Introduction
Pearson marrow-pancreas syndrome (PS; MIM #557000),
was initially described in 1979, and is a progressive multi-organ
disorder caused by deletions and duplications of mitochondrial DNA
(mtDNA) (1). The presenting
symptoms include hematological features, and/or growth retardation,
secondary to pancreatic exocrine dysfunction (1). The onset of PS generally occurs in
early infancy. The hematological features include macrocytic
sideroblastic anemia and vacuolization of bone marrow (BM)
precursors, which are sometimes associated with neutropenia or
thrombocytopenia (1). Other
symptoms include tubulopathy and aminoaciduria, hepatomegaly,
cytolysis and cholestasis, endocrine gland disturbances,
neuromuscular manifestations, cardiac involvement or splenic
atrophy, which may develop simultaneously or during the course of
the disease (2). PS is often fatal
in infancy, and the majority of the reported patients with PS
succumb to the disease before reaching three-years-of-age, due to
septicemia, metabolic acidosis or hepatocellular insufficiency
(2). Previous studies have
demonstrated that surviving infants exhibit hematological
improvement; however, they were eventually shown to develop the
typical features of Kearns-Sayre syndrome, which is characterized
by a triad of symptoms: Progressive external ophthalmoplegia,
pigmentary degeneration of the retina and cardiomyopathy (3–5).
Numerous studies have identified genetic defects associated with
PS, which include the presence of the common 4.977 kb deletion,
de novo large-scale deletions or rare duplications in the
mitochondrial genome (6–8), which lead to impaired production of
ATP (9).
The present report describes the case of a
four-month-old infant who presented with the clinical features of
PS. Molecular analysis identified a novel 5.712 kb deletion between
nucleotides 8,011 and 13,722 in mtDNA.
Case report
Patient and clinical findings
The female patient was the first-born child of
healthy non-consanguineous Korean parents. She was born following
an uncomplicated pregnancy and was delivered at term (40+4 weeks).
Her birth weight was 2,400 g and the first months of her life were
uneventful. Family members typically did not show signs and
symptoms associated with PS. At the age of four months, the patient
visited Seoul St. Mary’s Hospital (Seoul, South Korea) presenting
with severe pallor. Mucocutaneous pallor was observed upon physical
examination. Physical and psychomotor development was shown to be
within the normal range and no clinical signs of neuromuscular
