Introduction
Complex I is the first and largest component of the
mitochondrial respiratory chain. Impairment of complex I accounts
for most cases of respiratory chain deficiency, and results in a
wide range of clinical manifestations (1,2),
including Leigh syndrome (LS) (3).
Complex I deficiency is also the most common cause of LS (4). In LS patients with complex I
deficiency, complex I activity may be reduced up to 66% (4), whereas the activity of other
complexes is normal.
LS is a subacute, necrotizing encephalopathy,
characterized by bilateral symmetrical necrotic lesions of gray
matter nuclei in the basal ganglia, diencephalon, cerebellum, or
brainstem (5). Onset of LS
typically occurs at early infancy. Patients present psychomotor
retardation, seizures, nystagmus, ophthalmoparesis, optic atrophy,
ataxia, dystonia, or respiratory failure, due to the progressive
decline of the central nervous system functions. In most cases, LS
patients die before age 5 (6).
The mutation m.10197 G>A in the mitochondrial
NADH dehydrogenase, subunit 3 (ND3) gene was found to cause
LS. This mutation, causing the typical manifestations of LS, is
considered to lead to a severe phenotype. In this study, we
describe the clinical and molecular features of the first Chinese
LS pedigree with the mutation m.10197 G>A, and compare the
phenotype of these LS patients to those previously described for LS
patients with the same mutation. We also describe follow-up results
of treatment with the coenzyme Q10.
Materials and methods
Pedigree and ethics
The study was approved by the Ethics Committee of
Fujian Medical University Union Hospital. Informed consent was
obtained from all patients included in the study.
A two-generation Han Chinese family living in
southeast China near the Min River with 4 family members was
studied (Fig. 1A). The proband, a
16-year-old girl (40 kg weight, 157 cm height), was admitted in the
Union Hospital in 2012 with slight facial paralysis. She had been
delivered with mild asphyxia, due to a 6-h-long second stage of
