Introduction
The spinal cord is a part of the central nervous
system in humans and other vertebrates (1). Spinal cord injury (SCI) is damage to
the spinal cord, which is categorized according to the extent of
loss of function, loss of sensation and the inability of the
individual to stand or walk (2).
It often results in confinement to a wheelchair and a lifetime of
medical comorbidity (3). SCI may
result from serious accidents, including road traffic accidents or
sports injuries, but may also occur accompanying serious diseases,
including developmental disorders, neurodegenerative diseases or
demyelinating diseases. Multiple sclerosis, transverse myelitis
resulting from stroke or inflammation and vascular malformations
can all result in severe consequences and high-disability due to
SCI (4).
Several genes and signaling pathways are involved in
spinal cord injury (5). Expression
of nerve growth factor, brain-derived neurotrophic factor (BDNF),
neurotrophin-3 (NT-3), p75 low-affinity nerve growth factor
receptor, transforming tyrosine kinase B and interleukin (IL)-6
have been reported to increase in non-neuronal cells and neuronal
cells, suggesting that these molecules may be involved in promoting
axonal sprouting in the injured spinal cord (6). Furthermore, it has been demonstrated
that upregulation of IL-1β, BDNF and NT-3 in the injured spinal
cord is attenuated by treatment with high-dose glucocorticoids,
with the suggestion that the downregulation of BDNF and NT-3 may be
disadvantageous to the survival and axonal sprouting of spinal
neurons (7). As for the pathways
involved, a previous study revealed that the Rho signaling pathway
may be a potential target for therapeutic interventions following
SCI (8). In addition, apoptosis
signal-regulating kinase l and stress-activated mitogen-activated
protein kinase pathways, have also been reported to be involved in
the transmission of apoptotic signals following SCI (9). However, identification and evaluation
of specific and associated genes of SCI, which assist in the
clinical diagnosis and treatment of SCI, remain to be
elucidated.
In the present study, bioinformatics methods were
used to assess the abnormal gene expression in SCI to determine the
associated feature genes. Critical genes were screened using
expression profiling microarray data. Pathway analysis and
protein-protein interaction (PPI) network analysis were performed
on the proteins involved in SCI to investigate their function. The
aim of the present study was to explore the molecular mechanisms of
SCI and identify potential therapeutic target genes for the
treatment of SCI.
Materials and methods
Data preprocessing and differential
expression analysis
The transcription profile of GSE2599 was downloaded
from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/), which was based on
the Affymetrix Rat Genome U34 array (Affymetrix, Santa Clara, CA,
USA) and deposited by Aimone et al (10). A total of six tissue specimens were
available for further analysis, including three SCI samples,
obtained from female Fischer 344 rats (165–200 g) 35 days after
SCI, and three normal tissues, as described in the original
experiment (10). The annotation
information of all probe sets was provided by Affymetrix, where the
raw data (CEL) file was downloaded.
Initially, the probe-level data in the CEL files
were converted into expression measures. For each sample, the
expression values of all the probes for a particular gene were
reduced to a single value by calculating the average expression
value. Probes corresponding to more than one gene were discarded.
Subsequently, the data with the low signal strength was missing
data and the missing data was imputed using the K-nearest neighbor
averaging (KNN) method (11) and
the complete data were standardized (12). The Samr package in R language
(13) was used to identify
differentially expressed genes (DEGs) between three samples in the
control group (normal specimen) and three samples in the
experimental group (samples with SCI). In order to circumvent the
multi-test problem, which may induce an excess of false positive
results, the Benjamini-Hochberg procedure (14) was used to adjust the raw P-values
into false discovery rate (FDR). FDR<0.05 and |logFC|>1.5
were used as the cut-off criteria for DEG identification.
Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway and Gene Ontology (GO) enrichment analysis
Based on the deficiency of individual gene analysis,
gene set enrichment analysis evaluates differential expression
patterns of gene groups to distinguish whether their biological
functions and characteristics differ (15). In the present study, the P-value
indicated the probability that a gene was randomly endowed a GO
function and it was usually used as the criterion for assigning a
certain function to a module. A lower P-value increased the
probability that the function of a module had not been assigned
randomly, but with the purpose of performing a certain biological
function, and it has important biological significance (16). The Database for Annotation,
Visualization, and Integrated Discovery (DAVID) (17) bioinformatics resource consists of
an integrated biological knowledge base and analytical tools aimed
at systematically extracting biological meaning from lists of genes
or proteins (18). The functional
enrichment analysis for the screened DEGs was performed using
DAVID, and FDR<0.01 and P<0.05 were selected as the cut-off
criteria. Subsequently, KEGG pathway analysis was performed on the
upregulated and downregulated genes, obtained using DAVID, to
screen for disease-associated pathways.
Uniprot (UP) tissue analysis
GO analysis has become a commonly used approach for
functional investigations of large-scale genomic or transcriptomic
data (19). DAVID, a
high-throughput and integrated data-mining environment, analyzes
gene lists derived from high-throughput genomic experiments
(20).
In the present study, UP tissue analysis was
performed on DEGs in disease-associated metabolic pathways to
identify the genes associated with spinal cord tissue. Therefore,