disease were present. Blood examination detected severe normocytic
normochromic anemia. Complete blood count data were as follows:
White blood cell count, 4,390/mm3; hemoglobin, 5.0 g/dl;
platelets, 180,000/mm3; mean corpuscular volume, 95.7
fl; mean corpuscular hemoglobin concentration, 33.1%; and
reticulocytes 0.79%. The results of other routine laboratory tests,
including blood glucose, pH, blood gas analysis, electrolytes,
blood urea nitrogen, creatinine, aspartate transaminase, alanine
transaminase, creatine kinase, lactate dehydrogenase, prothrombin
time, activated partial thromboplastin time and urinalysis were all
within the normal ranges. No viral pathogens were detected, in
particular, infections with parvovirus B19, Epstein-Barr virus or
cytomegalovirus were excluded. A BM aspirate was performed at 4.5
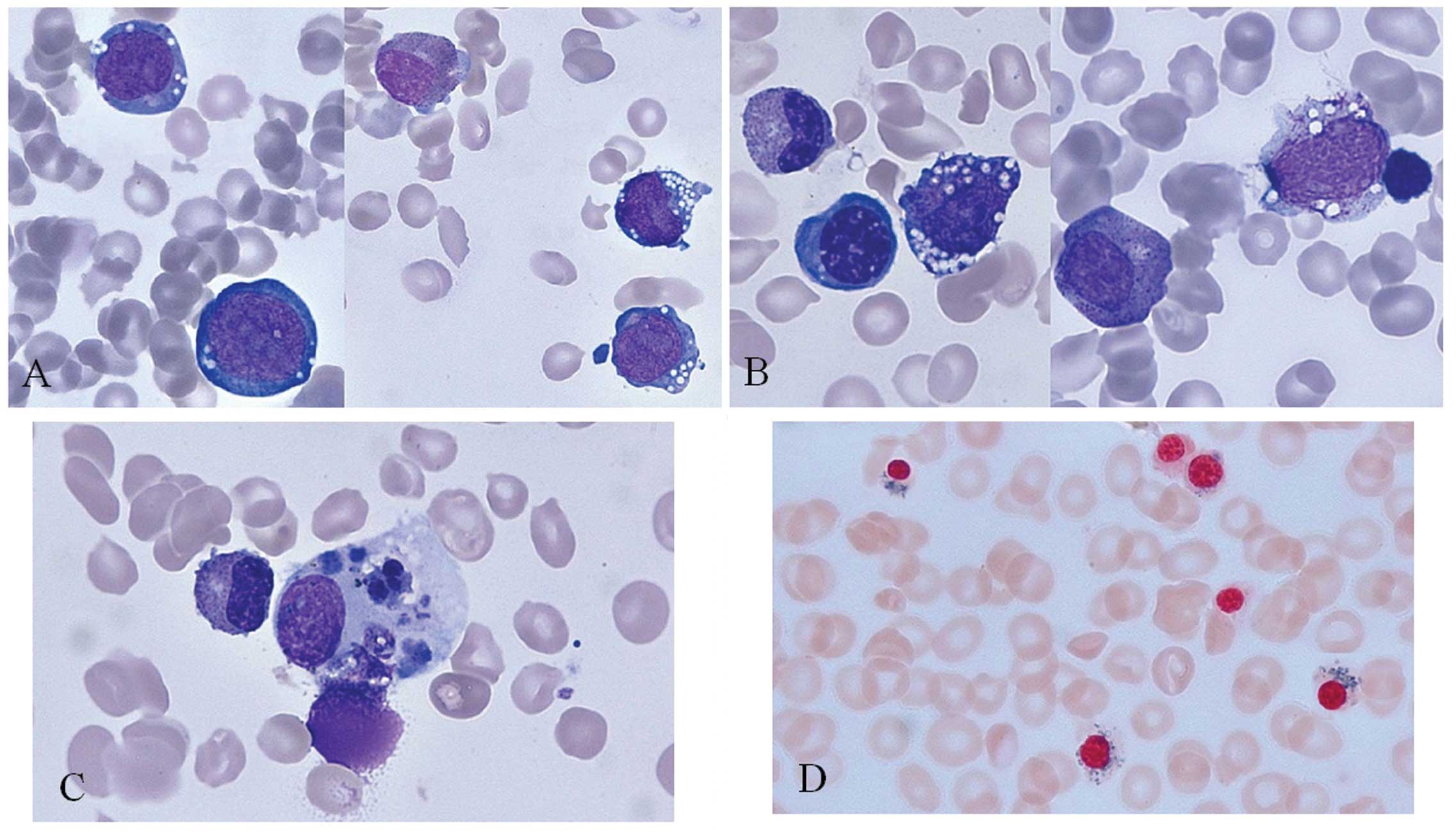
months. Erythroid and myeloid precursors exhibited numerous
cytoplasmic vacuoles (Fig. 1A and
B). Histiocytes exhibited erythrophagocytosis (Fig. 1C) and ringed sideroblasts were
observed following Prussian blue staining (Fig. 1D). Karyotyping and a chromosome
instability assay revealed normal results. Echocardiography
demonstrated normal ventricular systolic and diastolic function,
without any definite structural anomalies. Abdominal
ultrasonography demonstrated mild pelviectasia in both kidneys.
Brain magnetic resonance imaging detected no abnormal signal in the
brain parenchyma.
At the age of 23 months, biochemical analyses
detected metabolic acidosis with a high pyruvic acid concentration
of 1.07 mg/dl (reference range 0.3–0.9). A blood organic acid
profile revealed increased 3-hydroxybutyric acid (512.3 μmol/l;
reference range, 22–213 μmol/l) and palmitic acid (267.9 μmol/l;
reference range, 27–222 μmol/l) levels. A urinary organic acid
profile revealed an elevated excretion of 3-hydroxybutyric acid
(224.7 mmol/mol creatinine; normal <62.6 mmol/mol),
3-methylglutaconic acid (61.3 mmol/mol creatinine; normal <6.0
mmol/mol), 3-hydroxyglutaric acid (11.6 mmol/mol creatinine;
normal: Not detected), 4-hydroxyphenylacetic acid (105.7 mmol/mol
creatinine; normal <69.9 mmol/mol), and 4-hydroxyphenyllactic
acid (73.3 mmol/mol creatinine; normal <19.6 mmol/mol). These
clinical features indicated the presence of a mitochondrial
disease, such as PS.
mtDNA deletion analysis
A molecular analysis was performed in order to
confirm the diagnosis of PS. Written informed consent was obtained
and mtDNA was extracted from the peripheral blood of the patient
using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany).
