Introduction
Dilated cardiomyopathy (DCM) is a disease that
causes injury to the heart muscle; the heart may become enlarged
and weakened, therefore restricting its ability to effectively pump
blood (1). In addition, DCM may
affect numerous other organs, including the liver and lungs, and
may even affect entire body systems. DCM is one of the leading
causes of mortality in the world (2); however, the underlying molecular
mechanisms of DCM remain to be elucidated.
H3K9 histone methyltransferase G9a is localized in
euchromatin and catalyzes the methylation of histone (3). G9a has an important role in the
silencing of numerous genes. For example, Oct-3/4 is a homeobox
gene with a Pit-Oct-Unc domain, which is expressed in the process
of producing cells for sexual reproduction and during the embryonic
period (4); during early
embryogenesis, G9a mediates the inactivation of Oct-3/4 (5). G9a has been previously characterized,
although its functions in DCM are not yet understood.
G9a was reported to silence cell adhesion molecules
(CAMs) in the development of lung cancer (6). CAMs exhibit pleiotropic biological
effects and have been reported to be overexpressed in patients with
DCM (7), suggesting an association
between the CAM levels and the severity of the disease (8). CAMs are elevated in patients with
heart failure and may have an important role in the pathogenesis of
heart failure via mediating the cell-cell interactions of the
immune response (9); therefore,
G9a may reduce the progression of DCM via silencing the CAM gene,
therefore reducing the enhanced levels of CAM in heart disease
(6,10). Furthermore, the promotion of DCM
epigenetics research is essential for elucidation of the underlying
molecular mechanism of DCM.
Numerous animal models are available for the study
of DCM. For example, a rat model of myocarditis has been created
through immunization with porcine cardiac myosin, which resulted in
severe heart failure characterized by increased cardiac fibrosis
and left ventricular (LV) dilation (11). In addition, furazolidone,
N-(5-nitro-2-furfurylidene)-3-amino-2-oxazolidone (FZ), is a
nitrofuran that has been successfully used for the treatment of
certain bacterial and protozoal infections in animals (12); however, FZ also induces CAM
expression and has been successfully used for the construction of
DCM models (13–15). These models provide the convenience
to investigate the molecular mechanism of DCM. In the present
study, the rat DCM model was established via FZ treatment. The
association between G9a and CAM expression was then explored in
order to investigate the biological function of G9a in the
development of DCM.
Materials and methods
Establishment of a rat model of DCM
All protocols of the present study were approved by
the animal committee of the First Affiliated Hospital of Harbin
Medical University (Harbin, China). A total of 60 Wistar rats
(male/female, 1:1; aged two weeks; weight, ~300 g) were purchased
from the Shanghai Animal Resource Center (Shanghai, China). Rats
were fed a commercial diet (KeAoXieLi Feed Co., Ltd., Beijing,
China) and were kept at a controlled temperature (22±2°C) in a 12-h
light/dark cycle. Rats were divided into a control and a DCM group.
Rats in the DCM group were administered 20 ml FZ solution (700
mg/l) each day for 4, 8 or 12 weeks (16). A pathophysiological study of the
hearts was performed as previously described (17), following anesthetization of rats
using intraperitoneal injections of 10% chloral hydrate (30 μl/kg;
Beijing Dingguo Biotechnology Co., Ltd, Beijing, China).
Two-dimensional transthoracic echocardiography was used to examine
the left ventricular end-diastolic dimensions (LVEDD), left
ventricular end-systolic dimensions (LVESD), fraction shortening
(FS) and left ventricular ejection fraction (LVEF) (18).
Measurement of aortic pressure (AOP) and
right atrium pressure (RAP)
AOP was measured as previously described (19), Four-dimensional phase contrast
magnetic resonance imaging (ECG100C-MRI Electrocardiogram
Amplifier; Biopac Systems, Inc., Goleta, CA, USA) was used for
computation of the AOP waveform. The AOP waveform was then
calculated using a segmentation algorithm. RAP was examined using
the combination of 2-dimensional echocardiographic parameters and a
3-dimensional right atrial volume index (RA volume, 11±2.8
ml/m2) (20). The mean
AOP and RAP were recorded.
Angiotensin (Ang) I, II and renin
assay
Ang I and II concentration was measured using a
modification of a previously described method (21). In brief, blood was collected from
rats in a tube containing 2 mg/ml K3EDTA, 3 mM
1,10-phenanthroline and 4 μM enalaprilat, which were all purchased
from Beijing Dingguo Biotechnology Co., Ltd. Samples were spiked
with Iodine (I)125-Ang I and II (Beijing Dingguo
Biotechnology Co., Ltd) as the internal controls. Samples were then
analyzed using preconditioned C8 Bond Elute columns (GE Healthcare,
Little Chalfont, UK). Angiotensins were eluted with 2 ml water and
acetonitrile (1:1; Beijing Dingguo Biotechnology Co., Ltd), and
0.1% trifluoroacetic acid (Beijing Dingguo Biotechnology Co., Ltd).
The samples were then dried, resuspended in 20% acetonitrile/0.1%
trifluoroacetic acid and filtered.
Filtered samples were purified using reverse-phase
high-performance liquid chromatography with a C8 column (300 Å, 4.6
mm internal diameter × 25 cm; Rainin Dynamax, Woburn, MA, USA) at a
rate of 1 ml/min. Angiotensin peptides were separated by a linear
gradient from 20–35% acetonitrile containing 0.1% trifluoroacetic
acid. Retention times for angiotensins were as follows: Ang I, 16.5
min; Ang II, 11.9 min; I125-Ang I, 22 min and
I125-Ang II, 16 min. The purified fractions were dried,
reconstituted with a buffer containing 10 mM phosphate-buffered
saline (PBS), 1 mM EDTA, 0.25 mM thimerasol and 0.1% gelatin (pH
7.3), which were all purchased from Beijing Dingguo Biotechnology
Co., Ltd. Ang I and II were quantitatively measured using
Angiotensin I and II (125I) Radioimmunoassay (RIA) kits
(Amersham Plc, Amersham, UK). The RIA reaction mixture was
incubated for 12 h and treated with dextran-coated charcoal
(Beijing Dingguo Biotechnology Co., Ltd) to separate Ang I and II.