the abnormally expressed genes in the injured spinal cord 35 days
after injury were selected to distinguish these genes from those,
which were expressed not solely in injured spinal cord.
Construction of the PPI network
PPI analysis was performed on the DEGs using Search
Tool for the Retrieval of Interacting Genes/Proteins (STRING;
http://www.string-db.org/) online database
(21). Combined_score was used to
measure the strength of the interaction of protein pairs and only
the interaction with combined_score > 0.4 was selected as
significant. Subsequently, critical genes, which exhibited >45
interactions with other genes, were selected. The feature genes
associated with SCI were identified by comparing the critical genes
with the DEGs 35 days after SCI. Finally, the PPI network was
constructed using Cytoscape software (http://cytoscape.org/) (22,23),
based on the STRING database, to determine the association between
feature genes and the interacting genes, which may trigger SCI.
Protein domain analysis of specific
genes
Coding area prediction of the critical genes
associated with SCI was performed using the GENSCAN (http://genes.mit.edu/GENSCAN.html) online
software programme (24).
Subsequently, the Pfam (25)
database was used to examine the protein domain for further protein
domain analysis.
Results
Data pre-processing and screening for
DEGs
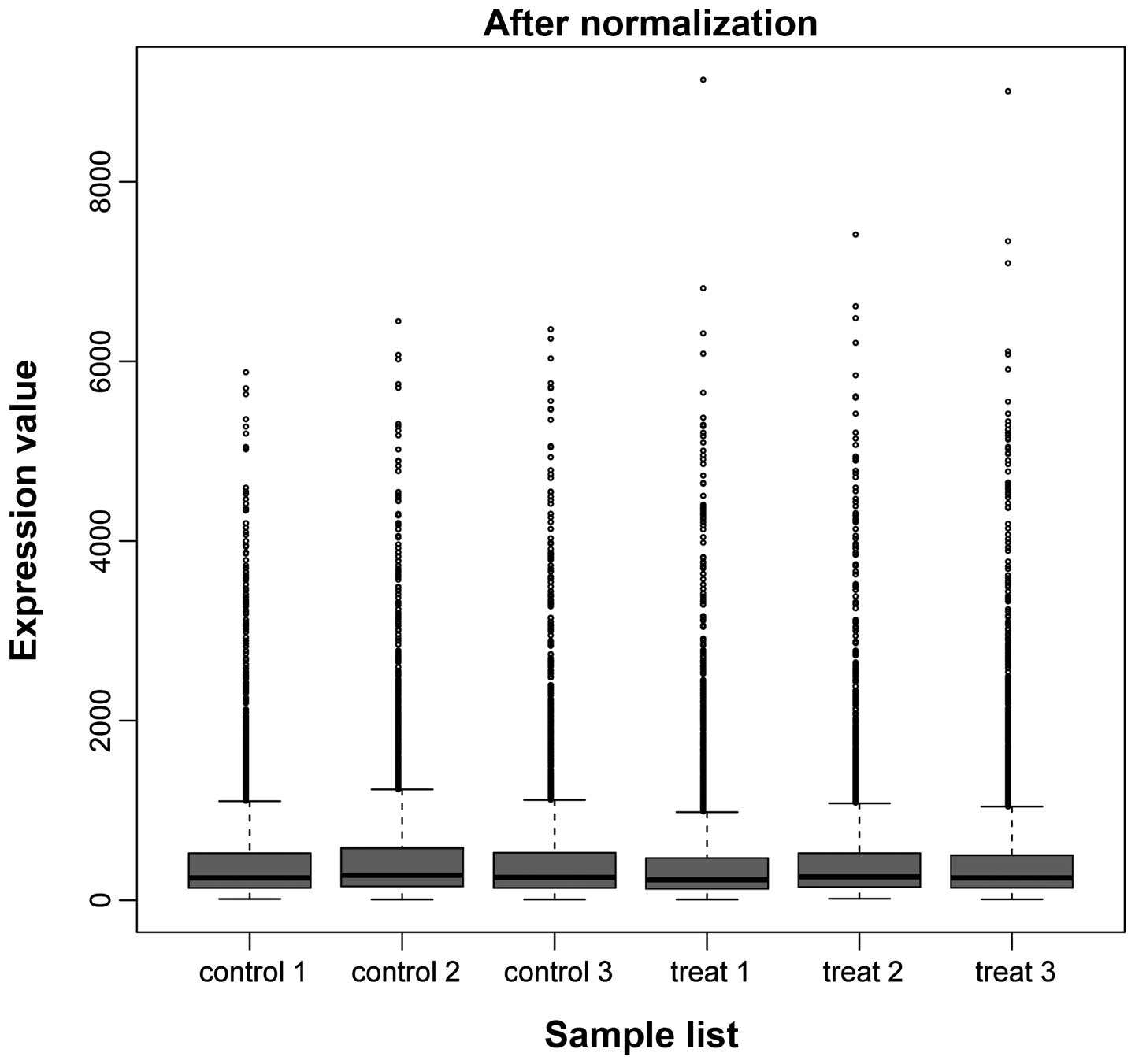
The results of data pre-processing are shown in
Fig. 1. Following data
pre-processing, the median was almost identical between the
samples, indicating good normalization and that the data was
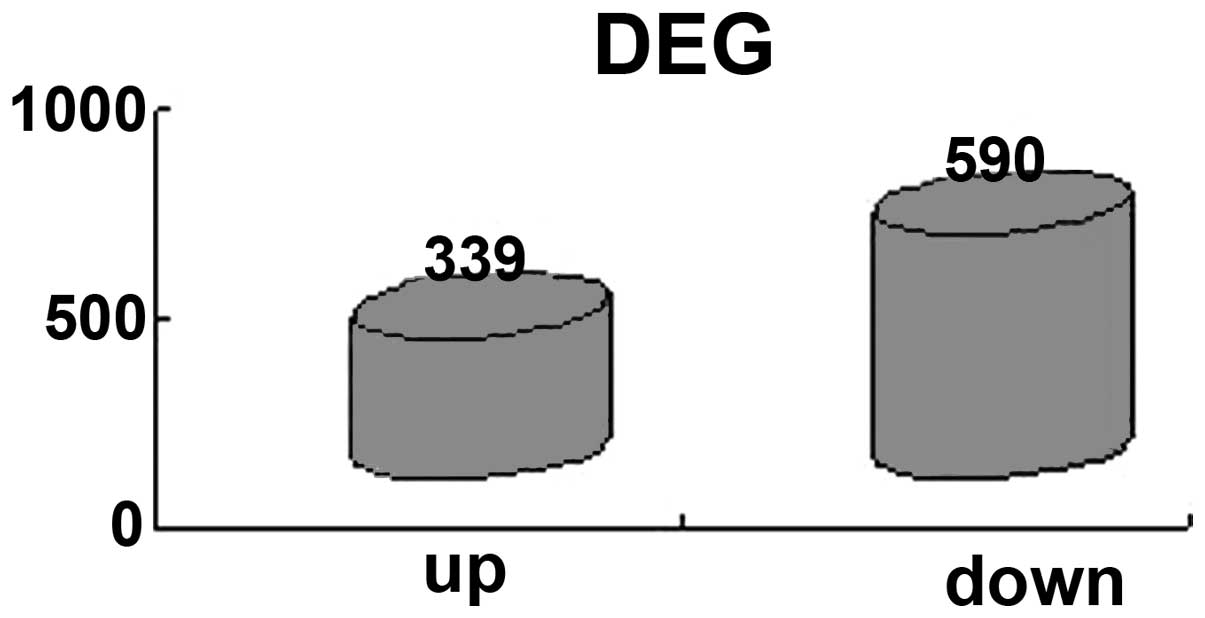
suitable for further analysis. A total of 929 DEGs were screened
for, including 339 upregulated genes and 590 downregulated genes
(Fig. 2).
