Introduction
Renal ischemia/reperfusion (I/R) is a common risk
factor for acute renal failure (1). Inflammatory responses were also
reported to be partially responsible for renal damage following I/R
injury. Previous studies have demonstrated that acute renal I/R
injury may initiate the inflammatory cascade; in addition, the
inhibition of inflammation was demonstrated to protect kidneys
against I/R injury (2,3).
Endothelin-1 (ET-1) is a protein primarily produced
by endothelial cells (4); in the
kidneys, ET-1 is produced by renal glomerular endothelial cells
(RGECs), in which it is predominantly located within the cytoplasm
(5). However, in nephrotic states,
ET-1 may be produced by and localized in other cells, such as
tubular epithelial cells (5). ET-1
production contributes to vascular tone and other cellular
processes via activation of its receptors, which include type A
(ETA) and type B (ETB) (4). Previous studies have shown that ET-1
production in kidneys was increased with the development of chronic
renal diseases (6), and the
increase of ET-1 was also found to inversely accelerate the
progress of chronic renal diseases (7). In addition, the infusion of exogenous
ET-1 was reported to lead to functional and morphological
alterations of rat kidneys (7).
ETA, a G protein-coupled receptor, is one
of the primary receptors of ET-1. In the majority of cases,
ETA and ETB have opposing actions (8); however, upregulation of both
receptors has been observed in chronic renal diseases (9). Selective inhibition of ETA
using its antagonists, such as atrasentan, was demonstrated to have
beneficial effects on renal injury progression (10). Previous studies have shown that
ET-1 was upregulated in rat kidneys following chronic I/R (11,12);
however, the underlying mechanisms of action of this remain to be
elucidated. In the present study, the expression of ET-1 and
ETA in mouse kidneys and human renal glomerular
endothelial cells (HRGECs) was investigated following acute renal
I/R and hypoxia/reoxygenation (H/R); in addition, the associations
between inflammation and ET-1/ETA expression were
examined.
Materials and methods
Animal preparation
A total of 18 male C57BL/6 mice (eight-week-old;
weight, 23±0.6 g) were obtained from the Animal Center of Xinxiang
Medical University (Henan, China) and randomly divided into three
groups: Sham ischemia, ischemia and ischemia/reperfusion. The mice
were housed in a temperature-controlled room (22–24°C) under a 12-h
light/dark cycle, with access to standard mouse chow and water.
Animals were anesthetized by intraperitoneal injection of sodium
pentobarbital (60 mg/kg; Beijing Propbs Biotechnology Co., Ltd,
Beijing, China) and maintained on a 37.5°C Homeothermic Blanket for
Rodents (Shanghai Youer Equipment Scientific Co., Ltd, Shanghai,
China). Kidneys and renal arteries were exposed following abdominal
incisions. Renal arteries in the ischemia group were closed using
bulldog clamps (Dongxiyi Science and Technology Co. Ltd, Beijing,
China) for 1 h; mice were then sacrificed by cervical dislocations
under anesthesia (60 mg/kg sodium pentobartital), and plasma and
kidneys were collected and frozen at −80°C until further use. Renal
arteries in the I/R group were temporarily closed using bulldog
clamps for 1 h and subsequently reopened by removal of the bulldog
clamps to allow for reperfusion for 1 h; mice were then sacrificed,
and plasma and kidneys were collected. Mice in the sham ischemia
group received a sham surgery, without closing the renal arteries.
The use of animals and study protocols of the present study were
approved by the Ethics Committee of Xinxiang Medical University
(Xinxiang, China).
Culture of human RGECs (HRGECs)
HRGECs were purchased from ScienCell Research
Laboratories (Carlsbad, CA, USA) and cultured in special
Endothelial Cell Medium (ScienCell Research Laboratories). HRGECs
were exposed to hypoxia for 3 h using a hypoxia C-chamber
(Biospherix, Ltd, Lacona, NY, USA) and then placed in a cell
incubator for 1 h for reoxygenation; the mRNA and protein
expression levels of tumor necrosis factor (TNF)-α and interleukin
(IL)-6 were then measured using reverse transcription quantitative
polymerase chain reaction (RT-qPCR) and western blot analyses. In
order to elucidate the effect of inflammation on ET-1 and
ETA production, HRGECs were exposed to 0, 1, 5, 10 and
50 U/ml TNF-α (Sigma-Aldrich, Shanghai, China) for 6 h; ET-1 and
ETA expression levels were then measured using RT-PCR
and western blot. In parallel, another group of HRGECs were
incubated with the TNF-α inhibitor CAY10500 (5 μM; Sigma-Aldrich)
30 min prior to and during hypoxic treatments.