Each RIA mixture was centrifuged at 1,500 × g at 4°C for 30 min.
The supernatant was counted and the Ang I and II values were read
off a standard curve constructed using the solutions of known Ang
concentrations. For renin activity assays, blood was collected in
syringes containing 1.5 mg/ml EDTA and immediately centrifuged at
4°C. Plasma was frozen in 1-ml aliquots at −20°C until further use.
Renin was determined as previously described (22).
Immunohistochemistry
Three rats from each group were sacrificed following
30 min anaesthesia with sodium pentobarbitone (50 mg/kg; Sihuan
Pharmaceutical Holdings Group Ltd., Beijing, China) by cervical
dislocation at weeks 4, 8 or 12. Immunohistochemistry was performed
as previously described (23). In
brief, paraffin-embedded heart tissues were sectioned into 2-μm
serial sections and then deparaffinized by incubating in xylene
(Beijing Chemical Plant, Beijing, China), which was repeated four
times for 5 min each; slides were then rehydrated by immersion in
normal saline for 30 sec. Slides were treated with citric acid
buffer (pH 6.0; Beijing Dingguo Biotechnology Co., Ltd) with 0.2%
Tween 20 (Beijing Dingguo Biotechnology Co., Ltd) and boiled for 10
min in order to retrieve the antigen. Slides were then rinsed in
10% H2O2 solution and blocked with methanol
for 10 min. Slides were incubated with mouse anti-human G9a
monoclonal antibodies (1:1,000; Abcam, Cambridge, MA, USA) and
mouse anti-human CAM monoclonal antibodies (1:1,000; Abcam)
overnight at 4°C. Samples were then rinsed in PBS and incubated for
1 h with secondary antibodies labeled with
streptoavidin-biotin-peroxidase (Shanghai Sangon Biotechnology Co.
Ltd., Shanghai, China). The bound complexes were then visualized
using diaminobenzidine liquid chromogen and counterstaining with
hematoxylin.
RNA interference
Primary neonatal cardiomyocytes (PNCs) from
three-day-old Wistar rats, which were obtained from Beijing Vital
River Laboratories, Co., Ltd (Beijing, China) were prepared as
previously described (24). In
order to inhibit G9a expression, small interfering (si)RNA
targeting G9a (si-G9a) was used (Shanghai GeneChem Co. Ltd,
Shanghai, China). The cardiomyocytes were grown in Dulbecco’s
modified Eagle’s medium (DMEM; Beijing Dingguo Biotechnology Co.,
Ltd) supplemented with 10% fetal bovine serum (Beijing Dingguo
Biotechnology Co., Ltd) and agitated at 10 min intervals.
Transfection of G9a RNAi was performed using HiperFect (Qiagen,
Limburg, Netherlands) with 3′-fluorescein isothiocyanate
(FITC)-labeled control siRNA (AUAUAAUUGUGGCCGUUGCdTdT) or
3′-FITC-labeled si-G9a (GUCUCUACUAUGAUUCCUACUDTDT) according to the
manufacturer’s instructions. Following incubations of 36 h,
transfection efficiencies were determined using fluorescence
microscopy (IX 50; Olympus, Tokyo, Japan).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Hearts were excised in ice-cold 30 mM KCl (Beijing
Dingguo Biotechnology Co., Ltd). Tissues and PNCs were snap-frozen
in liquid nitrogen for RNA analysis. First-strand complementary
(csNA was synthesized using the PrimeScript 1st strand
cDNASynthesis kit (Takara Bio, Inc., Dalian, China). Primers
specific to CAM forward, (5′-CCATC TACAACGCCAACATCG-3′ and reverse,
5′-CACAATGACC TGAATGTCCTTG-3′) and G9a cDNA forward, (5′-CCAATG
ACACGTCTTCACTGG-3′ and reverse, 5′-GTGGGGACA GAAGACCATCCC-3′) were
used to detect the gene transcripts as described previously
(25). GAPDH (forward, 5′-CGGAGTC
AACGGATTTGGTCGTAT-3′ and reverse, 5′-AGCCTT CTCCATGGTGGTGAAGAC-3′)
was used as a loading control. All primers were synthesized by
Beijing BGI-GBI Biotech Co., Ltd (Beijing, China).
Western blot analysis
Grounded tissues and PNCs were lysed using a buffer
[2% SDS, 100 mM Tris (pH 6.8), 10% glycerol and 10 mM
dithiothreitol; Beijing Dingguo Biotechnology Co., Ltd]. Samples
were boiled and subjected to 10% SDS-PAGE (Beijing Dingguo
Biotechnology Co., Ltd). Proteins were then transferred to
polyvinylidene fluoride membranes (HMA-0200; NTR Systems, Seattle,
WA, USA). Membranes were then immersed in 5% non-fat milk in
Tris-buffered saline buffer (pH 7.6; Beijing Dingguo Biotechnology
Co., Ltd). Blots were then incubated for 15 min with mouse
anti-human monoclonal G9a antibody (1:1,000; Abcam), followed by
30-min incubation with the secondary antibody
alkaline-phosphatase-conjugated goat anti-mouse immunoglobulin G
(1:1,000; Beijing Dingguo Biotechnology Co., Ltd). Molecular
weights were determined using known markers (PageRuler Plus
Prestained Protein Ladder; #26619; Thermo Fisher Scientific,
Beijing, China) and assays were performed in duplicate.