KEGG pathway enrichment analysis
As shown in Table
I, the pathways associated with SCI included Huntington’s
disease (rno 05016), Parkinson’s disease (rno 05012) and
Alzheimer’s disease (rno 05010). A total of 39 mutual genes were
identified between these pathways and all of these genes were
downregulated, as shown in Table
II.
 | Table IKyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis. |
Table I
Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis.
| Term | P-value | False discovery
rate | Up/downregulated |
|---|
| rno03010:
Ribosome | 3.22 E-12 | 3.79 E-09 | Upregulated |
| rno04612: Antigen
processing and presentation | 4.13 E-08 | 4.85 E-05 | Upregulated |
| rno00190: Oxidative
phosphorylation | 2.50 E-23 | 2.98 E-20 | Downregulated |
| rno05016:
Huntington’s disease | 1.94 E-21 | 2.31 E-18 | Downregulated |
| rno05012: Parkinson’s
disease | 9.73 E-21 | 1.16 E-17 | Downregulated |
| rno05010:
Alzheimer’s disease | 7.00 E-20 | 8.33 E-17 | Downregulated |
| Table IIDownregulated genes in Huntington’s
disease, Parkinson’s disease and Alzheimer’s disease. |
Table II
Downregulated genes in Huntington’s
disease, Parkinson’s disease and Alzheimer’s disease.
| Uqcrc2 | Atp5o | Ndufb5 | Cox7a2 |
| Atp5d | Atp5j | Ndufb6 | Ndufa3 |
| Atp5b | Ndufb10 | Ndufb8 | Ndufa8 |
| Cyc1 | Cycs | Ndufb9 | Ndufa9 |
| Ndufab1 | Ndufc2 | Cox7b | Ndufa6 |
| Cox5a | Cox4i1 | Atp5g1 | Sdha |
| Uqcrfs1 | Uqcr | Ndufb2 | Ndufv2 |
| Cox5b | Atp5c1 | Ndufa4 | Cox6a1 |
| Ndufs7 | Ndufb3 | Loc688869 | Atp5a1 |
| Ndufs5 | Ndufb4 | Ndufa5 | |
GO enrichment analysis
DAVID was used to identify over-represented GO
categories among the genes (Table
II) and P<0.05 and FDR<0.01 were selected as thresholds.
The most markedly enriched five terms among these genes in the PPI
network were all associated with the chondriosome (Table III). GO terms associated with the
mitochondria, which were enriched in the network, included the
‘mitochondrial inner membrane’, ‘organelle inner membrane’ and
‘mitochondrial envelope’.
| Table IIIThe five most enriched genes in Gene
Ontology enrichment analysis. |
Table III
The five most enriched genes in Gene
Ontology enrichment analysis.