ELISA
Protein levels of TNF-α and IL-6 in mouse plasma
following the induction of ischemia and ischemia/reperfusion were
measured using a mouse TNF-α Quantikine ELISA kit (R&D Systems
Inc., Minneapolis, MN, USA) and a mouse IL-6 Quantikine ELISA kit
(R&D Systems Inc.), according to the manufacturer’s
instructions. Absorbance was measured at 450 nm using an
xMarkTM microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
RT-qPCR
Total RNA was extracted from the frozen kidneys and
HRGECs using RNeasy Mini kits (Invitrogen Life Technologies,
Carlsbad, CA, USA) and complementary (c)DNA was synthesized using a
SuperScript II 1st Strand DNA Synthesis kit (Invitrogen Life
Technologies) according to the manufacturer’s instructions. qPCR
reactions were performed using 2X PCR reaction solution
(Sigma-Aldrich, St. Louis, MO, USA) with 100 ng cDNA and 0.3 μM
primers. The primers used for PCR reactions are listed in Table I.
 | Table IPrimers for reverse transcription
quantitative polymerase chain reaction. |
Table I
Primers for reverse transcription
quantitative polymerase chain reaction.
| A, Mouse primers |
|---|
|
|---|
| Primers | Sense | Sequence | Product size
(bp) |
|---|
| ET-1 | Sense |
ggtggaaggaaggaaactac | 367 |
| Antisense |
caagaagaggcagaaaggca | |
| ETA | Sense |
aacaagtgtatgaggacggc | 325 |
| Antisense |
ggccaagatgaaggaaagaa | |
| TNF-α | Sense |
ccgatgggttgtaccttgtc | 352 |
| Antisense |
gggctgggtagagagaatggat | |
| IL-6 | Sense |
gatgctaccaaactggatataatc | 269 |
| Antisense |
ggtccttagccactccttctgtg | |
| β-actin | Sense |
ttctttgcagctccttcgttgccg | 458 |
| Antisense |
tggatggctacgtacatggctggg | |
|
| B, Human primers |
|
| Primers | Sense | Sequence | Product size
(bp) |
|
| ET-1 | Sense |
catcatttgggtcaacactcc | 281 |
| Antisense |
cttcctctcactaactgctg | |
| ETA | Sense |
ctcggtactcccataatcct | 332 |
| Antisense |
gaacattcaccagaactgcc | |
| TNF-α | Sense |
gagtgacaagcctgtagccca | 437 |
| Antisense |
gcaatgatcccaaagtagacc | |
| IL-6 | Sense |
gtagtgaggaacaagccaga | 245 |
| Antisense |
gagagccaaccaaccaaaca | |
| GADPH | Sense |
gggtgtgaaccatgagaagt | 225 |
| Antisense |
gtagaggcagggatgatgtt | |
Western blot analysis
Proteins were extracted from the frozen kidneys and
HRGECs, and separated using 12% SDS-PAGE (Zhengzhou
Bao-Biotechnology Co., Ltd, Zhengzhou, China). Following
electrophoresis, proteins were transferred onto polyvinylidene
fluoride membranes (Zhengzhou Jiuh-Bao-Biotechnology Co., Ltd). The
membranes were blocked with 5% non-fat milk (Zhengzhou
Jiuh-Bao-Biotechnology Co., Ltd) in Tris-buffered saline with
Tween-20 (TBS-T; Beijing Solarbio Science and Technology Co., Ltd,
Beijing, China) and then incubated with the following primary
antibodies: goat immunoglobulin (IgG) polyclonal anti-ET-1 (1:500;
Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit IgG
polyclonal anti-ETA (1:1,000; Thermo Fisher Scientific
Inc., Waltham, MA, USA), goat IgG polyclonal anti-TNF-α (1:1,000;
Santa Cruz Biotechnology Inc.), goat IgG polyclonal anti-IL-6
(1:500, Santa Cruz Biotechnology Inc.) and mouse IgG monoclonal
anti-β-actin (1:2,000; Santa Cruz Biotechnology Inc.) at 4°C
overnight. Subsequently, the membranes were washed three times with
TBS-T and incubated with donkey anti-goat IgG, donkey anti-rabbit
IgG or donkey anti-mouse IgG horseradish peroxidase-conjugated
secondary antibodies (1:10,000; Santa Cruz Biotechnology Inc.) for
1 h at room temperature. Immunoreactive bands were visualized using
an Enhanced Chemiluminescence Western Blotting Substrate kit
(Thermo Fisher Scientific Inc.).