Amplification of mtDNA was performed using long-distance polymerase
chain reaction (PCR) (Expand Long Template PCR system; Roche
Diagnostics, Basel, Switzerland) with various combinations of
primers, as previously described (10) (Table
I). Direct sequencing of the PCR products was performed using
the BigDye® Terminator v3.1 Cycle Sequencing kit
(Applied Biosystems Life Technologies, Foster City, CA, USA) and
the products were resolved on an ABI PRISM 3130 Genetic analyzer
(Applied Biosystems). Following long-distance PCR amplification
using M12F and M20R primers a large-scale mtDNA deletion was
detected in the patient, as compared with the normal control
(Fig. 2A). Sanger sequencing
identified the breakpoints of the 5.712 kb deletion, and the
deletion was shown to span between the nucleotide positions 8,011
and 13,722 of mtDNA (Fig. 2B). No
perfect or imperfect repeats were detected at the boundaries of the
mtDNA deletion (Fig. 2C). This
heteroplasmic deletion removes part of the COXII gene, the
entire tRNALys, ATPase 8, ATPase
6, COXIII, tRNAGly, ND3,
tRNAArg, ND4L, ND4,
tRNAHis,
tRNASer(AGY),
tRNALeu(CUN) genes and part of the
ND5 gene (Fig. 3). The
sequence homology in the 49 nt region (24 nt on each side of the
breakpoint nucleotide) of the proximal and distal sequences was
analyzed using Clustal Omega software (11), with default parameters (http://www.ebi.ac.uk/Tools/msa/clustalo/), and was