labor with an 8-h duration from full dilatation to delivery. She
presented symptoms of anorexia and fatigue following physical
activities. Therefore, she did not pass any physical agility test
at school. With regards to academic performance, she ranked middle
in her class. There was no evidence of mental or psychomotor
retardation. She scored 28 in both the Mini-Mental State
Examination and Montreal Cognitive Assessment. Neurological
examination revealed horizontal nystagmus, slightly shallow left
nasolabial fold, slight hypalgesia in the left low face, hypotonia,
and indications of bilateral Babinski sign. The levels of serum
ceruloplasmin, serum creatine kinase, serum creatine kinase-MB, and
antinuclear antibody were normal. An elevated plasma lactate level
was noted [4.0 mmol/l; normal level (N) <2.7].
Electroencephalogram and electromyography examinations revealed no
abnormality. Brain magnetic resonance imaging (MRI) showed
prolonged T1- and T2-weighted signals in the basal ganglia,
thalamus, midbrain, pons, and medulla oblongata (Fig. 1B–E). Magnetic resonance
spectroscopy showed a markedly increased lactate doublet in the
lesions (Fig. 1F and G). The
pressure, routine, biochemical, cytological, and IgG index
examinations of the cerebrospinal fluid (CSF) were normal, and
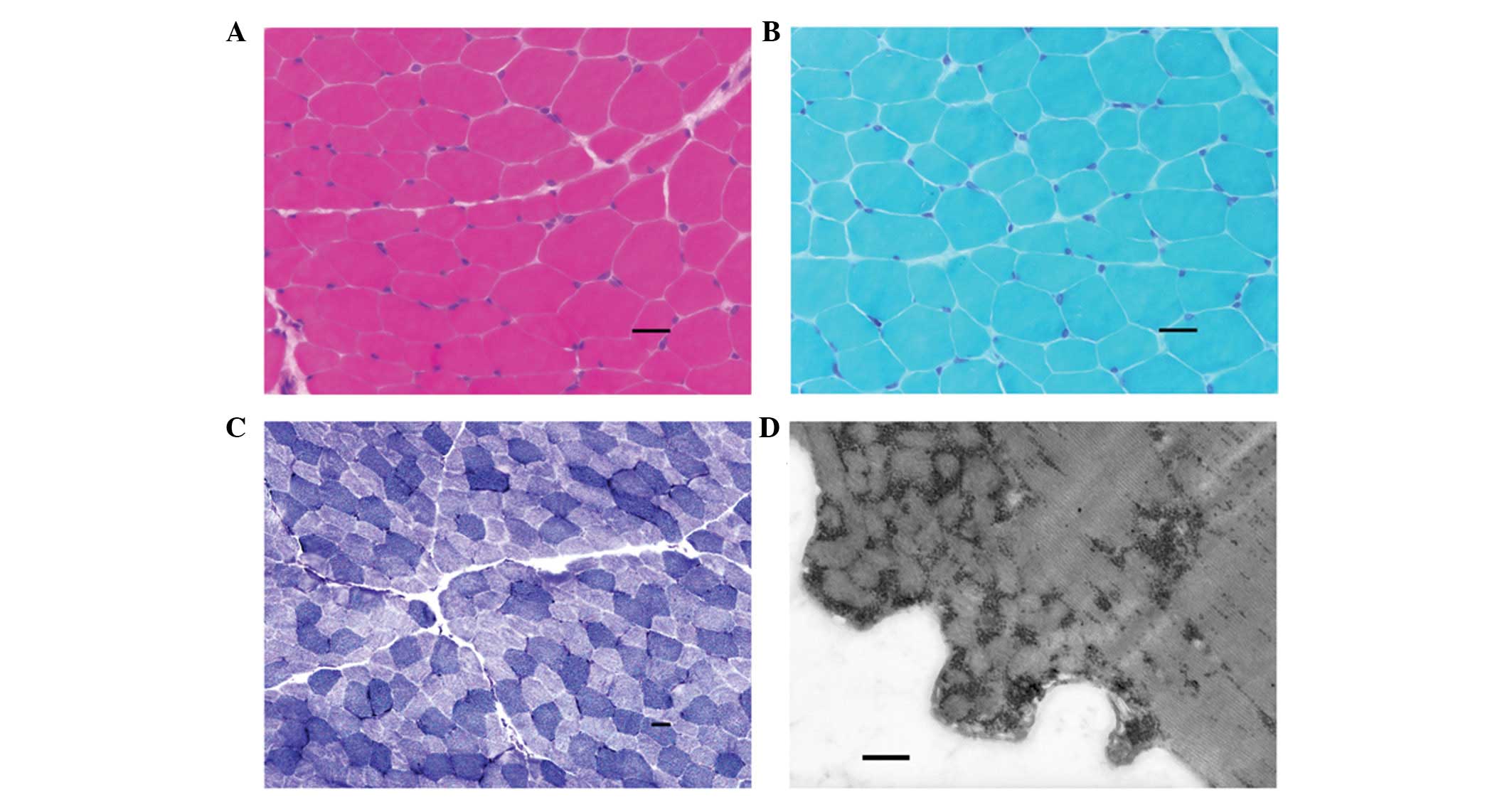
oligoclonal bands were absent. Histochemical examination of a
muscle biopsy specimen stained with hematoxylin and eosin (Fig. 2A), modified Gomori trichrome
(Fig. 2B), nicotinamide adenine
dinucleotide (Fig. 2C), as well as
electron microscopy examination (Fig.
2D) showed no characteristic abnormality. The mother of the
proband, and her 8-year-old brother were normal. There were no
abnormalities in their physical examination and MRI brain imaging.
The proband received treatment with 90 mg/day of the coenzyme
Q10 for a year. Physical examination, MRI brain imaging,
and plasma lactate level assessment were performed at the end of
the treatment.