Statistical analysis
Statistical analysis was performed using SPSS 11.5
software (SPSS Inc., Chicago, IL, USA). Values are presented as the
mean ± standard deviation. Univariate comparisons of the means were
evaluated using one-way analysis of variance with Tukey’s post-hoc
adjustment for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
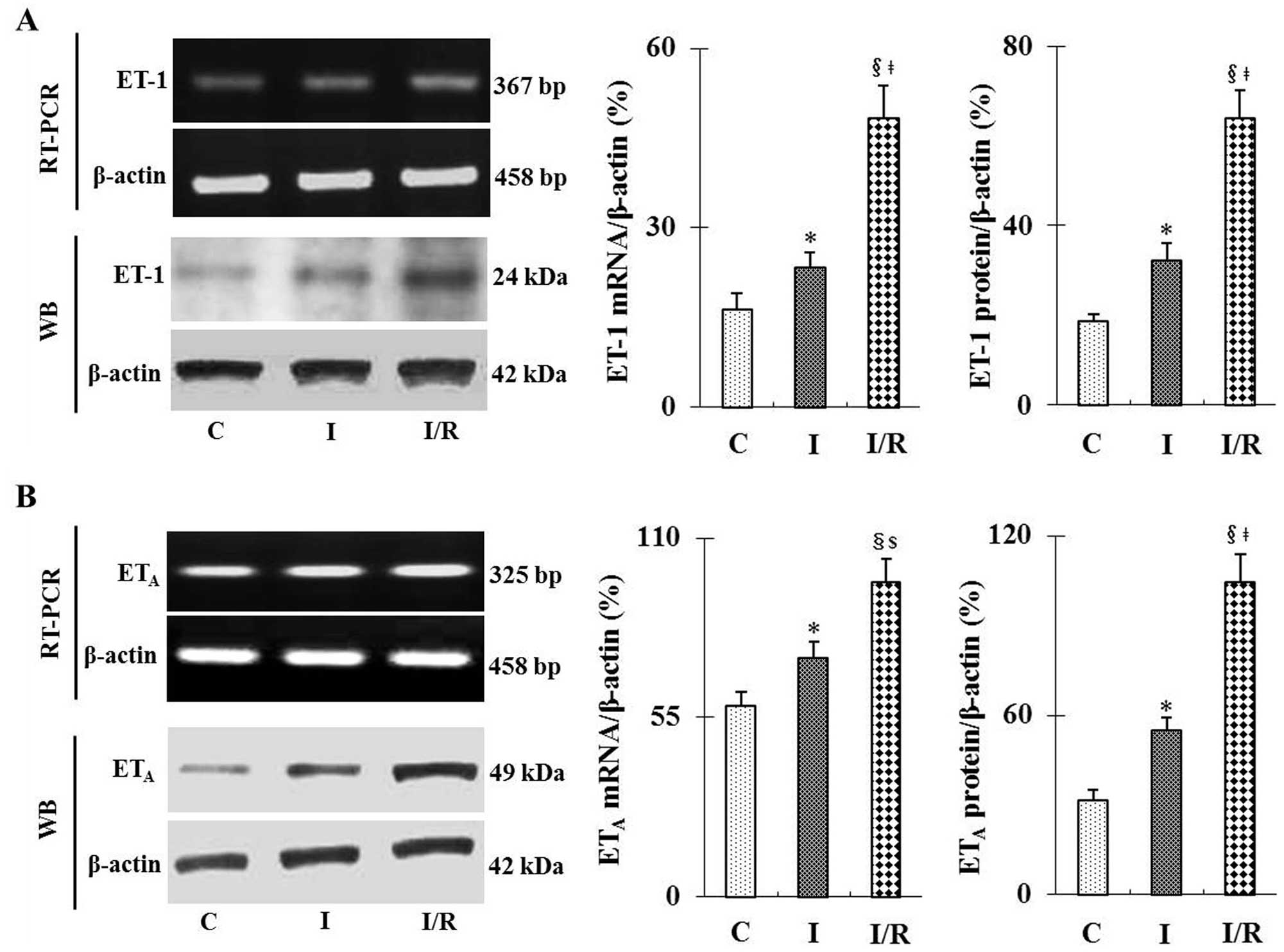
I/R increases mRNA and protein expression
levels of ET-1 and ETA in mouse kidneys
As shown in Fig. 1A and
B, RT-PCR and western blot analyses revealed that I/R
significantly increased the mRNA and protein expression levels of
ET-1 and ETA compared with those of the sham group
(P<0.01). Following ischemia only, a moderate but significant
increase was observed in ET-1 and ETA expression levels
compared with those of the sham group (P<0.05). In addition,
mRNA and protein expression levels of ET-1 and ETA in
the I/R group were significantly increased compared to those in the
ischemia only group (P<0.05 or P<0.01).
