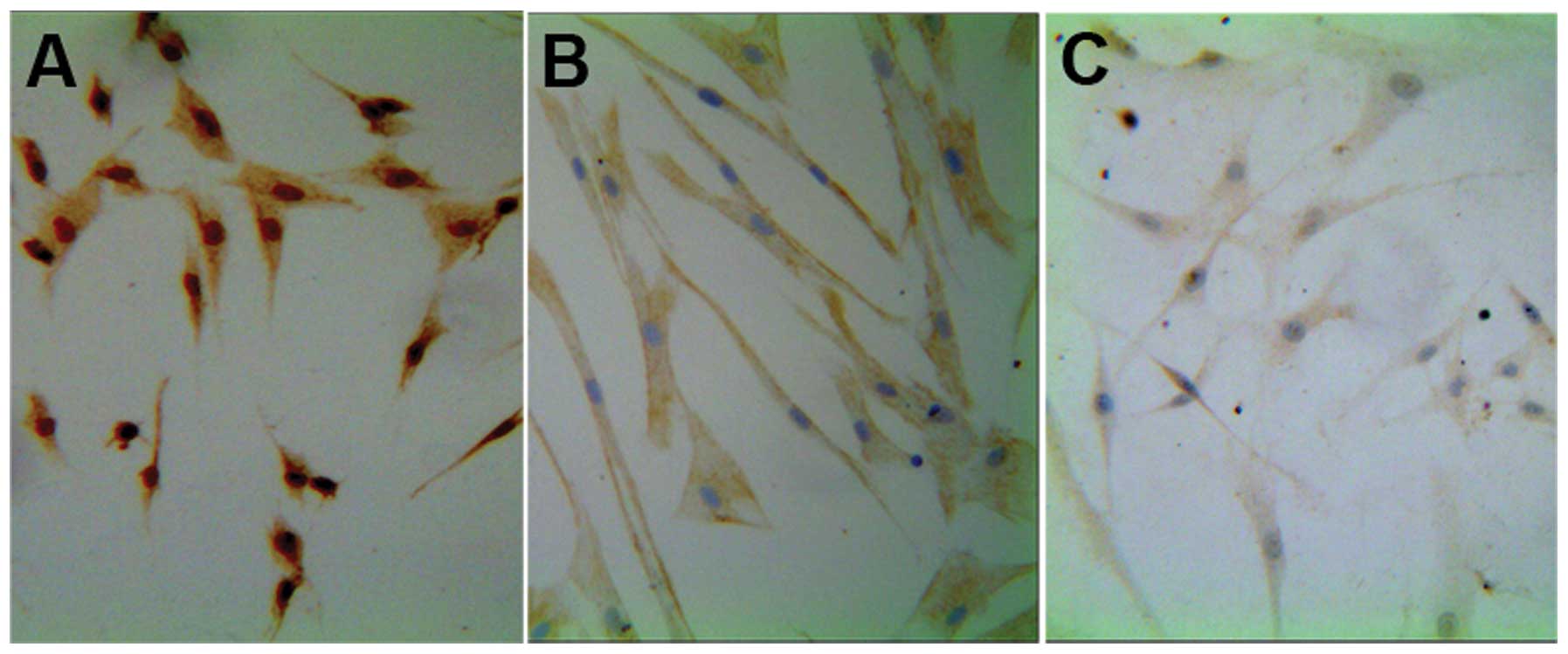
Almost all of the differentiated cells expressed the
glial cell markers S100b, GFAP and P75 (Fig. 5), the majority of which (~70%)
exhibited the classical dipolar fusiform morphology of SCs. Glial
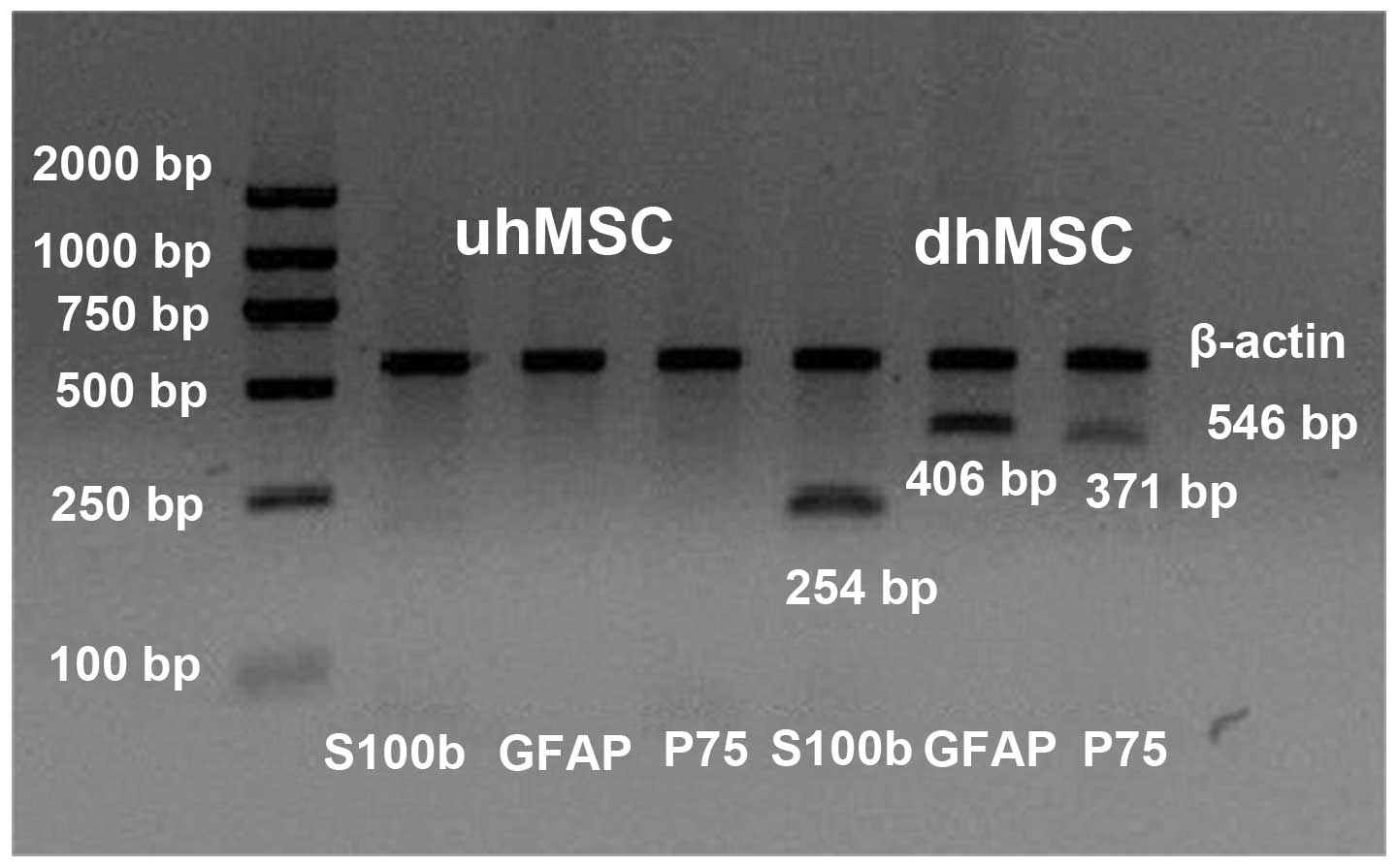
cell marker gene transcripts were detected by RT followed by PCR
(Fig. 6). Differentiated human
(dh) MSCs expressed the transcripts for S100b (254 bp), GFAP (406
bp) and p75 (371 bp) whilst the undifferentiated human (uh) MSCs
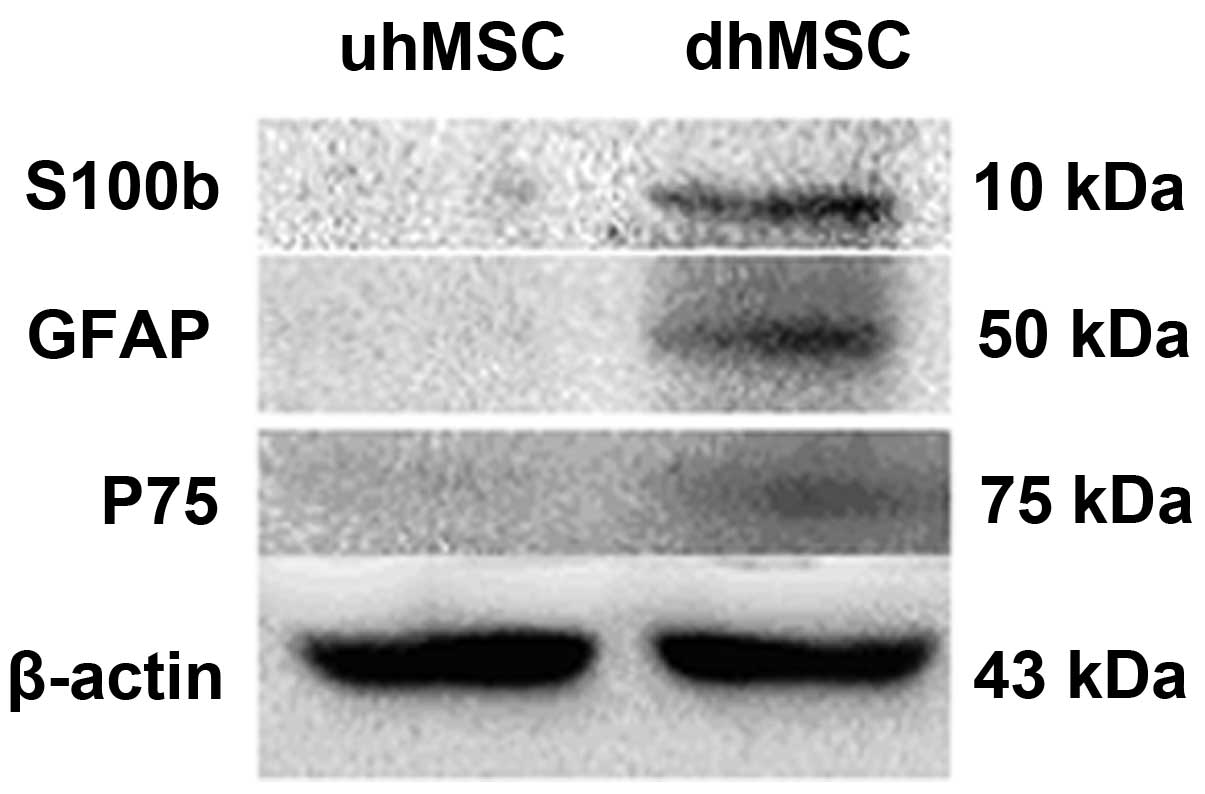
did not. Western blotting (Fig. 7)
indicated positive protein expression for the glial proteins S100b
(10 kDa), GFAP (50 Da) and p75 (75 kDa) in dhMSC, which were not
expressed in uhMSCs.
Discussion
Erices et al (7) described the generation of
fibroblast-like cells upon separation of umbilical cord blood.