compared with the updated consensus Cambridge sequence (GenBank
Accession no. NC_012920.1).
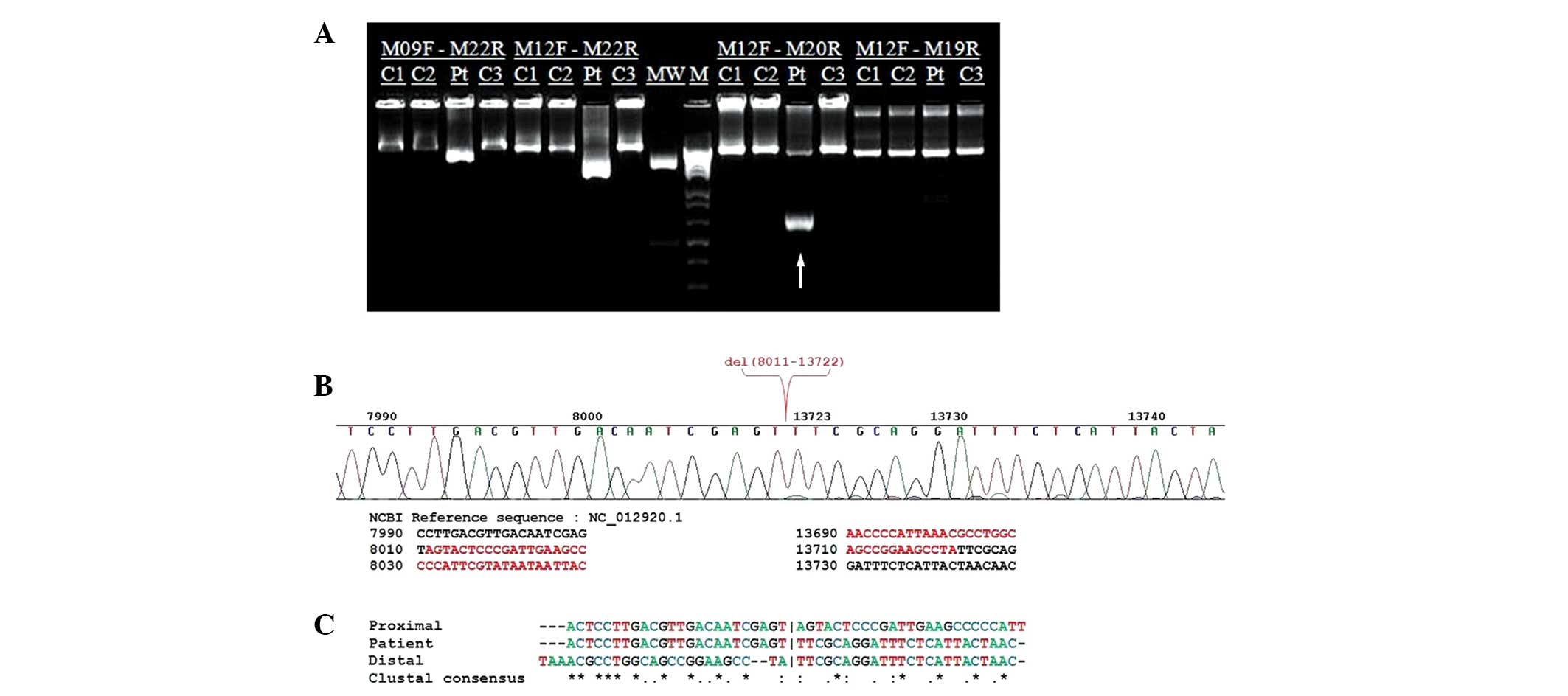 | Figure 2Molecular genetic analysis of the
mitochondrial (mt)DNA in a patient with Pearson syndrome (PS). (A)
Long-distance polymerase chain reaction amplification with M12F and
M20R primers detected the presence of a large-scale mitochondrial
deletion (arrow) in the Korean patient with PS. C1, normal control
1; C2, normal control 2; Pt, patient; C3, normal control 3; MW,
GeneRuler™ 1 kb DNA Ladder, 250–10,000 bp; M, molecular DNA ladder
100 bp. (B) Electropherogram from Sanger DNA sequencing,
representing the breakpoints of the 5.712 kb deletion spanning
nucleotides 8,011–13,722 of mtDNA. (C) Sequence homology analysis
of the 49 nucleotides surrounding the deletion junction revealed no
perfect (class I) or imperfect (class II) repeats at the deletion
boundaries. No base pair repeats were detected (class III).
Vertical bars (|) indicate the breakpoints. |
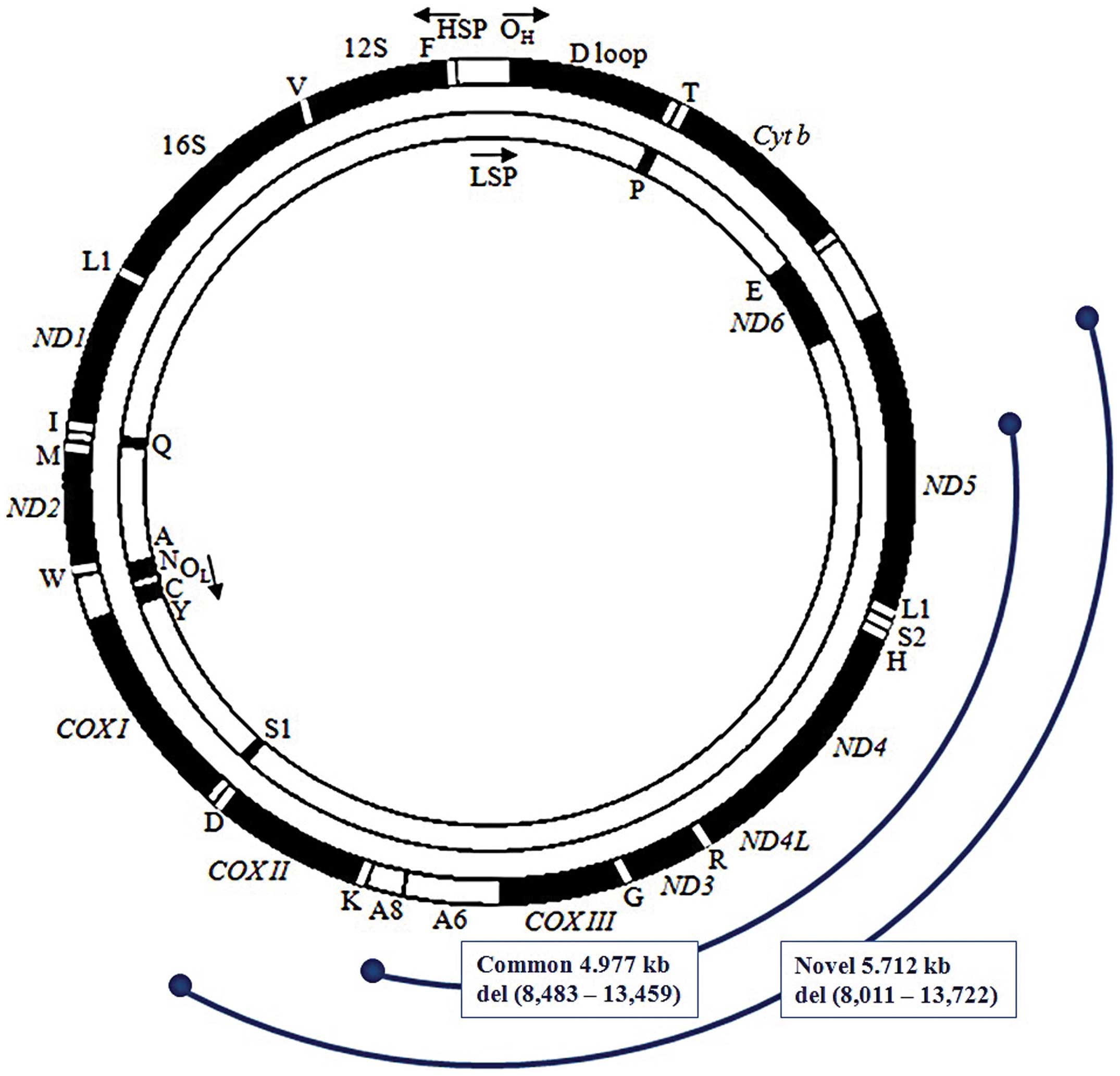 | Figure 3Comparison of the defective region of
mtDNA caused by common and novel large-scale deletions. The novel
large-scale deletion in a Korean patient with Pearson syndrome
spanned nucleotide position 8,011–13,722 of mtDNA, and removed part
of the COXII gene and part of the ND5 gene. The novel
deletion covered a larger defective region, particularly the first