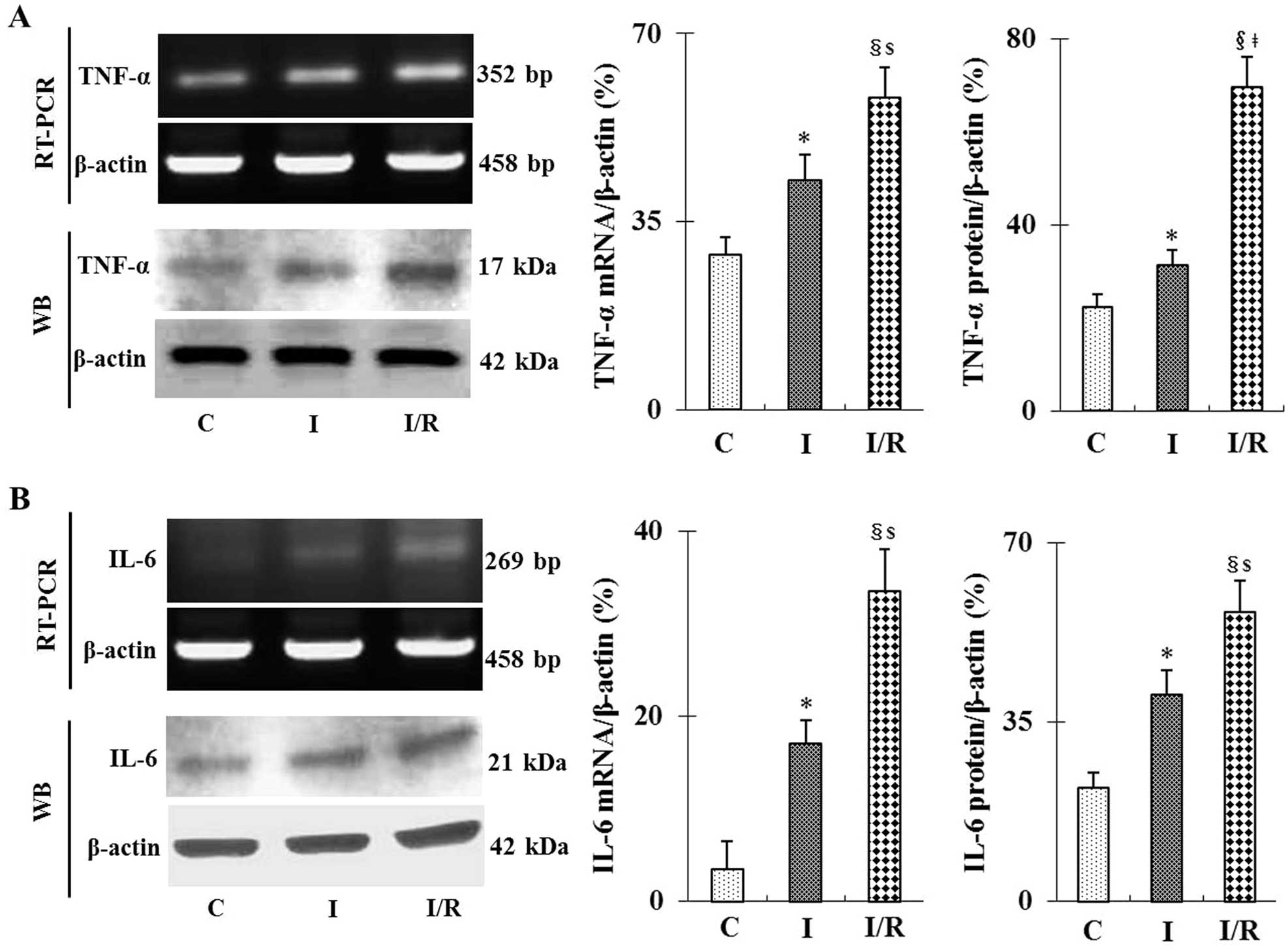
I/R increases TNF-α and IL-6 mRNA and
protein expression levels in mouse kidneys
As shown in Fig. 2,
RT-qPCR and western blot analyses demonstrated that following I/R,
mRNA and protein expression levels of the inflammatory cytokines
TNF-α and IL-6 were significantly increased compared to those in
the sham and ischemia groups (P<0.05 or P<0.01). However,
ischemia alone also caused a moderate but significant increase in
TNF-α and IL-6 expression levels compared with those of the sham
group (P<0.05). These data were further confirmed using ELISA
assays, which showed that TNF-α and IL-6 protein expression levels
in mouse plasma following ischemia and I/R were significantly
increased (P<0.05; data not shown).