Evaluation of the growth rate of
PNCs
PNCs were prepared from hearts of three-day-old
Wistar rats as previously described (26). In brief, following isolation from
non-myocyte fibroblasts via plating, PNCs were purified using a
Percoll gradient, plated at a density of 0.5×105
cells/well in 12-well plates coated with 1% gelatin and grown in
media (DMEM and Media 199, 4:1), supplemented with 2 mM glutamine,
100 U/ml penicillin, 100 mg/ml streptomycin and 0.1 mmol/l BrdUrd,
which were all purchased from Beijing Dingguo Biotechnology Co.,
Ltd. BrdUrd was used to inhibit the growth of cardiac fibroblasts.
The experimental group was treated with 10 pM FZ and the growth
rate of PNCs was evaluated in the G9a RNAi and FZ-treated PNCs.
Statistical analysis
Statistical analysis was performed using SPSS 20.0
software (International Business Machines, Armonk, NY, USA). Values
are presented as the mean ± standard deviation. The Student’s
t-test was used to compare each variable between the DCM and
control rats. P<0.05 was considered to indicate as statistically
significant difference between values.
Results
Cardiac function
Analysis of cardiac function in rats is challenging
due to their small heart size and rapid heartbeat; therefore, the
sensitivity of cardiac function measures is extremely low. In order
to reduce the error in the present study, multiple parameters of
DCM were analyzed and compared with those of wild-type rats. The
total incidence rate of DCM in the experimental group was 80%
(24/30). DCM is defined as the dilatation and impaired
contractility of the LV; in addition, the LVEDD and LVESD
contribute to the LV dilatation and cardiac dysfunction (27). In the present study, the LVEDD and
LVESD were significantly increased in the FZ-treated group compared
with those in the control group (P<0.05 or P<0.01) (Table I). FS was used to measure the pump
function of the heart; it was calculated using the ratio of the
diameter of the left ventricle when it was relaxed and contracted,
with normal values commonly observed to be >26% (6). The LVEF represents the volumetric
fraction of blood pumped out of the ventricle with each heart beat
and may be applied to the left ventricle which ejects via the
aortic valve into the systemic circulation (28). In the present study, the FS and
LVEF values for the FZ-treated group were significantly decreased
compared with those in the control group (P<0.01) (Table I). The AOP is the blood pressure at
the root of the aorta, which was used to assess the efficacy of
antihypertensive treatment with respect to cardiovascular risk
factors (29). The RAP is the
blood pressure in the thoracic vena cava, which was used as a major
determinant of the right ventricular end diastolic volume (30). In the present study, the AOP was
not significantly altered in the FZ-treated group compared with
that in the control group (P>0.05) (Table I). However, compared with the
control group, the RAP increased significantly in the FZ-treated
group (P<0.01) (Table I).
 | Table IAnalysis of multiple independent
parameters of dilated cardiomyopathy. |
Table I
Analysis of multiple independent
parameters of dilated cardiomyopathy.
| Weeks | Control group | FZ-treated
group |
|---|
| LVEDD (cm) | 0 | 0.50±0.17 | 0.50±0.15 |
| 4 | 0.51±0.13 | 0.59±0.12a |
| 8 | 0.53±0.22 | 0.66±0.18b |
| 12 | 0.49±0.19 | 0.69±0.20b |
| LVESD (cm) | 0 | 0.28±0.08 | 0.27±0.06 |
| 4 | 0.29±0.10 | 0.35±0.11b |
| 8 | 0.31±0.12 | 0.39±0.12b |
| 12 | 0.33±0.15 | 0.48±0.19b |
| FS (%) | 0 | 45.51±4.10 | 45.89±3.91 |
| 4 | 46.63±4.51 | 38.57±2.21b |
| 8 | 42.48±3.62 | 30.17±1.18b |
| 12 | 44.02±3.19 | 28.74±1.48b |
| LVEF (%) | 0 | 84.25±6.78 | 84.62±5.81 |
| 4 | 82.21±7.73 | 79.12±3.87a |
| 8 | 83.96±5.98 | 65.19±4.16b |
| 12 | 80.01±6.69 | 60.99±3.39b |
| AOP (mmHg) | 0 | 84.06±6.12 | 84.93±6.55 |
| 4 | 89.45±7.18 | 88.99±7.62 |
| 8 | 90.45±5.69 | 91.03±5.23 |
| 12 | 88.87±5.23 | 90.12±6.21 |
| RAP (mmHg) | 0 | 5.53±1.17 | 5.78±2.15 |
| 4 | 4.79±1.11 | 13.77±4.42b |
| 8 | 6.47±1.01 | 19.98±5.53b |
| 12 | 5.81±1.36 | 20.91±3.27b |
| Renin | 0 | 2.38±0.25 | 2.35±0.19 |
| 4 | 2.41±0.28 | 3.61±0.28b |
| 8 | 2.39±0.29 | 3.45±0.65b |
| 12 | 2.35±0.21 | 3.59±0.86b |
| Angiotensin I | 0 | 17.07±2.28 | 17.55±2.76 |
| 4 | 16.89±1.97 | 10.99±2.31b |
| 8 | 18.01±2.69 | 11.69±3.24b |
| 12 | 16.63±2.43 | 11.22±2.22b |
| Angiotensin II | 0 | 65.13±7.99 | 64.31±8.22 |
| 4 | 69.52±9.11 |
108.30±11.49b |
| 8 | 70.14±7.95 |
110.23±13.70b |
| 12 | 71.19±8.75 |
113.56±15.77b |
Renin is an enzyme that participates in the
renin-angiotensin system which mediates extracellular volume and
arterial vasoconstriction, therefore regulating the body’s mean
arterial blood pressure (31).