| Category | GO term | P-value | FDR |
|---|
| CC | 0005743:
Mitochondrial inner membrane | 4.40 E-53 | 4.14 E-50 |
| CC | 0019866: Organelle
inner membrane | 4.34 E-52 | 4.09 E-49 |
| CC | 0031966:
Mitochondrial membrane | 3.90 E-49 | 3.67 E-46 |
| CC | 0005740:
Mitochondrial envelope | 4.49 E-48 | 4.22 E-45 |
| CC | 0044429:
Mitochondrial part | 1.43 E-43 | 1.34 E-40 |
Identification of feature genes in
SCI
Downregulated genes, which were abnormally expressed
following SCI, were also involved in several known nerve disease
pathways, including Huntington’s disease (rno 05016), Parkinson’s
disease (rno 5012) and Alzheimer’s disease (rno 05010) KEGG
pathways. Combined with the annotation information of
spinal-cord-specific expressed genes from the Uniprot database, a
candidate set of SCI-associated feature genes was obtained,
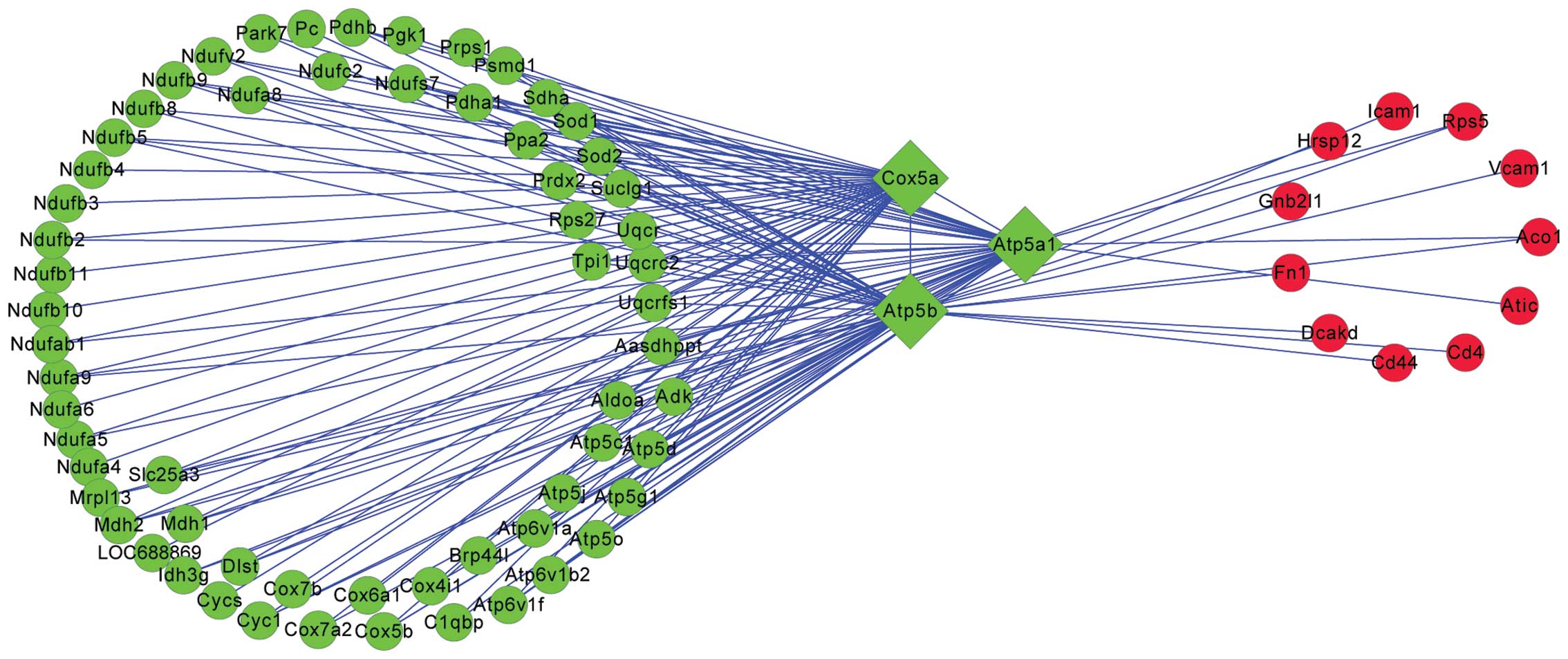
including Sdha, Uqcrc2, Ndufa5, Atp5b, Atp5a1 and Cox5a.
PPI network analysis
Feature genes were obtained by further analysis of
abnormally expressed genes in the injured spinal cord.
Subsequently, a PPI network was constructed, as shown in Fig. 3, which revealed that Atp5b, Atp5a1
and Cox5a, all downregulated genes, were closely associated with
the SCI when examined 35 days after the SCI. Additionally, the
majority of genes interacting with these three genes were also
downregulated.