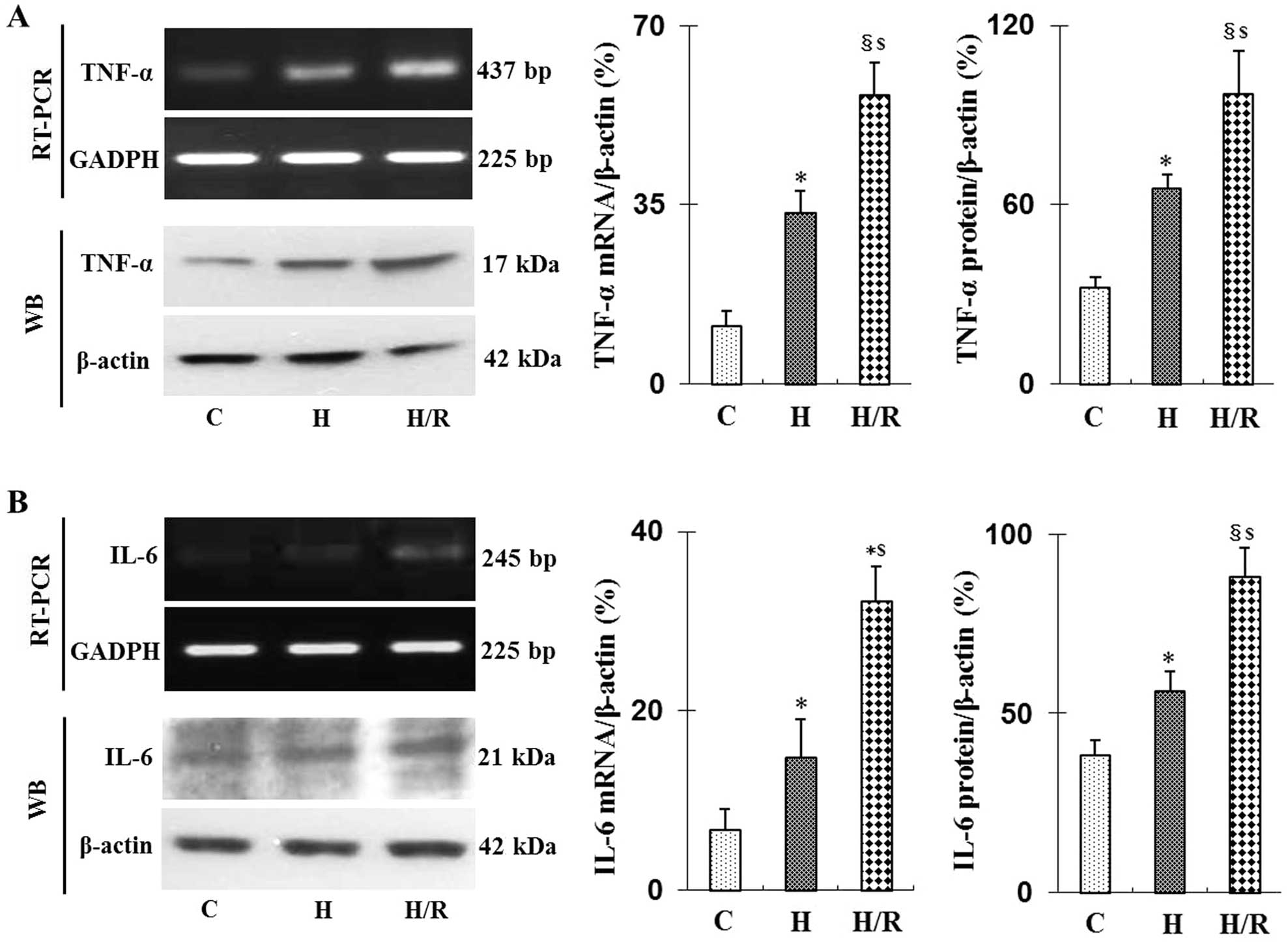
Effect of hypoxia/reoxygenation on TNF-α
and IL-6 mRNA and protein expression in HRGECs
Hypoxia is a primary mediator of tissue damage
following I/R (13). In kidneys,
ET-1 is predominantly produced by glomerular endothelial cells
(5). In the present study, HRGECs
were subjected to hypoxia for 3 h followed by reoxygenation for 1
h. As shown in Fig. 3, hypoxia
alone as well as H/R significantly increased TNF-α and IL-6 mRNA
and protein expression levels in HRGECs (P<0.05 or P<0.01) in
a comparable pattern to that of TNF-α and IL-6 expression in mouse
kidneys following ischemia and I/R.
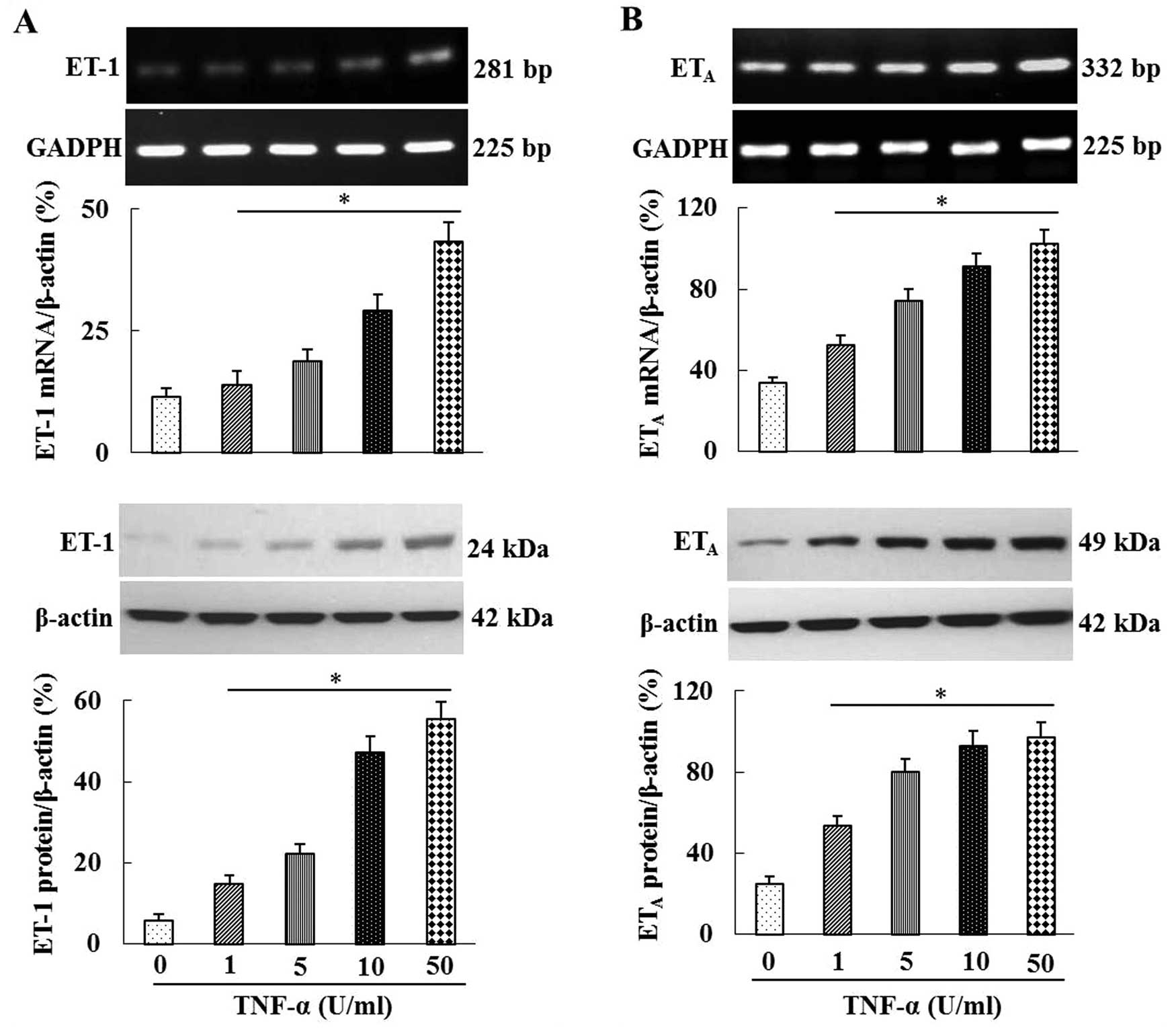
TNF-α increases ET-1 and ETA
mRNA and protein expression in HRGECs
In order to elucidate the role of inflammation in
ischemia- and hypoxia-induced ET-1 and ETA expression,
HRGECs were treated with various concentrations of TNF-α. As shown
in Fig. 4, TNF-α increased the
mRNA and protein expression of ET-1 and ETA in HRGECs in
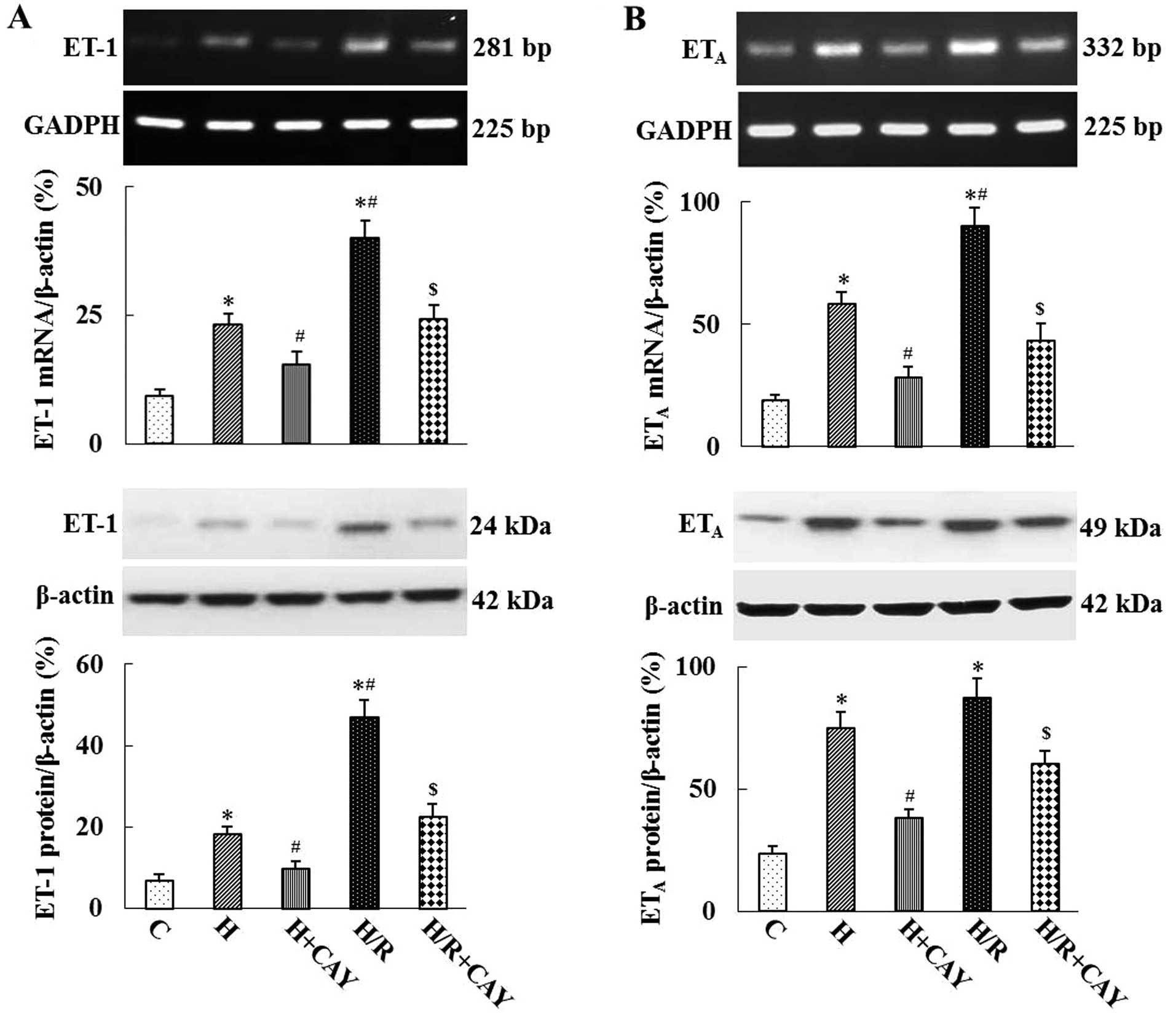
a dose-dependent manner. In addition, as shown in Fig. 5, hypoxia and H/R significantly
increased the mRNA and protein expression levels of ET-1 and
ETA in HRGECs compared with those in the control group.
Furthermore, the TNF-α inhibitor CAY10500 significantly inhibited
hypoxia- and H/R-induced ET-1 and ETA expression