Angiotensin is a peptide hormone which causes vasoconstriction and
an increase in blood pressure (32). Angiotensin also stimulates the
release of aldosterone from the adrenal cortex, which promotes
sodium retention in the distal nephron of the kidney and, in turn,
increases the blood pressure (33). Angiotensin I is formed in the liver
following the interaction of renin with α-2-glycoprotein (34). Renin is produced in the kidneys in
response to renal sympathetic activity, reduced intrarenal blood
pressure or decreased delivery of NaCl to the macula densa
(35). Angiotensin I is converted
to angiotensin II through the removal of two C-terminal residues by
angiotensin-converting enzyme (36). Angiotensin II acts as an endocrine,
autocrine/paracrine and intracrine hormone (37). In the present study, protein
expression levels of renin, angiotensin I levels were markedly
decreased, while angiotensin II levels were significantly increased
in the FZ-treated group compared with those in the control group
(P<0.01) (Table I).
Immunohistochemical analysis of the DCM
model
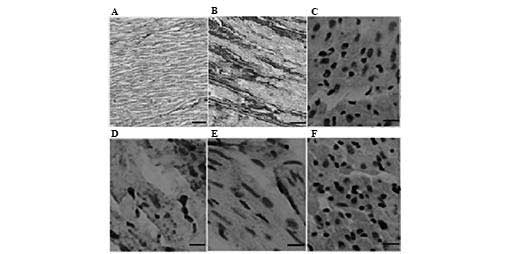
As shown in Fig.
1A, the heart muscle of rats in the control group showed
markedly less fibrosis compared with that in the experimental group
(Fig. 1B), which was consistent
with the characteristics of DCM (38). In addition, compared with that of
the control group, CAM was highly expressed in the DCM muscles
(Fig. 1C and D, respectively).
Conversely, G9a expression was markedly higher in the control group
compared with that in the DCM model (Fig. 1E and F, respectively). These
results therefore suggested that G9a affected the expression levels
of CAM in the development of DCM.
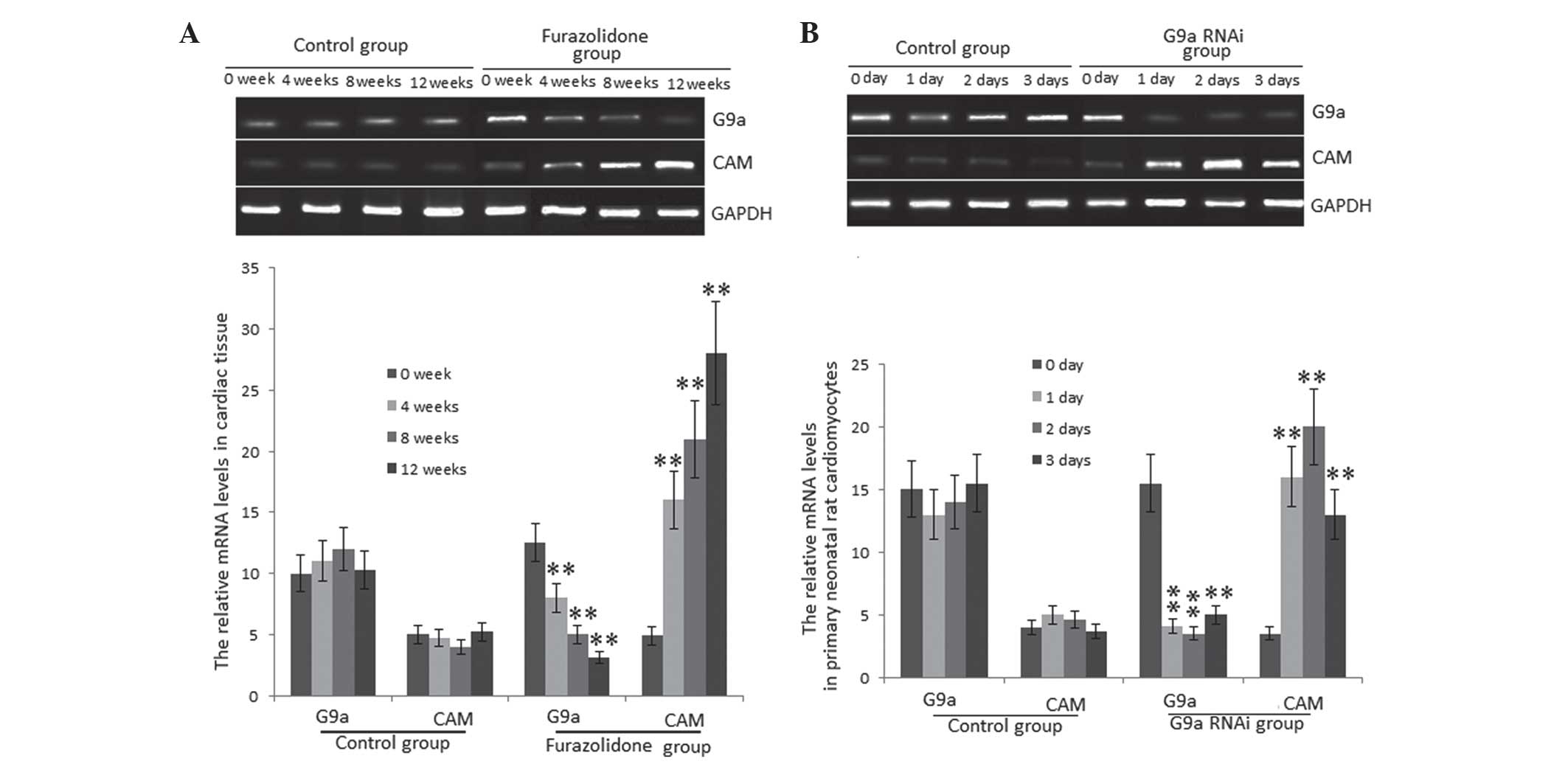
mRNA expression levels of G9a and CAM in
the DCM model
G9a is a mammalian histone methyltransferase which
contributes to the epigenetic silencing of tumor suppressor genes.