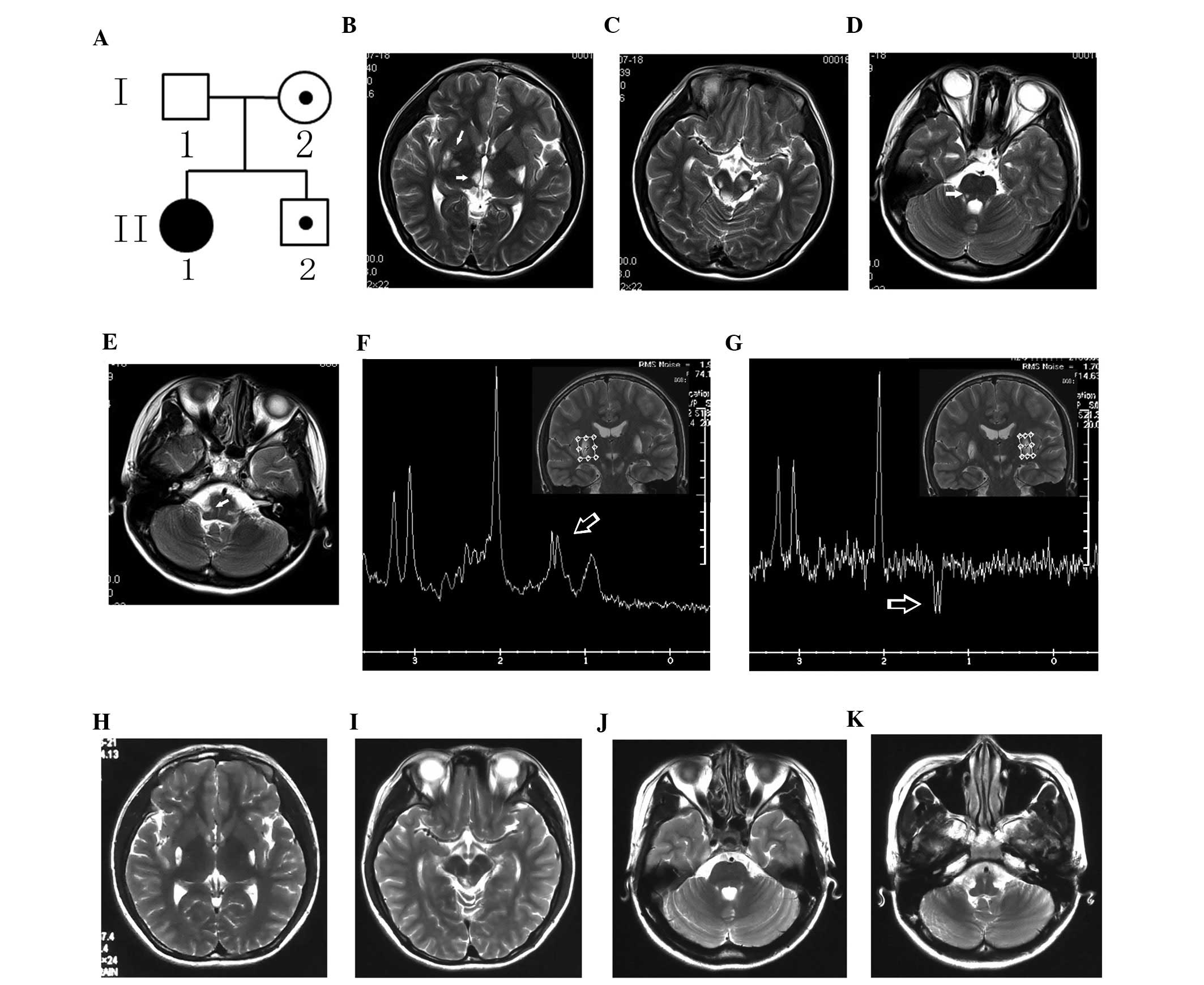 | Figure 1Pedigree chart of the family with the
Leigh syndrome (LS) and neuroimaging of the proband. (A) Pedigree
chart. I and II denote generation number, and 1–2 individual
number; the black circle denotes the proband. (B–E) T2-weighted
images (T2WI) from magnetic resonance imaging (MRI) of the proband
show prolonged signals (arrows) in the (B) basal ganglia and
thalamus, (C) midbrain, (D) pons, and (E) medulla oblongata. (F and
G) As shown in the magnetic resonance spectra, there is a markedly
increased lactate doublet (arrows) in the prolonged signal region
in the bilateral basal ganglia. Following one-year treatment, the
lesions disappeared in the (H) thalamus, (I) bilateral cerebral
peduncle, (J) pons and (K) medulla oblongata, and partially reduced
in the (H) basal ganglia and (I) the dorsal midbrain, as shown by
T2WI. |
Mutational analysis
Genomic DNA was extracted from blood lymphocytes and
skeletal muscle of the pedigree using QIAamp DNA Mini kit (Qiagen,
Dusseldorf, Germany). Full-length mitochondrial DNA (mtDNA) was
amplified by polymerase chain reaction using 24 pads of overlapping
primers (Sangon Biotech, Shanghai, China). PCR was performed in a
total volume of 25 μl, containing 100 ng of genomic DNA, 0.25 M of
each primer, 10 M deoxynucleotide triphosphates, 1.25 U of Taq
enzyme and 2.5 μl of 10X buffer. PCR amplification was performed
using an Applied Biosystems 9700 thermocycler (Life Technologies,
Carlsbad, CA, USA) with an initial denaturation step at 94°C for 5
min followed by 35 cycles of denaturation at 94°C for 30 sec,
annealing at 59°C for 45 sec, and extension at 72°C for 1 min, with
a final extension step at 71°C for 6 min. Sequencing was performed
on an Applied Biosystems 3730 DNA automatic sequencer (Life
Technologies) and resulting sequences were compared to the revised
Cambridge reference sequence (rCRS) of the human mtDNA (7,8).