(P<0.05 or P<0.01; Fig.
5).
Discussion
The results of the present study demonstrated that
ET-1 and its receptor ETA were significantly upregulated
in mouse kidneys following ischemia and I/R; in addition, the
expression of inflammatory cytokines TNF-α and IL-6 was markedly
increased in ischemic and I/R kidneys. Furthermore, mRNA and
protein expression levels of ET-1, ETA, TNF-α and IL-6
were markedly increased in HRGECs following exposure to hypoxia and
H/R. These data indicated that inflammation may be involved in
ischemia- and hypoxia-induced ET-1 and ETA expression in
kidneys and renal endothelial cells. In order to further elucidate
the role of inflammation in ischemia- and hypoxia-induced ET-1 and
ETA expression, HRGECs were treated with TNF-α; as
expected, TNF-α increased mRNA and protein expression levels of
ET-1 and ETA in HRGECs in a dose-dependent manner. Of
note, following treatment with the TNF-α inhibitor CAY10500, the
hypoxia- and H/R-induced increase in ET-1 and ETA
expression was significantly attenuated in HRGECs. These results
therefore suggested that ischemia and hypoxia may enhance the
upregulation of ET-1 and ETA, which is, at least in
part, dependent on the production of inflammatory cytokines.
ET-1 is an amino acid peptide primarily produced by
endothelial cells (4). In kidneys,
ET-1 is produced by renal glomerular endothelial cells (5). ET-1 is a pro-inflammatory factor,
which has an important role in maintaining normal renal functions
(14). Upregulation of ET-1 and
its receptors have also been reported to be involved in the
development of acute and chronic renal diseases (6,7).
Inflammation is a typical response and indicator of I/R injury; in
addition, inflammation results in the influx of inflammatory
mediators, which are responsible for tissue injury in organs
subjected to ischemia and/or I/R (15,16).
Inhibition of inflammatory responses has been demonstrated to
attenuate I/R-induced tissue damage (17,18).
The results of the present study demonstrated that inflammatory
mediators were involved in the expression of ET-1 and its receptor
ETA in kidneys subjected to acute ischemia and/or I/R.
Furthermore, these results also indicated that
inflammation-mediated ET-1 and ETA expression in kidneys
may, at least in part, be responsible for acute renal injury
following I/R.
The present study further investigated the role of
inflammation on ET-1 and ETA using an in vitro
study on HRGECs, which were previously reported to produce ET-1 and
express ETA receptor in kidneys (5,19).
The results of the present study revealed that the mRNA and protein
expression levels of ET-1, ETA, TNF-α and IL-6 were
markedly increased in HRGECs following H/R or hypoxia alone.