Emerging evidence suggested that G9a was required in order to
maintain a malignant phenotype (6). However, the biological function of
G9a in DCM has remained to be elucidated. CAMs were reported to be
higly expressed in DCM (10),
whereas G9a repressed the expression of CAMs through CAM gene
silencing. In order to explore the underlying molecular mechanisms
of this in the present study, mRNA expression levels of G9a and CAM
were investigated using RT-qPCR. The results showed that mRNA
levels of G9a were significantly decreased in the FZ-treated group
compared with those in the control group (P<0.01) (Fig. 2A). Conversely, mRNA levels of CAMs
significantly increased in the FZ-treated group (P<0.01)
(Fig. 2A). In order to further
determine the negative association of mRNA expression between G9a
and CAM, the G9a gene was silenced in PNCs via G9a RNAi. As shown
in Fig. 2B, mRNA levels of G9a
significantly decreased following one-day RNAi compared with those
in the control group. Conversely, mRNA levels of CAM significantly
increased in PNCs in the G9a RNAi group (P<0.01) (Fig. 2B). This therefore indicated that
G9a inhibited the progression of DCM through silencing the CAM
gene.
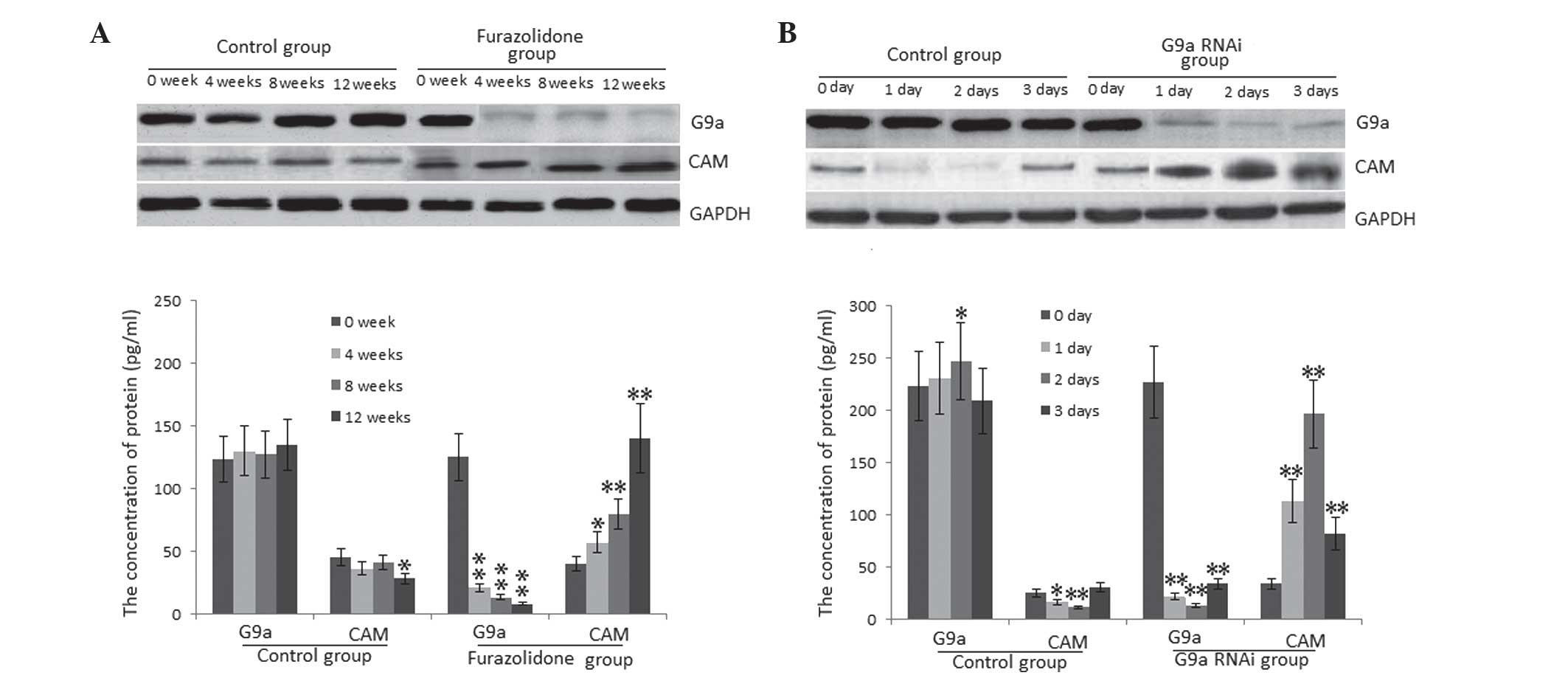
Protein expression levels of G9a and CAM
in DCM
DCM and wild-type control rats were sacrificed for
analysis following 0, 4, 8 or 12 weeks of treatment. CAM and G9a
expression in heart tissue was assessed using western blot
analysis. Significantly decreased expression levels of G9a were
observed in the cardiac tissues of DCM model rats compared with
those in the control rats at 4, 8 and 12 weeks (P<0.01)
(Fig. 3A). However, protein
expression levels of CAM were significantly increased in the DCM
model group compared with those in the control group at 4
(P<0.05), 8 and 12 weeks (P<0.01) (Fig. 3A). Furthermore, significantly
higher levels of CAM were found in PNCs transfected with G9a RNAi
compared with those in the the wild-type PNC group (Fig. 3B). These results suggested that G9a
inhibited the progression of DCM through repressing CAM
expression.