Quantification of heteroplasmy
The mutant load of m.10197 G>A was determined by
restriction fragment length polymorphism analysis. A 476-bp
fragment was amplified using the fluorescent-labeled FAM forward
primer 5′FAM-TTTGTAGCCACAGGCTTCC-3′ and the reverse primer
5′-AAGGAGGGCAATTTCTAGAT-3′. The fragment was then digested with the
restriction enzyme Cac8I (restriction site: GCN*NGC). This
restriction site is present only in the wild-type mtDNA, and is
thus expected to give 402- and 74-bp products upon wild-type mtDNA
digestion. The digested fragments were subjected to electrophoresis
and DNA sequencing, and the fluorescent fragments were detected
using the Applied Biosystems GeneMapper® software V3.3
(Life Technologies). Heteroplasmy or mutant load was calculated by
dividing the 476-bp peak area by the sum of the 476- and 402-bp
peak areas.
Haplogroup identification in the
pedigree
MtDNA haplogroup analysis was performed by
full-length mitochondrial DNA sequencing, using the same method
described above for mutational analysis. The results were confirmed
by referring to Kivisild’s study (9).
Results
Mutational analysis
Direct sequence analysis revealed our proband with
the m.10197 G>A mutation in the ND3 gene (Fig. 3A). This mutation was also found in
the proband’s mother and young brother, with the exception of the
father. Previously reported common mutations such as T8993C,
T9176G, and A3243G were not found (10). The mutation m.10197 G>A is a
substitution at codon 47 of the ND3 gene, causing an
alanine-to-threonine amino acid change. This mutation has been
reported (11,12), and based on Ensembl (www.ensembl.org), was not a polymorphism.
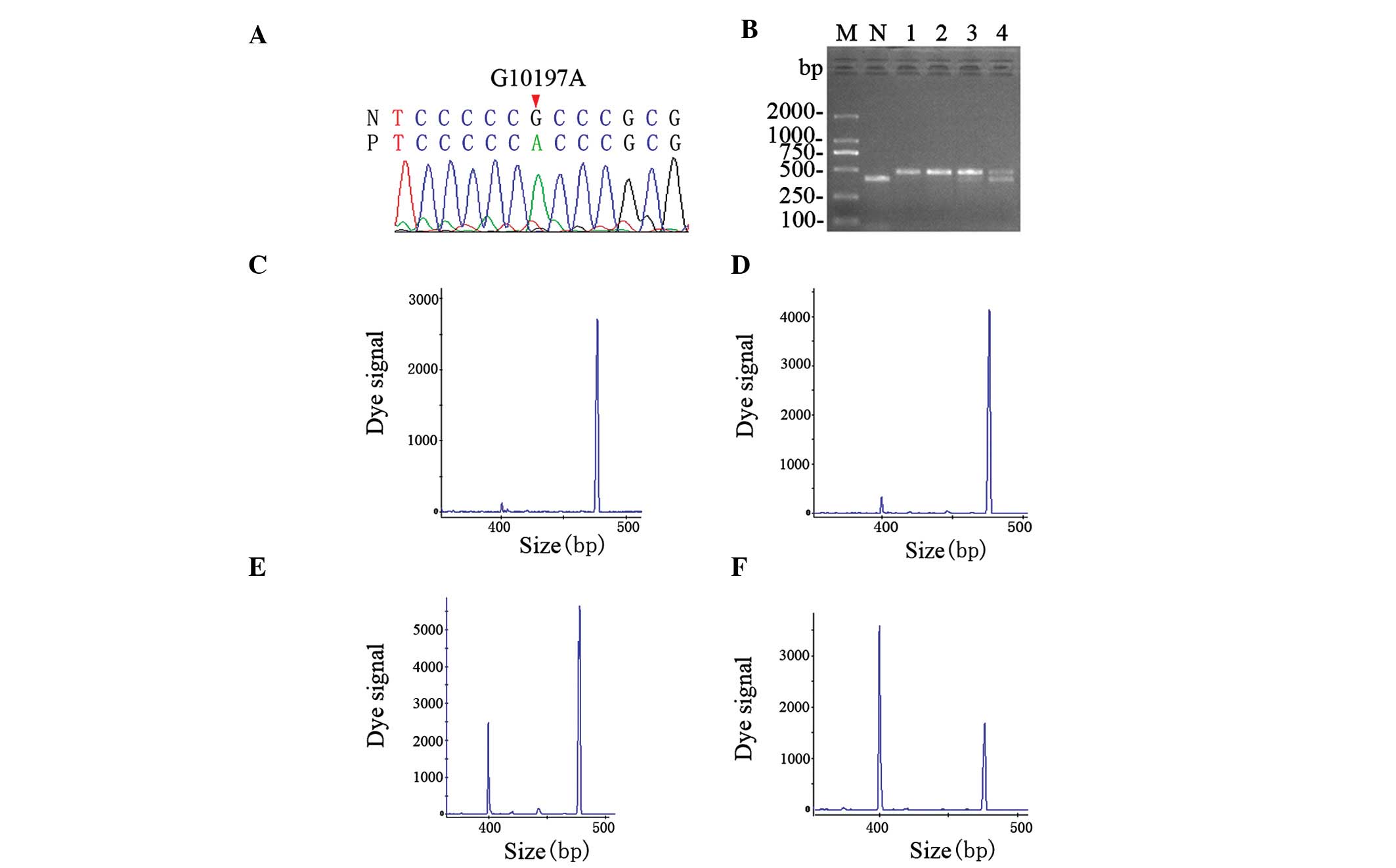 | Figure 3Identification of the mutation and
analysis of heteroplasmy. (A) The mutation m.10197 G>A (red
arrow) in the NADH dehydrogenase, subunit 3 (ND3) gene as
shown in the sequencing chromatogram. (B) Gel image of digestion.
Only the wild-type mtDNA sequence could be digested into two
fragments of 402 and 74 bp. The latter was absent in the gel. The
proband and the proband’s brother contained a higher proportion of
mutant sequence than their mother. M, DNA ladder; N, normal
individual; lane 1, proband skeletal muscle DNA; lane 2, proband
leukocyte DNA; lane 3, leukocyte DNA of proband’s brother; lane 4,
