Introduction
Sulforaphane,
[1-isothiocyanato-4-(methylsulfinyl)-butane; (SFN)] is an
isothiocyanate compound found in cruciferous vegetables, including
broccoli and cauliflower. SFN is a well-known cancer
chemopreventive agent, which may also influence normal cells
(1). Two putative mechanisms of
action underlying the anti-cancer effect of isothiocyanates have
been identified. The first mechanism is based on the activation of
the phase 2 detoxification enzymes, such as glutathione
S-transferase, and/or the simultaneous inhibition of the phase 1
enzymes, such as cytochrome P450 isoenzyme 2E1, which may be
responsible for the anti-carcinogenic activity (2,3).
Upregulation of phase 2 enzymes, including glutathione
transferases, protects cells against carcinogens and has previously
been shown to prevent carcinogen-induced tumors in various animal
models (1,2,4). The
second mechanism refers to the suppression of tumor development by
the induction of apoptosis, growth inhibition and cell cycle arrest
(5–8). At the molecular level, the protective
properties of SFN rely on activation of the proapoptotic B-cell
lymphoma 2 (Bcl-2)-associated X protein (Bax), resulting in
apoptosis of endothelial progenitor cells and consequently reducing
tumor growth (9). In addition, SFN
has been shown to inhibit histone deacetylase activity and cause
histone hyperacetylation in cancer cells, including human colon
cancer (10).
The induction of apoptosis by SFN has previously
been observed in numerous cancer models, including the prostate
cancer cell lines PC-3 and LNCaP (8) and the human colon cancer cell line
HT29 (5). However, the exact
mechanism underlying the induction of apoptosis by SFN remains to
be elucidated. SFN-induced apoptosis in human prostate cancer cell
lines was shown to be initiated by the production of reactive
oxygen species (ROS) (7,11). In addition, caspase 8 is associated
with SFN-induced apoptosis and its activity depends on ROS
production (7).
SFN was previously reported to induce autophagy in
prostate cancer cell lines, which provided protection against the
induction of apoptosis by SFN (8).
The physiological function of autophagy is to maintain a natural
balance between existing and forthcoming synthesized proteins and
organelles. Autophagy literally means ‘self-eating’ and is a
process of cellular self-digestion which may lead to cell death
(12). The process of digesting
organelles and molecules occurs in double-membrane autophagosomes
(13). The protein beclin-1
interacts with a class III phosphatidyl-inositol 3-kinase (PI3K)
and initiates autophagosomal membrane assembly (14). Furthermore, microtubule-associated
protein light chain 3 (MAP-LC3 or LC3) is incorporated into the
autophagosome membrane, which may be tracked until fusion with the
lysosome vesicle (15). MAP-LC3
exists in two forms: cytoplasmic LC3-I (18 kDa) and
autophagosome-associated LC3-II (16 kDa), which has undergone
proteolytic cleavage (16). In the
PC-3 prostate cancer cell line, SFN causes redistribution of LC3
into autophagosomes and 3-methyladenine (3-MA), an inhibitor of
autophagy, is able to restrain this process (8). The adenine analogue blocks class III
PI3K complex activities and the mammalian target of rapamycin
pathway, leading to the inhibition of autophagy (17).
The present study used the BE(2)-C human neuroblastoma cell line
containing the amplified MYCN gene (responsible for high
levels of aggressiveness) and P53 mutation, to characterize
the effects of synthetic SFN on cells in vitro. The effects
of SFN on caspase activity, the mitochondrial membrane potential
and Bax levels were also investigated. The results of the present
study confirmed the effectiveness of SFN in the induction of
apoptosis in the BE(2)-C cell line
as well as its ability to induce early autophagy. The present study
showed that the combined treatment of the cells with SFN with 3-MA
enhanced cell death.
Materials and methods
Reagents
D, L-Sulforaphane was synthesized at the University
of Agriculture (Kraków, Poland), according to the method by Schmidt
and Karrer (18). Purification of
the compound was performed by high-performance liquid
chromatography and the identity of the SFN was verified by nuclear
magnetic resonance and ultraviolet spectroscopy. A stock solution
of SFN (7.2 M in serum-free medium) was stored at −20°C until
further use. SFN was diluted with culture medium immediately prior
to use. 3-MA, staurosporine (STS) and antibodies against GAPDH
(G8795) were purchased from Sigma-Aldrich (Poznań, Poland). Trypsin
(0.25%) was obtained from Gibco Life Technologies (Carlsbad, CA,
USA), and (6-3H) thymidine was from GE Healthcare Life
Sciences (Little Chalfont, UK). Rabbit antibodies against LC3B
(3868) and beclin-1 (3495) were purchased from Cell Signaling
Technology (Danvers, MA, USA). An antibody against Bcl-2 (sc-783)
was obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and an
antibody against Bax (556467) was obtained from BD Pharmingen (San
Diego, CA, USA). Secondary horseradish peroxidase (HRP)-conjugated
goat anti-rabbit immunoglobulin G (IgG) (A0545) and anti-mouse IgG
antibodies (A9044) were obtained from Sigma-Aldrich. The secondary
anti-rabbit IgG HRP-linked antibodies (7074) were from Cell
Signaling Technology. All of the other chemicals and reagents used
in the experiments of the present study were of analytical
grade.