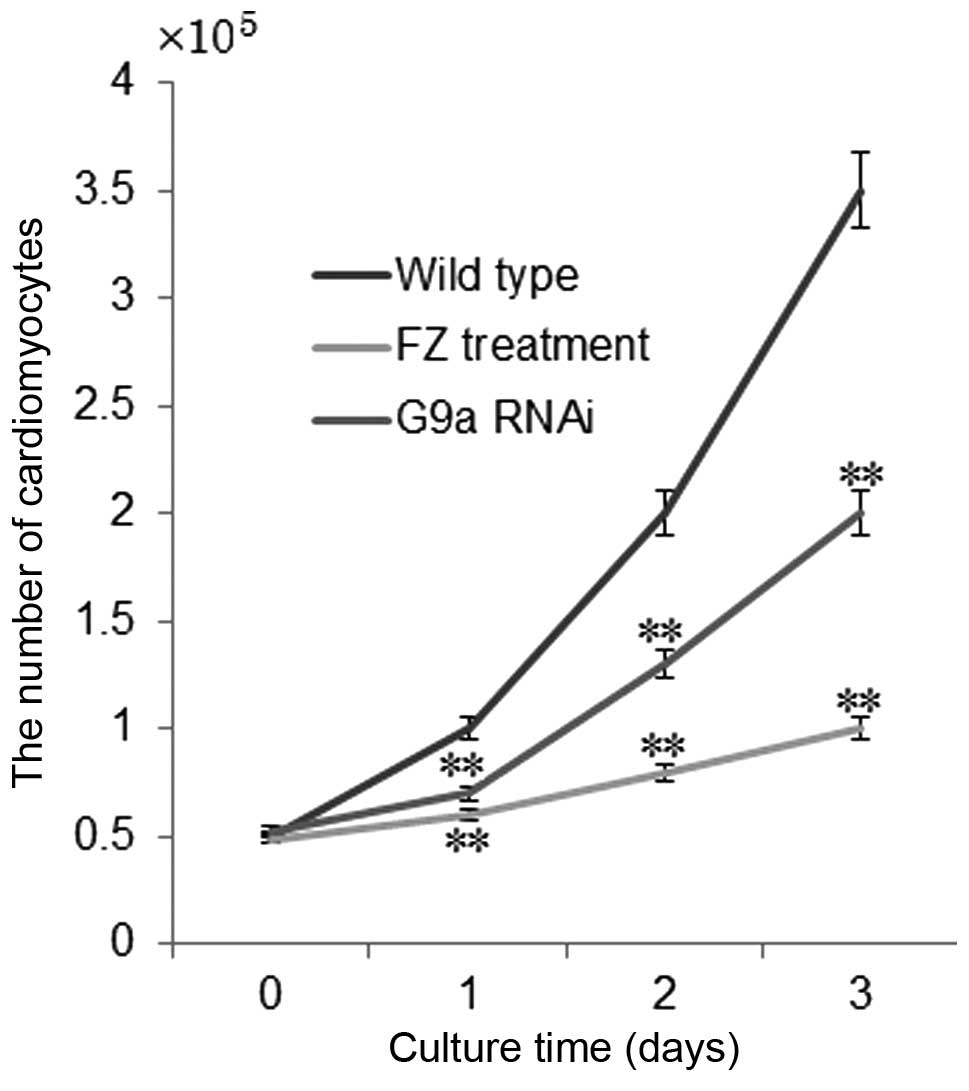
Growth rate of PNCs
The growth rate of PNCs was significantly inhibited
following treatment with FZ or G9a RNAi. Following three-day
culture with FZ or G9a RNAi, the growth rate of PNCs was inhibited
by 70 and 35%, respectively, compared to that in the control group
(P<0.01) (Fig. 4). The
inhibitory mechanism of repression of PNC growth by FZ proceeds via
the induction of apoptosis in cardiomyocytes (39). G9a RNAi reduced the growth
inhibition of PNCs in FZ-induced DCM through silencing CAM gene
expression, therefore enhancing the growth of cardiomyocytes.
Discussion
In the present study, in order to assess whether the
DCM model was successfully constructed, multiple parameters of DCM
were evaluated. The results showed that the reconstructed model
exhibited all the parameters consistent with the characteristics of
DCM compared with those of the wild-type rats. LVEDD, LVESD, RAP,
renin and angiotensin II levels were significantly increased in DCM
models compared with those in the wild-type animals, while
angiotensin I levels were significantly decreased. Conversely, the
parameters FS and LVEF were significantly decreased in DCM model
rats. However, AOP was not significantly different between the DCM
model and wild-type rats. These results therefore indicated that FZ
successfully induced the DCM model in rats; these results were in
concurrence with a previous study involving FZ-induced DCM in
turkeys and rats (40). These
models provided convenient methods for exploring the molecular
mechanism for the risk factors and pathogenesis of DCM.