leukocyte DNA of proband’s mother. (C–F) Quantification of
heteroplasmy. The digested segments were separated by sequencer and
heteroplasmy was calculated by dividing the 476-bp peak area by the
sum of the 476- and 402-bp peak area. The proportion of mutant
mtDNA of the (C) proband’s muscle, (D) proband’s leukocytes, (E)
leukocytes of the proband’s mother and (F) leukocytes of the
proband’s brother were 97, 95, 39 and 80%, respectively. |
Quantification of heteroplasmy
Digestion of the mtDNA with the restriction enzyme
Cac8I and electrophoresis (Fig.
3B) showed that the mutant load of the proband and her brother
are higher than that of their mother. Quantification of the dye
signal showed that the proband is heteroplasmic with 97% mutant
load in the skeletal muscle (Fig.
3C), and 95% in the leukocytes (Fig. 3D). Leukocytes of her mother had 39%
mutant load (Fig. 2E) and those of
her brother 80% (Fig. 2F).
Haplogroup identification in the
pedigree
By analysis of direct sequence, the mtDNA haplogroup
for our pedigree was determined. The proband, her mother and young
brother belong to the mitochondrial haplogroup N9a.
Following results of treatment with
coenzyme Q10
After a 3-month treatment with the coenzyme
Q10, neurological examination of the proband revealed no
marked alteration. However, the proband’s exercise tolerance and
anorexia were markedly improved. Her body weight increased by 10
kg, and she passed all the physical agility tests. One year later,
the proband maintained this status. Reexamination by brain MRI
demonstrated that certain lesions detected one year earlier had
disappeared (Fig. 1H–K). The
plasma lactate level was decreased to a normal level (2.5 mmol/l,
N<2.7).
Discussion
LS is a severe, progressive, metabolic
neurodegenerative, mitochondrial disorder. The molecular defects
and pathogenetic background of LS would provide considerable
information to the patients in terms of management, genetic
counseling, and prognosis. In this study, we described the clinical
and molecular features of the first Chinese LS pedigree with the
m.10197 G>A mutation, which leads to a severe phenotype, and
report promising effects of treatment with the coenzyme
Q10, as assessed by both clinical feature assessment and
brain imaging.