Hypoxia commonly occurs following tissue ischemia and results in
tissue and organ damage due to oxygen deficiency. However,
reoxygenation or reperfusion following hypoxia or ischemia may
further increase tissue damage due to the increased production of
reactive oxygen species (ROS) (20). The results of the present study
also showed that inhibition of inflammation through treatment with
a TNF-α inhibitor significantly repressed hypoxia-induced ET-1 and
ETA expression in RGECs. This therefore confirmed an
association between inflammation and ET-1/ETA
expression.
In conclusion, the results of the present study
demonstrated that inflammatory responses were involved in ischemia
and hypoxia-induced ET-1 and ETA expression in kidneys
and HRGECs. In addition, the present study may provided evidence
for a potential therapeutic target for the prevention of renal
failure following ischemia/reperfusion.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 31401246;
Beijing, China).
References
|
1
|
Thurman JM: Triggers of inflammation after
renal ischemia/reperfusion. Clin Immunol. 123:7–13. 2007.
View Article : Google Scholar
|
|
2
|
Kher A, Meldrum KK, Wang M, et al:
Cellular and molecular mechanisms of sex differences in renal
ischemia-reperfusion injury. Cardiovasc Res. 67:594–603. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen YT, Tsai TH, Yang CC, et al:
Exenden-4 and sitagliptin protect kidney from ischemia-reperfusion
injury through suppressing oxidative stress and inflammatory
reaction. J Transl Med. 11:2702013. View Article : Google Scholar
|
|
4
|
Marasciulo FL, Montagnani M and Potenza
MA: Endothelin-1: the yin and yang on vascular function. Curr Med
Chem. 13:1655–1665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vlachojannis JG, Tsakas S, Petropoulou C,
et al: Endothelin-1 in the kidney and urine of patients with
glomerular disease and proteinuria. Clin Nephrol. 58:337–343. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vlachojannis J, Tsakas S, Petropoulou C
and Kurz P: Increased renal excretion of endothelin-1 in nephrotic
patients. Nephrol Dial Transplant. 12:470–473. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen HC, Guh JY, Chang JM, et al: Plasma
and urinary endothelin-1 in focal segmental glomerulosclerosis. J
Clin Lab Anal. 15:59–63. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan RJ, Zhou L, Zhou D, et al: Endothelin
receptor a blockade is an ineffective treatment for adriamycin
nephropathy. PLoS One. 8:e799632013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ong AC, Newby LJ and Dashwood MR:
Expression and cellular localisation of renal endothelin-1 and
endothelin receptor subtypes in autosomal-dominant polycystic
kidney disease. Nephron Exp Nephrol. 93:e802003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watson AM, Li J, Schumacher C, et al: The
endothelin receptor antagonist avosentan ameliorates nephropathy
and atherosclerosis in diabetic apolipoprotein E knockout mice.
Diabetologia. 53:192–203. 2010. View Article : Google Scholar
|
|
11
|
Wihelm SM, Simonson MS, Robinson AV, et
al: Endothelin up-regulation and localization following renal
ischemia and reperfusion. Kidney Int. 55:1011–1018. 1999.
View Article : Google Scholar
|
|
12
|
Abu-Salen N, Ovcharenko E, Awad H, et al:
Involvement of the endothelin and nitric oxide systems in the
pathogenesis of renal ischemic damage in an experimental diabetic
model. Life Sci. 91:669–675. 2012. View Article : Google Scholar
|
|
13
|
Adhami F, Liao G, Morozov YM, et al:
Cerebral ischemia-hypoxia induces intravascular coagulation and
autophagy. Am J Pathol. 169:566–583. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richter CM: Role of endothelin in chronic
renal failure-developments in renal involvement. Rheumatology
(Oxford). 45(Suppl 3): iii36–iii38. 2006. View Article : Google Scholar
|
|
15
|
Daemen MA, van’t Veer C, Denecker G, et
al: inhibition of apoptosis induced by ischemia-reperfusion
prevents inflammation. J Clin Invest. 104:541–549. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lutz J, Thürmel K and Heemann U:
Anti-inflammatory treatment strategies for ischemia/reperfusion
injury in transplantation. J Inflamm (Lond). 7:272010. View Article : Google Scholar
|
|
17
|
Du X, He S, Jiang Y, et al: Adiponectin
prevents islet ischemia-reperfusion injury through the
COX2-TNFα-NF-κB-dependent signal transduction pathway in mice. J
Endocrinol. 218:75–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Vries B, Matthijsen RA, Wolfs TG, et
al: Inhibition of complement factor C5 protects against renal
ischemia-reperfusion injury: inhibition of late apoptosis and
inflammation. Transplantation. 75:375–382. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wendel M, Knels L, Kummer W and Koch T:
Distribution of endothelin receptor subtypes ETA and ETB in the rat
kidney. J Histochem Cytochem. 54:1193–1203. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leonard MO, Kieran NE, Howell K, et al:
Reoxygenation-specific activation of the antioxidant transcription
factor Nrf2 mediated cytoprotective gene expression in
ischemia-reperfusion injury. FASEB J. 20:2624–2626. 2006.
View Article : Google Scholar : PubMed/NCBI
|