G9a histone methyltransferase has a dominant role in
euchromatic histone H3 lysine-9 methylation, which is essential for
early embryogenesis (41). G9a was
reported to promote lung cancer invasion and metastasis through
silencing the cell adhesion molecule gene Ep-CAM (6). G9a has also been found to inhibit the
expression of numerous other genes (42); however, the biological function of
G9a in DCM remains to be elucidated. The results of the present
study demonstrated that G9a was expressed at low levels in DCM
model rats compared with those of the control rats; this therefore
indicated that G9a may decrease the risk of DCM through
downregulating the expression of CAM. However, the precise role of
G9a in the pathophysiology of DCM remains elusive, as previous
studies have not defined whether altered expression levels of G9a
in DCM were pathophysiologically important or simply an
epiphenomenon associated with DCM (43). Furthermore, G9a is only one member
of the histone H3 methyltransferases and numerous other members may
also be associated with the development of DCM. However, G9a was
found to repress the protein expression of CAM and may therefore
inhibit the progression of DCM.
It may be of interest for future studies to explore
the association of protein levels between cardiac CAMs and G9a in
human heart failure, which may then be compared with the present
results from DCM models of rats. The present study strongly
suggested an important pathophysiological role for low levels of
G9a in the development of DCM; in addition, the rat model used
provided the opportunity to evaluate the mechanisms which initiated
the expression of CAMs in the development of DCM. The DCM model
develops a primary phenotype consistent with congestive heart
failure, which resulted in cardiac dilatation and left ventricular
dysfunction. This model allowed for the analysis of physiological
and biochemical changes during development of end-stage heart
failure, therefore providing a potential unique platform for the
preclinical evaluation of G9a pharmacological therapies for
inhibiting the progression of DCM.
In conclusion, the rat model of DCM was successfully
induced through daily administration of FZ to rats. In the
DCM-induced rats, the parameters for LVEDD, LVESD, RAP, renin as
well as angiotensin I and II were significantly increased compared
to those in the wild-type group, whereas FS and LVEF were
significantly decreased in the DCM model, indicating that the rat
model closely reflected the pathophysiological characters of DCM.
The mRNA and protein expression levels of G9a were found to be
significantly lower in the DCM model compared with those in the
control group, while the expression levels of CAM were
significantly increased in the DCM model. In PNCs, the expression
of CAM was upregulated following G9a gene silencing by RNAi. These
results indicated that G9a reduced the risk of DCM through
downregulation of CAMs in a rat model of DCM; this therefore
indicated the potential novel therapeutic use of G9a in the
treatment of DCM.
References
|
1
|
Linke WA and Bücker S: King of hearts: a
splicing factor rules cardiac proteins. Nat Med. 18:6602012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meyer L, Stubbs B, Fahrenbruch C, et al:
Incidence, causes and survival trends from cardiovascular-related
sudden cardiac arrest in children and young adults 0 to 35 years of
age a 30-year review. Circulation. 126:1363–1372. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tachibana M, Sugimoto K, Fukushima T and
Shinkai Y: Set domain-containing protein, G9a, is a novel
lysine-preferring mammalian histone methyltransferase with
hyperactivity and specific selectivity to lysines 9 and 27 of
histone H3. J Biol Chem. 276:25309–25317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Albert M and Peters AH: Genetic and
epigenetic control of early mouse development. Curr Opin Genet Dev.
19:113–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feldman N, Gerson A, Fang J, et al:
G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during
early embryogenesis. Nat Cell Biol. 8:188–194. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen MW, Hua KT, Kao HJ, et al: H3K9
histone methyltransferase G9a promotes lung cancer invasion and
metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer
Res. 70:7830–7840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Devaux B, Scholz D, Hirche A, Klövekorn WP
and Schaper J: Upregulation of cell adhesion molecules and the
presence of low grade inflammation in human chronic heart failure.
Eur Heart J. 18:470–479. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blankenberg S, Rupprecht HJ, Bickel C, et
al: Circulating cell adhesion molecules and death in patients with
coronary artery disease. Circulation. 104:1336–1342. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yin WH, Chen JW, Jen HL, et al: The
prognostic value of circulating soluble cell adhesion molecules in
patients with chronic congestive heart failure. Eur J Heart Fail.
5:507–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Noutsias M, Seeberg B, Schultheiss H-P and
Kühl U: Expression of cell adhesion molecules in dilated
cardiomyopathy evidence for endothelial activation in inflammatory
cardiomyopathy. Circulation. 99:2124–2131. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Watanabe K, Ohta Y, Nakazawa M, et al: Low
dose carvedilol inhibits progression of heart failure in rats with
dilated cardiomyopathy. Br J Pharmacol. 130:1489–1495. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chadfield MS and Hinton MH: In vitro
activity of nitrofuran derivatives on growth and morphology of
Salmonella enterica serotype enteritidis. J Appl Microbiol.
96:1002–1012. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jankus E, Noren G and Staley N:
Furazolidone-induced cardiac dilatation in turkeys. Avian Dis.
16:958–961. 1972. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao R, Nascimben L, Friedrich J, Gwathmey
JK and Ingwall JS: Decreased energy reserve in an animal model of
dilated cardiomyopathy relationship to contractile performance.
Circ Res. 78:893–902. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Czarnecki CM: Animal models of
drug-induced cardiomyopathy. Comp Biochem Physiol Part C. 79:9–14.
1984. View Article : Google Scholar
|
|
16
|
Huang RJ, Liu TW, Wu WF and Pang YS:
Furazolidone induces dilated cardiomyopathy in rats [J]. Chinese J
Pathophysiol. 11:2005.
|
|
17
|
Kubota T, McTiernan CF, Frye CS, et al:
Dilated cardiomyopathy in transgenic mice with cardiac-specific
overexpression of tumor necrosis factor-α. Circ Res. 81:627–635.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stein AB, Tiwari S, Thomas P, et al:
Effects of anesthesia on echocardiographic assessment of left
ventricular structure and function in rats. Basic Res Cardiol.