part of COXII (8,011–8,269), all of
tRNALys, the first part of ATPase 8
(8,369–8,482), and the last part of ND5 (13,460–13,722), as
compared with the defective area caused by the common deletion.
OH, the origin of the heavy strand replication; D loop,
the displacement loop or non-coding control region; OL, the origin
of the light strand replication. |
 | Table IList and sequence of primers used for
the long-distance polymerase chain reaction of mitochondrial
DNA. |
Table I
List and sequence of primers used for
the long-distance polymerase chain reaction of mitochondrial
DNA.
| Pair | Name | Primer sequence
(5′→3′) | Position | Region | Size (bp) |
|---|
| 1 | M9F |
GAGGCCTAACCCCTGTCTTT | 5835–5854 |
tRNATyr | 10162 |
| M22R |
AGCTTTGGGTGCTAATGGTG | 15997–15978 |
tRNAPro | |
| 2 | M12F |
ACGAGTACACCGACTACGGC | 7908–7927 | COII | 8089 |
| M22R |
AGCTTTGGGTGCTAATGGTG | 15997–15978 |
tRNAPro | |
| 3 | M12F |
ACGAGTACACCGACTACGGC | 7908–7927 | COII | 6379 |
| M20R |
ACGAGTACACCGACTACGGC | 14287–14269 | ND6 | |
| 4 | M12F |
ACGAGTACACCGACTACGGC | 7908–7927 | COII | 5618 |
| M19R |
TCGATGATGTGGTCTTTGGA | 13526–13507 | ND5 | |
At the time of initial organic acid analysis and
mtDNA evaluation, the patient had been admitted to hospital with a
fever, poor oral intake and decreased activity, which persisted
despite administration of fluids and broad spectrum antibiotics.