These cells expressed the MSC surface antigens SH2, SH3, SH4, ASMA,
MAB1470, CD13, CD29 and CD49e, as do bone marrow MSCs. Lee et
al (8) acquired MSCs following
cord blood separation by gradient centrifugation. A rudimentary
identification and description of these cells was provided, which
further confirmed the presence of MSCs in UCB. In the current
study, the plastic-adherent cells separated from UCB exhibited a
lack of, or reduced expression, of hematopoietic stem/progenitor
cell antigen CD34, and highly expressed the stem cell antigens CD44
and CD73. These results exclude the possibility of the presence of
hematopoietic stem cells. It is possible for the cells to be
differentiated to osteoblasts and adipocytes. According to the
minimal criteria described for MSCs (9), the presence of hUCBMSCs can be
confirmed by the results of the present study.
hUCBMSCs have become a potential substitution source
of human bone marrow MSCs through tissue engineering in artificial
nerve treatment of peripheral nerve defects. These cells have
significant advantages in their clinical application prospects, as
they are able to be induced to differentiate into glial cells. Lee
et al (8) acquired
GFAP-positive cells subsequent to generating hUCBMSCs with a
multistage induction method. Zhang et al (10) then produced Schwann-like cells
through improved methods of inducing hUCBMSC differentiation from
embryonic stem cells. In the present study, hUCBMSC differentiation
to Schwann-like cells was successfully induced, based on the
methods of Dezawa et al (11) and Wang and Liu (4). However, notable modifications were
made from their protocols with regards to the induction process.