102:28–41. 2007. View Article : Google Scholar
|
|
19
|
Delles M, Rengier F, Jeong YJ, et al:
Estimation of aortic pressure waveforms from 4D phase-contrast MRI.
Conf Proc IEEE Eng Med Biol Soc. 2013:731–734. 2013.PubMed/NCBI
|
|
20
|
Patel AR, Alsheikh-Ali AA, Mukherjee J, et
al: 3D echocardiography to evaluate right atrial pressure in
acutely decompensated heart failure correlation with invasive
hemodynamics. JACC Cardiovasc Imaging. 4:938–945. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goldberg MR, Tanaka W, Barchowsky A, et
al: Effects of losartan on blood pressure, plasma renin activity
and angiotensin II in volunteers. Hypertension. 21:704–713. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kodish ME and Katz FH: Plasma renin
concentration: comparison of angiotensinase inhibitors and
correlation with plasma renin activity and aldosterone. J Lab Clin
Med. 83:705–715. 1974.PubMed/NCBI
|
|
23
|
Takano S, Yoshitomi H, Togawa A, et al:
Apolipoprotein C-1 maintains cell survival by preventing from
apoptosis in pancreatic cancer cells. Oncogene. 27:2810–2822. 2008.
View Article : Google Scholar
|
|
24
|
Guo Z, Wang S, Jiao Q, Xu M and Gao F:
RNAi targeting ryanodine receptor 2 protects rat cardiomyocytes
from injury caused by simulated ischemia-reperfusion. Biomed
Pharmacother. 64:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carleton KL: Quantification of transcript
levels with quantitative RT-PCR. Methods Mol Biol. 772:279–295.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Purcell NH, Tang G, Yu C, Mercurio F,
DiDonato JA and Lin A: Activation of NF-kappa B is required for
hypertrophic growth of primary rat neonatal ventricular
cardiomyocytes. Proc Nat Acad Sci USA. 98:6668–6673. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moon C, Krawczyk M, Ahn D, et al:
Erythropoietin reduces myocardial infarction and left ventricular
functional decline after coronary artery ligation in rats. Proc Nat
Acad Sci. 100:11612–11617. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paulus WJ, Tschöpe C, Sanderson JE, et al:
How to diagnose diastolic heart failure: a consensus statement on
the diagnosis of heart failure with normal left ventricular
ejection fraction by the heart failure and echocardiography
associations of the european society of cardiology. Eur Heart J.
28:2539–2550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Avolio A: Central aortic blood pressure
and cardiovascular risk a paradigm shift? Hypertension.
51:1470–1471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bombardini T, Cini D, Arpesella G and
Picano E: WEB downloadable software for training in cardiovascular
hemodynamics in the (3-D) stress echo lab. Cardiovasc Ultrasound.
8:482010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vander AJ: Control of renin release.
Physiol Rev. 47:359–382. 1967.PubMed/NCBI
|
|
32
|
Gigante B, Rubattu S, Russo R, et al:
Opposite feedback control of renin and aldosterone biosynthesis in
the adrenal cortex by angiotensin II AT1-subtype receptors.
Hypertension. 30(3 Pt 2): 563–568. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarafidis PA and Ruilope LM: Aggressive
blood pressure reduction and renin-angiotensin system blockade in
chronic kidney disease: time for re-evaluation? Kidney Int.
85:536–546. 2014. View Article : Google Scholar
|
|
34
|
Podwysocki B, Simon K and Gladysz A:
Activity of angiotensin converting enzyme I and levels of acid
alpha glycoprotein in selected liver and biliary tract diseases.
Pol Tyg Lek. 48:268–270. 1993.(In Polish). PubMed/NCBI
|
|
35
|
Aoyagi T, Izumi Y, Hiroyama M, et al:
Vasopressin regulates the renin-angiotensin-aldosterone system via
V1a receptors in macula densa cells. Am J Physiol Renal Physiol.
295:F100–F107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Erdös EG and Skidgel RA: Renal metabolism
of angiotensin I and II. Kidney Int Suppl. 30:S24–S27.
1990.PubMed/NCBI
|
|
37
|
Johnston CI: Renin-angiotensin system: a
dual tissue and hormonal system for cardiovascular control. J
Hypertens. 10:S271992. View Article : Google Scholar
|
|
38
|
Ohtani K, Yutani C, Nagata S, Koretsune Y,
Hori M and Kamada T: High prevalence of atrial fibrosis in patients
with dilated cardiomyopathy. J Am Coll Cardiol. 25:1162–1169. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang M, Shan H, Zhu Y-h, Lin L and Wei J:
Attenuating effect of estrogen on furazolidone induced
cardiomyocyte apoptosis is dependent on p66shc adapter protein.
cardiology karger allschwilerstrasse 10, ch-4009 Basel,
Switzerland: pp. 34. 2013
|
|
40
|
Genao A, Seth K, Schmidt U, Carles M and
Gwathmey JK: Dilated cardiomyopathy in turkeys: an animal model for
the study of human heart failure. Lab Anim Sci. 46:399–404.
1996.PubMed/NCBI
|
|
41
|
Tachibana M, Sugimoto K, Nozaki M, et al:
G9a histone methyltransferase plays a dominant role in euchromatic
histone H3 lysine 9 methylation and is essential for early
embryogenesis. Genes Dev. 16:1779–1791. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roopra A, Qazi R, Schoenike B, Daley TJ
and Morrison JF: Localized domains of G9a-mediated histone
methylation are required for silencing of neuronal genes. Mol Cell.
14:727–738. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hohl M, Wagner M, Reil JC, et al: HDAC4
controls histone methylation in response to elevated cardiac load.
J Clin Invest. 123:1359–1370. 2013. View
Article : Google Scholar : PubMed/NCBI
|