Protein-domain analysis
The protein domain in the coding area of the Atp5b,
Atp5a1 and Cox5a feature genes, among genes that may be associated
with the disease at 35 days after the spinal cord injury are shown
in Table IV.
| Table IVProtein domain in coding areas of
feature genes associated with diseases 35 days after spinal cord
injury. |
Table IV
Protein domain in coding areas of
feature genes associated with diseases 35 days after spinal cord
injury.
| Gene | Family | Description | P-value |
|---|
| Cox5a | COX5A | Cytochrome c
oxidase subunit Va | 2.50 E-58 |
| Atp5a1 | ATP-synt_ab_N | ATP synthase α/β
family, β-barrel domain | 3.60 E-17 |
| Atp5b | ATP-synt_ab | ATP synthase α/β
family, nucleotide-binding domain | 2.50 E-72 |
| ATP-synt_ab_C | ATP synthase α/β
chain, C terminal domain | 1.50 E-26 |
As shown in Table
IV, the protein domain in the coding areas of Cox5a belonged to
the COX5A family, 13 sub-unit complex, EC: 1.9.3.1, which is the
terminal oxidase in the mitochondrial electron transport chain
(26). By contrast, the protein
domain in the coding areas of Atp5a and Atp5b belong to the ATP
synthase α and β family, including ATP-synt_ab_N, ATP-synt_ab and
ATP-synt_ab_C. The ATP synthase α/β family includes the ATP
synthase α and β subunits and ATP synthase, associated with
flagella (27).
Discussion
In the present study, it was demonstrated that the
three feature genes, Cox5a, Atp5al and Atp5b, in the injured spinal
cord, were rapidly downregulated 35 days after the onset of injury,
resulting in the destruction of the mitochondrial electron
transport chain and membrane-bound enzyme complexes/ion
transporters. These genes have been reported to be involved in the
pathways of several types of neurological disease, including
Huntington’s disease, Parkinson’s disease and Alzheimer’s disease
(28,29). Since these processes are associated
with the transportation of energy in biological bodies, changes to
these processes 35 days after SCI may result in disruption in the
transport of energy.
COX5A is a protein-coding gene. It is a
multi-subunit enzyme complex, which couples the transfer of
electrons from cytochrome c to molecular oxygen and
contributes to a proton electrochemical gradient across the inner
mitochondrial membrane (26).
Diseases associated with COX5A include acquired idiopathic
sideroblastic anemia and cardioencephalomyopathy (30). Its associated super-pathways
include the electron transport chain and metabolic pathways. GO
annotations associated with this gene include electron carrier
activity and cytochrome c oxidase activity (31). This indicates that COX5A may be
important in the regulation and assembly of the complex in the
human mitochondrial respiratory chain enzyme, thus, affecting
energy supply in SCI. A previous study revealed that COX5A is
associated with the migration, invasion and prediction of distant
metastasis (32). In the present
study, COX5A was markedly downregulated in SCI, therefore, it was
hypothesized that the downregulation of COX5A in SCI caused the
interdiction of energy transportation, interrupting the metabolic
process.
ATPases, or ATP synthases, are membrane-bound enzyme
complexes/ion transporters, which combine ATP synthesis and/or
hydrolysis with the transport of protons across a membrane. ATPases
harness the energy from a proton gradient, using the flux of ions
across the membrane via the ATPase proton channel, to drive the
synthesis of ATP (33). Atp5a1
(34) and Atp5b (35) are also protein-coding genes.
Super-pathways associated with the genes include the electron
transport chain and adenosine ribonucleotides de novo
biosynthesis. GO annotations associated with ATP5A1 include
eukaryotic cell surface binding and ATPase activity (36), while those for ATP5B include
transmembrane transporter activity and transporter activity
(37,38). Deregulated energy metabolism is a
marker of malignant disease, which offers possible future targets
for treatment (39). Polymorphism
and association analysis has revealed that mutations in Atp5a1 and
Atp5b genes may be potential markers of diseases associated with
the destruction of energy transport (40). Atp5a1 and Atp5b, which are involved
in energy transportation in mitochondria, may be critical genes and
certain variations of these genes may lead to increased risk in SCI
(40).
In addition, the results obtained from GO enrichment
analysis of the PPI network in the present study demonstrated that
most enriched GO terms of the DEGs in SCI were associated with
mitochondria, including ‘mitochondrial electron transport chain’,
‘mitochondrial membrane’ and ‘mitochondrial envelope’. This
suggested that the majority of DEGs in SCI were associated with
energy transportation and that the progression of SCI may be
affected by the genes expressed differently in the tissue.