Intermittent time was included for 24 h following pre-induction by
β-ME and bFGF, the dosage of β-ME was reduced, and the induction
time with RA was shortened. This approach improved cell viability
and cytoactivity, and also reduced cell death by minimizing damage
from chemical induction and allowing more time for the recovery of
damaged cells. Additionally, the number of typical bipolar fusiform
SCs increased dramatically subsequent to induction with NGF.
Immunocytochemistry analysis suggested that almost
all the differentiated cells expressed glial cell markers, similar
to the result of Tohill et al (12), who induced the differentiation of
rat MSCs. In addition, the present study provided genetic and
molecular evidence that these cells exhibit the characteristics of
SCs, in addition to morphological similarity. The differentiation
protocol of hUCBMSC to SC was investigated based on the preliminary
understanding of differentiation mechanisms. During induction, β-ME
activates cell surface channels or receptors directly, leading to
cell contraction and phenotypic changes, including cytoplasmic
neurite elongation and alteration of cytoskeletal structure within
the cell (13). bFGF can
accelerate the transition from SC precursors to SCs (14). A combination of β-ME and bFGF was
used pre-induction to accelerate the transition from MSCs to SCs in
the preliminary experiments. RA, derived from vitamin A, strongly
induces differentiation, and its biological effect is mediated by
the RA receptor (15). Generally,
RA forms a complex with cell RA binding protein in the cytoplasm
subsequent to entering cells. It then forms a complex with the
chromatin receptor upon entering the nuclear core leading to
alteration of cellular phenotype through regulation of specific
gene expression (15). As potent
mitogens, bFGF and PDGF are able to activate MAP kinase (MAPK) in
MSCs to stimulate DNA synthesis in SCs (14). HRG and neuregulin, which are growth
factors of the same family, are able to encourage SC proliferation.
HRG is regarded as the pivotal signal that controls SC progression
at each stage of the lineage (14,16).
It has been reported (17) that
HRG is able to induce neural crest cell differentiation to SCs by
activating MAPK signaling. FSK is commonly used to activate
adenylate cyclase and elevate cAMP levels in cells (18). Additionally, FSK is able to
increase the expression of growth factor receptors and, when
combined with bPGF, PDGF-BB and HRG, enhances the synergism of SC
differentiation (19).
NGF is a type of neurocyte growth regulatory factor
that has dual biological functions in neuron nutrition and the
promotion of neurite growth, and it is also able to enhance the
metabolism of various types of neurocytes. NGF has the ability to
protect and nourish normal neurocytes after maturation, to maintain
the sensation of the pars affecta and subsistence of the
sympathetic neurons, and to promote axon growth in order to repair
damaged nerve fibers. In addition, NGF is able to promote the
differentiation of neural precursor cells to mature neurons and
glial cells in vitro (20).
When bound to the TrkA receptor, NGF imparts its biological effect
through the ERK/MAPK and PI3K/AKT signal transduction pathways,
initiating a series of reactions that regulate structures on target
cells or gene expression of functional proteins (21). However, co-culturing with DRGs does
not support previous evidence that NGF can upregulate FSK (22). Regardless, in the present study,
the number of induced cells presenting the classical morphology of
SCs increased after adding NGF to the induction medium.
In conclusion, the current study established a
modified method of inducing directional differentiation of hUCBMSCs
to Schwann-like cells, and verified the morphological and
phenotypic similarity between the induced cells with SCs. These
data suggest that HUCBMSCs are able to proliferate substantially
in vitro and also that they exhibit similar surface markers
and a similar multidirectional differentiation potential to bone
marrow MSCs. Future studies should aim to determine the
physiological function of Schwann-like cells in vivo,
providing experimental support for their application in the
clinical repair of peripheral nerve defects through formation of
tissue engineered artificial nerves on scaffold materials.
Acknowledgements
The current study was supported by the Key Medical
Research Project of Department of Health of Anhui Province
(2010A015).
References
|
1
|
Heath CA: Cells for tissue engineering.
Trends Biotechnol. 18:17–19. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brohlin M, Mahay D, Novikov LN, et al:
Characterisation of human mesenchymal stem cells following
differentiation into Schwann cell-like cells. Neurosci Res.