Blood culture was negative and urine culture was positive for
Pseudomonas aeruginosa infection, which was treated with
ceftazidime. The patient’s fever did not remit and her overall
condition failed to show improvement, therefore she was transferred
to another institution for further care.
Discussion
The present report describes the case of a female
patient with PS harboring a large-scale mitochondrial deletion,
which is to the best of our knowledge, the first case reported in
South Korea. Molecular investigation of mtDNA extracted from a
peripheral blood sample detected a novel large-scale heteroplasmic
mitochondrial deletion without direct repeats. This deletion was
shown to affect numerous tRNA and protein-coding genes, which may
lead to defects in mitochondrial polypeptide synthesis, and
impaired oxidative phosphorylation and energy metabolism in the
respiratory chain.
Usually, mtDNA in humans is ~16.6 kb in length, and
encodes numerous enzymes of the respiratory chain and oxidative
phosphorylation system, ribosomal RNAs, and various tRNAs (12). Impairment of the mitochondrial
respiratory chain may lead to lactic acidemia, high plasma
lactate/pyruvate molar ratios, and even fatal metabolic acidosis
(13). Previous studies have
reported various mitochondrial deletions in patients with PS
(4–10). A common 4.9 kb deletion spanning
the ATPase 8 gene to the ND5 gene, between 13 bp
direct repeats has previously been frequently observed (14). This deletion has been identified in
>80% of affected children; however, numerous other mtDNA
deletions have also been detected (www.mitomap.org).
Mitochondrial deletions differ in size and location; however, they
are confined to a region delineated by the heavy- and light-strand
origins of replication, which is the major arc of mtDNA (14). The known deletions include numerous
tRNA and protein-coding genes (15), and are often flanked by direct
repeats (16). In the majority of
cases, mtDNA deletions are spontaneous events that occur either in
the oocyte or during early stages of embryonic development
(17).
The underlying mechanisms regarding the regulation
of tissue distribution of deleted mtDNA molecules, or why the rate
of disease progression and the degree of disease severity is
variable, even for the same deletions, remains to be elucidated. No
strict correlations have been observed between heteroplasmy in
blood cells and the severity of hematopoietic features.
Furthermore, no obvious genotype-phenotype correlation has been
identified regarding the appearance of hematological manifestations
in PS and the mtDNA deletion (18). The different phenotypic expression
of the same mtDNA defects may be associated with nuclear modifier
genes, polymorphisms and environmental factors (19).
No specific treatment is currently available for
patients with PS, therefore awareness of possible complications,
and early intervention may minimize PS-associated mortality and
morbidity. Red blood cell transfusions are often required to manage
macrocytic anemia, and patients may be dependent on these
transfusions (20). Pancreatic
enzyme replacement is required for patients with malabsorption due
to exocrine pancreatic dysfunction (20). Intermittent metabolic crises are
managed with hydration, correction of electrolyte abnormalities,
and correction of acidosis (20).
Bicarbonate supplementation and dichloroacetic acid have also been
used to treat persistent metabolic acidosis. Previous studies have
reported the case of two patients with PS, who were treated with
allogeneic hematopietic stem cell transplantation (HSCT) (21,22).
The procedure was shown to correct the hematological and metabolic
abnormalities in the two patients; therefore, allogeneic HSCT may
be a viable treatment option for patients with PS whose prognosis
is uniformly fatal if left untreated.
In conclusion, to the best of our knowledge, the
present study was the first to report a novel mtDNA deletion
causing Korean PS. It is necessary to consider mitochondrial
disorder in infants who present with persistent hematological
abnormalities, such as hypoplastic anemia. Due to the often fatal
course of PS, molecular analysis of possible mtDNA deletions may be
beneficial for the diagnosis and prognosis of PS, and may also aid
in the genetic counseling of relatives.
Acknowledgements
The present study was supported by a grant from the
Korea Health Technology R&D Project, Ministry of Health &
Welfare, Republic of Korea (grant no. A120175).