Therefore, the 39 mutual genes in Huntington’s disease, Parkinson’s
disease and Alzheimer’s disease, which coordinate with genes in
SCI, may assist in defining the origins of malignancies and offer
promise for earlier diagnosis and improved treatment of SCI.
In conclusion, the results of the present study
presented a comprehensive bioinformatics analysis of genes and
pathways, which may be involved in the progression of SCI. A total
of 929 DEGs were identified from GSE2599, and PPI networks were
constructed using these DEGs. Furthermore, the Cox5a, Atp5al and
Atp5b genes, which were downregulated in SCI, were found to result
in the destruction of the mitochondrial electron transport chain
and membrane-bound enzyme complexes/ion transporters, thus
affecting the normal function of nerves. These genes can be
identified as feature genes of SCI and assist in the early
diagnosis and improved treatment of SCI.
References
|
1
|
Richards JS, Kewman DG, Pierce CA, Frank R
and Elliott T: Spinal cord injury. Handbook of rehabilitation
psychology. 11–27. 2000. View Article : Google Scholar
|
|
2
|
Karimi MT: Evidence-Based Evaluation of
Physiological Effects of Standing and Walking in Individuals with
Spinal Cord Injury. Iran J Med Sci. 36:2422011.
|
|
3
|
Mcdonald JW and Sadowsky C: Spinal-cord
injury. The Lancet. 359:417–425. 2002. View Article : Google Scholar
|
|
4
|
Bareyre FM and Schwab ME: Inflammation,
degeneration and regeneration in the injured spinal cord: insights
from DNA microarrays. Trends Neurosci. 26:555–563. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fraser A and Edmonds-Seal J: Spinal cord
injuries. Anaesthesia. 37:1084–1098. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xia T, Ni S, Li X, et al: Sustained
delivery of dbcAMP by poly (propylene carbonate) micron fibers
promotes axonal regenerative sprouting and functional recovery
after spinal cord hemisection. Brain Res. 1538:41–50. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hayashi M, Ueyama T, Nemoto K, Tamaki T
and Senba E: Sequential mRNA expression for immediate early genes,
cytokines, and neurotrophins in spinal cord injury. J Neurotrauma.
17:203–218. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dergham P, Ellezam B, Essagian C,
Avedissian H, Lubell WD and Mckerracher L: Rho signaling pathway
targeted to promote spinal cord repair. J Neurotrauma.
22:6570–6577. 2002.
|
|
9
|
Nakahara S, Yone K, Sakou T, et al:
Induction of apoptosis signal regulating kinase 1 (ASK1) after
spinal cord injury in rats: possible involvement of ASK1-JNK
and-p38 pathways in neuronal apoptosis. J Neuropathol Exp Neurol.
58:442–450. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aimone JB, Leasure JL, Perreau VM and
Thallmair M: Spatial and temporal gene expression profiling of the
contused rat spinal cord. Experimental neurology. 189:204–221.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Troyanskaya O, Cantor M, Sherlock G, et
al: Missing value estimation methods for DNA microarrays.
Bioinformatics. 17:520–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujita A, Sato JR, de Rodrigues LO,
Ferreira CE and Sogayar MC: Evaluating different methods of
microarray data normalization. BMC Bioinformatics. 7:4692006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smyth GK: Limma: linear models for
microarray data. Bioinformatics and Computational Biology Solutions
using R and Bioconductor. Springer; New York: pp. 397–420. 2005,
View Article : Google Scholar
|
|
14
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: a practical and powerful approach to
multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300.