The mutation m.10197 G>A was first reported in
2007 in 3 French pedigrees, as present in totally 8 LS patients
(11). As shown in Table I, patients generally showed
early-onset LS with a severe clinical outcome. Seventy-five percent
of the patients died 1 year after the onset. A number of studies
have provided evidence for the pathogenic role of the mutation
m.10197 G>A. The mutation involves the codon 47 of the
ND3 gene. This codon encodes alanine, a highly conserved
residue in higher eukaryotes such as Homo sapiens, Bos
Taurus, Equus caballus, Mus musculus, and
Gallus gallus (Fig. 4). The
conservation index of the mutation m.10197 G>A based on 39
species (conservation in 36 species except Xenopus laevis,
Drosophila melanogaster and Sciurus vulgaris) is
92.3% (11). This value reached to
the average level (93±13%) of conservation index of 22
well-characterized human pathogenic mtDNA mutation studied in the
same 39 species (12). The
mutation identified in the proband is a substitution from an
alanine to a threonine. These two residues are quite different in
structure and chemical property. Alanine is a hydrophobic amino
acid without any side-chain. By contrast, threonine is a
hydrophilic amino acid with a hydroxyl side-chain. These
differences in amino acid properties may underlie the pathogenic
role of the m.10197 G>A mutation in causing complex I deficiency
and a severe LS phenotype.
 | Table IClinical features of Leigh syndrome
patients with the m.10197 G>A mutation, summary of present and
literature data. Information on the patient’s mother in each
pedigree is not shown. |
Table I
Clinical features of Leigh syndrome
patients with the m.10197 G>A mutation, summary of present and
literature data. Information on the patient’s mother in each
pedigree is not shown.
| Country (Refs.) | Patient no. | Gender | Age at onset | Manifestation | Mutant load (%) | Duration/Death
age | Cerebral imaging | Lactate level
(mM) | Muscle biopsy |
|---|
| France (11) | 1 | Ma | 1 m | Growth retardation,
hypotonia, psychomotor regression, seizures | 100 (muscle) | 4 m/5 m | Brainstem | CSF: 3.5 | NA |
| 2 | Ma | 2 m | Anorexia, nystagmus,
hypotonia | 100 (liver) | 0 m/2 m | Lentiform nuclei | Blood: 4.27
(N<2.8) | NA |
| 3 | Fe | 1 m | Hypotonia, growth
retardation, liver enlargement and slight muscle atrophy | 100 (muscle) | 7 m/8 m | NA | Blood: 2.8 | NA |
| 4 | Ma | 4 m | Trunk hypotonia,
pyramidal syndrome, psychomotor retardation, myoclonic
epilepsy | NA | 7 m/11 m | Lentiform nuclei,
thalamus and red nuclei | Blood: 4.6
CSF: 2.1 | NA |
| 5 | Ma | 5 m | Trunk hypotonia,
peripheral hypertonia, plagiocephalia, severe psychomotor delay,
strabismus, epilepsy, dystonia, pyramidal syndrome | 100 (muscle) | 5 y/No | Basal ganglia | Blood: N
CSF: 2.35 | NA |
| 6 | Fe | 5 m | Seizures and
developmental delay | 100
(leukocytes) | 5 y/No | NA | NA | NA |
| 7 | Ma | NA | Severe progressive
encephalopathy, | NA | NA/2 y | NA | Blood: N | NA |
| 8 | Ma | 5 m | Motor milestones,
hypotonia, seizures | 100 (muscle) | 6 m/11 m | Basal ganglia,
thalamus and brain stems | Blood: N | N |
| Korea (12) | 1 | Fe | 7 y | Right-hand
weakness, emotional lability, progressive gait ataxia, dystonia,
poor coordination, dysarthria, cerebellar ataxia | 98 (muscle) | NA/No | Globus
pallidus | Blood: 1.5
(N<2.5) | Moderate SPM |
| 2 | Ma | 4 y | Gait abnormality,
dysarthria, impaired fine motor coordination, ataxic, dystonia | 86 (muscle) | NA/No | Globus
pallidus | Blood: 2.5 | Mild SPM |
| China (present
study) | Proband | Fe | 16 y | Slight facial
paralysis, dystonia | 97
(muscle)
95 (leukocytes) | 1 y/No | Basal ganglia,
thalamus, midbrain, pons and medulla oblongata | Blood: 4
(N<2.7) | N |
It is notable that the individuals carrying the
mutation in our study did not show the typical clinical
manifestations, as previously reported (9,14).