References
|
1
|
Pearson HA, Lobel JS, Kocoshis SA, et al:
A new syndrome of refractory sideroblastic anemia with
vacuolization of marrow precursors and exocrine pancreatic
dysfunction. J Pediatr. 95:976–984. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rötig A, Cormier V, Blanche S, et al:
Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial
disorder in infancy. J Clin Invest. 86:1601–1608. 1990. View Article : Google Scholar
|
|
3
|
Holt IJ, Harding AE and Morgan-Hughes JA:
Deletions of muscle mitochondrial DNA in patients with
mitochondrial myopathies. Nature. 331:717–719. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McShane MA, Hammans SR, Sweeney M, et al:
Pearson syndrome and mitochondrial encephalomyopathy in a patient
with a deletion of mtDNA. Am J Hum Genet. 48:39–42. 1991.PubMed/NCBI
|
|
5
|
Rötig A, Bourgeron T, Chretien D, Rustin P
and Munnich A: Spectrum of mitochondrial DNA rearrangements in the
Pearson marrow-pancreas syndrome. Hum Mol Genet. 4:1327–1330. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blaw ME and Mize CE: Juvenile Pearson
syndrome. J Child Neurol. 5:187–190. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rotig A, Colonna M, Bonnefont JP, et al:
Mitochondrial DNA deletion in Pearson’s marrow/pancreas syndrome.
Lancet. 1:902–903. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van den Ouweland JM, de Klerk JB, van de
Corput MP, et al: Characterization of a novel mitochondrial DNA
deletion in a patient with a variant of the Pearson marrow-pancreas
syndrome. Eur J Hum Genet. 8:195–203. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santorelli FM, Barmada MA, Pons R, Zhang
LL and DiMauro S: Leigh-type neuropathology in Pearson syndrome
associated with impaired ATP production and a novel mtDNA deletion.
Neurology. 47:1320–1323. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ayed IB, Chamkha I, Mkaouar-Rebai E, et
al: A Tunisian patient with Pearson syndrome harboring the 4.977kb
common deletion associated to two novel large-scale mitochondrial
deletions. Biochem Biophys Res Commun. 411:381–386. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sievers F, Wilm A, Dineen D, et al: Fast,
scalable generation of high-quality protein multiple sequence
alignments using Clustal Omega. Mol Syst Biol. 7:5392011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schapira AH: Primary and secondary defects
of the mitochondrial respiratory chain. J Inherit Metab Dis.
25:207–214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gibson KM, Bennett MJ, Mize CE, et al:
3-Methylglutaconic aciduria associated with Pearson syndrome and
respiratory chain defects. J Pediatr. 121:940–942. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kleinle S, Wiesmann U, Superti-Furga A, et
al: Detection and characterization of mitochondrial DNA
rearrangements in Pearson and Kearns-Sayre syndromes by long PCR.
Hum Genet. 100:643–650. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wallace DC, Shoffner JM, Trounce I, et al:
Mitochondrial DNA mutations in human degenerative diseases and
aging. Biochim Biophys Acta. 1271:141–151. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schon EA, Rizzuto R, Moraes CT, Nakase H,
Zeviani M and DiMauro S: A direct repeat is a hotspot for
large-scale deletion of human mitochondrial DNA. Science.
244:346–349. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harding AE and Hammans SR: Deletions of
the mitochondrial genome. J Inherit Metab Dis. 15:480–486. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Binder V, Steenpass L, Laws HJ, Ruebo J
and Borkhardt A: A novel mtDNA large-scale mutation clinically
exclusively presenting with refractory anemia: is there a chance to
predict disease progression? J Pediatr Hematol Oncol. 34:283–292.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Limongelli A, Schaefer J, Jackson S, et
al: Variable penetrance of a familial progressive necrotising
encephalopathy due to a novel tRNA(Ile) homoplasmic mutation in the
mitochondrial genome. J Med Genet. 41:342–349. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seneca S, De Meirleir L, De Schepper J, et
al: Pearson marrow pancreas syndrome: a molecular study and
clinical management. Clin Genet. 51:338–342. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faraci M, Cuzzubbo D, Micalizzi C, et al:
Allogeneic bone marrow transplantation for Pearson’s syndrome. Bone
Marrow Transplant. 39:563–565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hoyoux C, Dresse MF, Robinet S, et al:
Cord blood transplantation in a child with Pearson’s disease.
Pediatr Blood Cancer. 51:5662008. View Article : Google Scholar
|