64:41–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimizu S, Kitada M, Ishikawa H, Itokazu
Y, et al: Peripheral nerve regeneration by the in vitro
differentiated-human bone marrow stromal cells with Schwann cell
property. Biochem Biophys Res Commun. 359:915–920. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang J and Liu K: Method of
differentiation of adult human bone marrow mesenchymal stem cell
into Schwann-like cells in vitro. Beijing Da Xue Xue Bao.
35:202–206. 2003.(In Chinese). PubMed/NCBI
|
|
5
|
Sethe S, Scutt A and Stolzing A: Aging of
mesenchymal stem cells. Ageing Res Rev. 5:91–116. 2006. View Article : Google Scholar
|
|
6
|
Wang M, Yang Y, Yang D, et al: The
immunomodulatory activity of human umbilical cord blood-derived
mesenchymal stem cells in vitro. Immunology. 126:220–232. 2009.
View Article : Google Scholar :
|
|
7
|
Erices A, Conget P and Minguell JJ:
Mesenchymal progenitor cells in human umbilical cord blood. Br J
Haematol. 109:235–242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee OK, Kuo TK, Chen WM, et al: Isolation
of multipotent mesenchymal stem cells from umbilical cord blood.
Blood. 103:1669–1675. 2004. View Article : Google Scholar
|
|
9
|
Dominici M, Le Blanc K, Mueller I, et al:
Minimal criteria for defining multipotent mesenchymal stromal
cells. The International Society for Cellular Therapy position
statement. Cytotherapy. 8:315–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang HT, Cheng HY, Zhang L, et al:
Umbilical cord blood cell-derived neurospheres differentiate into
Schwann-like cells. Neuroreport. 20:354–359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dezawa M, Takahashi I, Esaki M, et al:
Sciatic nerve regeneration in rats induced by transplantation of in
vitro differentiated bone-marrow stromal cells. Eur J Neurosci.
14:1771–1776. 2001. View Article : Google Scholar
|
|
12
|
Tohill M, Mantovani C, Wiberg M and
Terenghi G: Rat bone marrow mesenchymal stem cells express glial
markers and stimulate nerve regeneration. Neurosci Lett.
362:200–203. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Croft AP and Przyborski SA: Formation of
neurons by non-neural adult stem cells: potential mechanism
implicates an artifact of growth in culture. Stem Cells.
24:1841–1851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuse D, Kitada M, Kohama M, et al:
Human umbilical cord-derived mesenchymal stromal cells
differentiate into functional Schwann cells that sustain peripheral
nerve regeneration. J Neuropathol Exp Neurol. 69:973–985. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmidt-Mende J, Gogvadze V,
Hellström-Lindberg E and Zhivotovsky B: Early mitochondrial
alterations in ATRA-induced cell death. Cell Death Differ.
13:119–128. 2006. View Article : Google Scholar
|
|
16
|
Xu Y, Liu Z, Liu L, et al: Neurospheres
from rat adipose-derived stem cells could be induced into
functional Schwann cell-like cells in vitro. BMC Neurosci.
9:212008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ogata T, Yamamoto S, Nakamura K and Tanaka
S: Signaling axis in schwann cell proliferation and
differentiation. Mol Neurobiol. 33:51–62. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jang S, Cho HH, Cho YB, et al: Functional
neural differentiation of human adipose tissue-derived stem cells
using bFGF and forskolin. BMC Cell Biol. 11:252010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin W, Chen X, Wang X, et al: Adult rat
bone marrow stromal cells differentiate into Schwann cell-like
cells in vitro. In vitro Cell Dev Biol Anim. 44:31–40. 2008.
View Article : Google Scholar
|
|
20
|
Benoit BO, Savarese T, Joly M, et al:
Neurotrophin channeling of neural progenitor cell differentiation.
J Neurobiol. 46:265–280. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song EJ and Yoo YS: Nerve growth
factor-induced neurite outgrowth is potentiated by stabilization of
TrkA receptors. BMB Rep. 44:182–186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mahay D, Terenghi G and Shawcross SG:
Schwann cell mediated trophic effects by differentiated mesenchymal
stem cells. Exp Cell Res. 314:2692–2701. 2008. View Article : Google Scholar : PubMed/NCBI
|