1995.
|
|
15
|
Nam D and Kim SY: Gene-set approach for
expression pattern analysis. Brief Bioinform. 9:189–197. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Allison DB, Cui X, Page GP and Sabripour
M: Microarray data analysis: from disarray to consolidation and
consensus. Nat Rev Genet. 7:55–65. 2006. View Article : Google Scholar
|
|
17
|
Huang Da Wei BTS and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2008. View Article : Google Scholar
|
|
18
|
Huang Dw SB and Lempicki Ra: Systematic
and integrative analysis of large gene lists using DAVID
Bioinformatics Resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
19
|
Hulsegge I, Kommadath A and Smits MA:
Globaltest and GOEAST: two different approaches for Gene Ontology
analysis. BMC Proc. 3(Suppl 4): S102009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang Da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Franceschini A, Szklarczyk D, Frankild S,
et al: STRING v9.1: protein-protein interaction networks, with
increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar :
|
|
22
|
Shannon P, Markiel A, Ozier O, et al:
Cytoscape: a software environment for integrated models of
biomolecular interaction networks. Genome Res. 13:2498–2504. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saito R, Smoot ME, Ono K, et al: A travel
guide to Cytoscape plugins. Nat Methods. 9:1069–1076. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Burge C and Karlin S: Prediction of
complete gene structures in human genomic DNA. J Mol Biol.
268:78–94. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bateman A, Coin L, Durbin R, et al: The
Pfam protein families database. Nucleic Acids Res. 32:D138–D141.
2004. View Article : Google Scholar :
|
|
26
|
Malström BG: Cytochrome c oxidase
Structure and catalytic activity. Biochim Biophys Acta.
549:281–303. 1979. View Article : Google Scholar
|
|
27
|
Sauer K, Cullen M, Rickard A, Zeef L,
Davies D and Gilbert P: Characterization of nutrient-induced
dispersion in Pseudomonas aeruginosa PAO1 biofilm. J Bacteriol.
186:7312–7326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang WS, Reiman EM, Valla J, et al:
Alzheimer’s disease is associated with reduced expression of energy
metabolism genes in posterior cingulate neurons. Proc Natl Acad
Sci. 105:4441–4446. 2008. View Article : Google Scholar
|
|
29
|
Emahazion T, Jobs M, Howell WM, Siegfried
M, Wyöni P-I, Prince JA and Brookes AJ: Identification of 167
polymorphisms in 88 genes from candidate neurodegeneration
pathways. Gene. 238:315–324. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Capaldi RA: Structure and function of
cytochrome c oxidase. Annu Rev Biochem. 59:569–596. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miller BR and Cumsky MG: An unusual
mitochondrial import pathway for the precursor to yeast cytochrome
c oxidase subunit Va. J Cell Biol. 112:833–841. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen W-L, Kuo K-T, Chou T-Y, et al: The
role of cytochrome c oxidase subunit Va in non-small cell lung
carcinoma cells: association with migration, invasion and
prediction of distant metastasis. BMC cancer. 12:2732012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rappas M, Niwa H and Zhang X: Mechanisms
of ATPases - a multi-disciplinary approach. Curr Protein and Pept
Sci. 5:89–105. 2004. View Article : Google Scholar
|
|
34
|
DeKloet SR: Loss of the Gene for the
Subunit of ATP Synthase (ATP5A1) from the W Chromosome in the
African Grey Parrot (Psittacus erithacus). J Mol Evol. 2:2001.
|
|
35
|
Zheng S-Q, Li Y-X, Zhang Y, Li X and Tang
H: MiR-101 regulates HSV-1 replication by targeting ATP5B.
Antiviral Res. 89:219–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jonckheere AI, Renkema GH, Bras M, et al:
A complex V ATP5A1 defect causes fatal neonatal mitochondrial
encephalopathy. Brain. 136:1544–1554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Doi K and Uetsuka K: Mechanisms of
mycotoxin-induced neurotoxicity through oxidative stress-associated
pathways. Int J Mol Sci. 12:5213–5237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sineshchekova OO, Kawate T, Vdovychenko OV
and Sato TN: Protein-trap version 2.1: screening for expressed
proteins in mammalian cells based on their localizations. BMC Cell
Biol. 5:82004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hjerpe E, Brage SE, Carlson J, et al:
Metabolic markers GAPDH, PKM2, ATP5B and BEC-index in advanced
serous ovarian cancer. BMC Clin Pathol. 13:302013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gunawan A, Sahadevan S, Cinar MU, et al:
Identification of the Novel Candidate Genes and Variants in Boar
Liver Tissues with Divergent Skatole Levels Using RNA Deep
Sequencing. PloS One. 8:e722982013. View Article : Google Scholar : PubMed/NCBI
|