Certain common hypotheses postulated to explain the phenotypic
variability of mtDNA diseases can be excluded based on the results
of the Chinese pedigree reported in our study. First, the proband
and her brother both presented a rather high mutant load, which has
been commonly considered the most important factor affecting the
severity of the disease (14).
Thus, it is unlikely that the phenotypic variability of the patient
may be due to mutant load. Second, the mutation was invariably
present in both the blood and muscle tissues of the proband. Thus,
it is unlikely that the mild clinical manifestation of the proband
might be due to a different tissue distribution of the mutation.
Third, the severe pathogenicity associated with certain mutations
causing LS is expected to affect the early development of the
affected individual. In Leshinsky-Silver’s study, brain lesions
could be generated even at the fetal stage, before clinical
manifestations of the syndrome become apparent (14). However, the normal brain image of
the proband’s brother suggests that this mutation may not generate
visible anatomical changes at the early stage.
Additional factors potentially contributing to the
variability of phenotypes of mtDNA diseases include ethnicity,
haplogroup and the environment. For instance, in the Leber
hereditary optic neuropathy also caused by a mutation, the risk of
visual loss is significantly increased in the haplogroups J2 in
Europeans and M7b1’2 in Han Chinese, in contrast to the respective
H and M8a haplogroups (16,17).
As Table I shows, the severity of
the disease was different between European patients and Asian ones,
although they carried the same mutation m.10197G>A. European
patients had an earlier onset than Asian ones (3.3±1.9 month v.s.
9±6.2 year) and the former progressed so rapidly that 75% of them
succumbed to the disease within 1 year. The haplogroup of
previously reported LS patients presented in Table I is regrettably not available. The
Chinese pedigree presented herein was characterized as haplogroup
N9a, which is not a common haplogroup in European populations.
Individuals of the N9a haplogroup were reported to be resistant or
protected against certain metabolic diseases such as type 2
diabetes (18,19) and metabolic syndrome (20). Thus, it is likely that N9a has an
additive effect to the insusceptibility to LS. It is notable that
another Han Chinese pedigree with the mutation m.10197 G>A, but
in a different genetic background (haplogroup D4b) presented a
different phenotype, of Leber hereditary optic neuropathy and
dystonia (LDYT) (21).
The prognosis of LS is generally poor, and no
effective treatment is available for this syndrome. The coenzyme
Q10 has a crucial role as the electron acceptor in
complexes I and II of the mitochondrial electron transport chain.
Coenzyme Q10 affects the apoptosis of mitochondria
though a number of mechanisms, including interference with
mitochondrial depolarization (22), protection against reactive oxygen
species (23), intervention in the
production of ceramide (24), and
mitochondrial protein uncoupling (25). Q10 is the most common drug used for
treatment of mitochondrial diseases. In the present study, the
condition of the patient was not serious. However, the typical
brain lesions, particularly lower brainstem lesions, indicated that
the LS patient would suffer from severe brainstem dysfunction in
the near future (26). Low-dose
coenzyme Q10 (2 mg/kg/day) treatment was applied on the
proband as an experimental treatment, although patients with LS
have rarely shown an improvement following administration of
Q10 (27).
Surprisingly, the patient showed a clear improvement in both
clinical manifestations and the brain image, while its plasma
lactate level also decreased down to a normal level. This is thus
the first report on successful treatment of an LS patient with
mutation m.10197 G>A. It is likely that the coenzyme
Q10 may attenuate the mitochondrial dysfunctions caused
by the m.10197 G>A mutation.
In conclusion, the present study reported the first
Chinese LS pedigree with mutation m.10197 G>A, and discussed a
number of factors potentially affecting the pathogenic potential of
the mutation m.10197 G>A and contributing to the phenotypic
variability of LS, such as molecular defects, ethnicity, haplogroup
and environment. Our study further indicated that treatment with
the coenzyme Q10 might be useful for LS patients with
the mutation m.10197 G>A.
References
|
1
|
Kirby DM, Crawford M, Cleary MA, et al:
Respiratory chain complex I deficiency: an under diagnosed energy
generation disorder. Neurology. 52:1255–1264. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Distelmaier F, Koopman WJ, van den Heuvel
LP, et al: Mitochondrial complex I deficiency: from organelle
dysfunction to clinical disease. Brain. 132:833–842. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morris AA, Leonard JV, Brown GK, et al:
Deficiency of respiratory chain complex I is a common cause of
Leigh disease. Ann Neurol. 40:25–30. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taylor RW, Morris AA, Hutchinson M and
Turnbull DM: Leigh disease associated with a novel mitochondrial
DNA ND5 mutation. Eur J Hum Genet. 10:141–144. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leigh D: Subacute necrotizing
encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry.
14:216–221. 1951. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bénit P, Slama A, Cartault F, et al:
Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh
syndrome. J Med Genet. 41:14–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anderson S, Bankier AT, Barrell BG, et al:
Sequence and organization of the human mitochondrial genome.
Nature. 290:457–465. 1981. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andrews RM, Kubacka I, Chinnery PF, et al:
Reanalysis and revision of the Cambridge reference sequence for
human mitochondrial DNA. Nat Genet. 23:1471999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kivisild T, Tolk HV, Parik J, et al: The
emerging limbs and twigs of the East Asian mtDNA tree. Mol Biol
Evol. 19:1737–1751. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Finsterer J: Leigh and Leigh-like syndrome
in children and adults. Pediatr Neurol. 39:223–235. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sarzi E, Brown MD, Lebon S, et al: A novel
recurrent mitochondrial DNA mutation in ND3 gene is associated with
isolated complex I deficiency causing Leigh syndrome and dystonia.
Am J Med Genet A. 143:33–41. 2007. View Article : Google Scholar
|
|
12
|
Chae JH, Lee JS, Kim KJ, et al: A novel
ND3 mitochondrial DNA mutation in three Korean children with basal
ganglia lesions and complex I deficiency. Pediatr Res. 61:622–624.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruiz-Pesini E, Mishmar D, Brandon M, et
al: Effects of purifying and adaptive selection on regional
variation in human mtDNA. Science. 303:223–226. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White SL, Collins VR, Wolfe R, et al:
Genetic counseling and prenatal diagnosis for the mitochondrial DNA
mutations at nucleotide 8993. Am J Hum Genet. 62:474–482. 1999.
View Article : Google Scholar
|
|
15
|
Leshinsky-Silver E, Lev D, Malinger G, et
al: Leigh disease presenting in utero due to a novel missense
mutation in the mitochondrial DNA-ND3. Mol Genet Metab. 100:65–70.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hudson G, Carelli V, Spruijt L, et al:
Clinical expression of Leber hereditary optic neuropathy is
affected by the mitochondrial DNA-haplogroup background. Am J Hum
Genet. 81:228–233. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ji Y, Zhang AM, Jia X, et al:
Mitochondrial DNA haplogroups M7b1′2 and M8a affect clinical
expression of leber hereditary optic neuropathy in Chinese families
with the m.11778G-->a mutation. Am J Hum Genet. 83:760–768.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hwang S, Kwak SH, Bhak J, et al: Gene
expression pattern in transmitochondrial cytoplasmic hybrid cells
harboring type 2 diabetes-associated mitochondrial DNA haplogroups.
PLoS One. 6:e221162011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fuku N, Park KS, Yamada Y, et al:
Mitochondrial haplogroup N9a confers resistance against type 2
diabetes in Asians. Am J Hum Genet. 80:407–415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka M, Fuku N, Nishigaki Y, et al:
Women with mitochondrial haplogroup N9a are protected against
metabolic syndrome. Diabetes. 56:518–521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang K, Takahashi Y, Gao ZL, et al:
Mitochondrial ND3 as the novel causative gene for Leber hereditary
optic neuropathy and dystonia. Neurogenetics. 10:337–345. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Papucci L, Schiavone N, Witort E, et al:
Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial
depolarization independently of its free radical scavenging
property. J Biol Chem. 278:28220–28228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishii N, Senoo-Matsuda N, Miyake K, et al:
Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative
stress. Mech Ageing Dev. 125:41–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Navas P, Fernandez-Ayala DM, Martin SF, et
al: Ceramide-dependent caspase 3 activation is prevented by
coenzyme Q from plasma membrane in serum-deprived cells. Free Radic
Res. 36:369–374. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Echtay KS, Winkler E and Klingenberg M:
Coenzyme Q is an obligatory cofactor for uncoupling protein
function. Nature. 408:609–613. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ostergaard E, Hansen FJ, Sorensen N, et
al: Mitochondrial encephalomyopathy with elevated methylmalonic
acid is caused by SUCLA2 mutations. Brain. 130:853–861. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Piao YS, Tang GC, Yang H and Lu DH:
Clinico-neuropathological study of a Chinese case of familial adult
Leigh syndrome. Neuropathology. 26:218–221. 2000. View Article : Google Scholar
|