Cell culture
The BE(2)-C human
neuroblastoma cell line was obtained from the American Type Culture
Collection (Manassas, VA, USA; ATCC number, CRL-2268). The
BE(2)-C cell line is a clone of
the SK-N-BE(2) neuroblastoma cell
line. The cells were cultured in a 1:1 mixture of Eagle’s minimum
essential medium and F12 medium, supplemented with 10% fetal calf
serum, 1% non-essential amino acid solution, 1 mM sodium pyruvate
and 50 μg/ml gentamicin (all from Sigma Aldrich) at 37°C in
an atmosphere containing 5% CO2.
MTT assay
The MTT assay is based on the ability of living
cells to reduce the MTT tetrazolium salt to MTT formazan by the
mitochondrial enzyme succinate dehydrogenase (19). The BE(2)-C cells (2.5×104/well) were
cultured in 96-well plates for 24 h and treated with increasing
concentrations of synthetic SFN for the indicated time periods. MTT
(Sigma-Aldrich) was added to each well to a final concentration of
0.5 mg/ml. Following a 4-h incubation at 37°C, the formazan
crystals were dissolved in isopropanol containing 5 mM HCl. The
absorbance was measured at wavelengths of 570 and 630 nm using the
SpectraMax 250 microplate reader (Molecular Devices, Sunnyvale, CA,
USA). The 50% maximal inhibitory concentration (IC50)
was defined as the concentration that resulted in a 50% decrease in
the absorbance of the SFN-treated cells, as compared with the
media-grown cells. In separate experiments, the BE(2)-C cells (2.5×104/well) were
cultured in 96-well plates for 24 h. The medium was then removed
and the cells were treated with either 40 μM synthetic SFN,
10 mM 3-MA or a combination of 40 μM synthetic SFN and 10 mM
3-MA. Untreated control cells were also included in the
experiments. After 48 h, the MTT assay was performed on the cells,
as described above. The measurements were run in triplicate and the
experiments were repeated three times.
Lactate dehydrogenase (LDH) activity
measurement
The BE(2)-C cells
(2.5×104/well) were cultured in 96-well plates for 24 h.
The medium was then removed and the cells were treated with either
40 μM synthetic SFN, 10 mM 3-MA or a combination of 40
μM synthetic SFN and 10 mM 3-MA. Untreated control cells
were also included in the experiments. After 48 h, the cell
cultures were centrifuged at 250 × g for 10 min at room
temperature. The medium was removed and the cells were lysed using
1% Triton-X 100 (Sigma-Aldrich) in serum-free culture medium. LDH
activity was measured using the Cytotoxicity Detecion kit (LDH)
from Roche Diagnostics (Warsaw, Poland) according to the
manufacturer’s instructions. The measurements were run in
triplicate and the experiments were repeated three times.
Crystal violet staining
The BE(2)-C cells
(2.5×104/well) were cultured in 96-well plates for 24 h.
The medium was then removed and the cells were treated with either
40 μM synthetic SFN, 10 mM 3-MA or a combination of 40
μM synthetic SFN and 10 mM 3-MA. Untreated control cells
were also included in the experiments. After 48 h, the cells were
collected and centrifuged at 250 × g for 10 min at room
temperature. The culture medium was then removed, the cells were
fixed with 0.1% crystal violet (Sigma-Aldrich) in 20% ethanol
(Sigma-Aldrich) for 10 min and washed once with distilled water.
The cells were observed using a Nikon Eclipse TS100 microscope
(Nikon Corooration, Tokyo, Japan). Images of the cells were
captured using a digital camera (Canon Powershot A1200; Canon,
Inc., Tokyo, Japan).
Thymidine incorporation
A thymidine incorporation assay was performed in
order to measure the rate of DNA synthesis. The BE(2)-C cells were seeded in a 24-well plate
(1.6×105 cells/well) and allowed to attach. After 24 h
the medium was refreshed and the indicated concentrations of SFN
(10-100 μM) were added. The cells treated with media alone
were used as control cells. Following a 24-h incubation, the media
was replaced with 400 μl isotope solution (activity, 1
μCurie/ml). The samples were prepared according to the
methods by Riss and Sirbasku (20). After 24 h, the cells were fixed
with 800 μl cold (4°C) methanol-acetic acid solution (3:1
proportion; POCH, Gliwice, Poland). The solution was then aspirated
and each well was washed with 80% methanol and dried for 30 min at
room temperature. A total of 125 μl trypsin was added to
each well and incubated for 30 min at 37°C. Following incubation,
125 μl 1% SDS solution (Sigma-Aldrich) was added and plates
were incubated for 30 min at 25°C. Finally, 20 μl of each
sample was mixed with 3 ml scintillator carrier (Akwascynt, POCH).
The measurements were performed using the 1211 Rackbeta Liquid
Scintillation Counter (LKB Wallac; LKB Instruments, Victoria,
Australia). Four cycles of measurements were performed and the
first measurement was discarded. The results are presented as plots
of counts per minute for 1 μg of whole-cell protein content.
The protein concentration was determined using the bicinchoninic
acid assay, according to the manufacturer’s instructions
(Sigma-Aldrich). The measurements were run in triplicate and the
experiment was repeated three times.
Caspase activation assay
The activation of caspases 2, 3, 6, 7, 8, 9 and 10
was determined using the Homogeneous Caspases Assay (Roche
Diagnostics) according to the manufacturer’s instructions. The
BE(2)-C cells (4×104)
were seeded onto 96-well plates with black sides and a transparent,
flat bottom (Corning-Costar, Corning, NY, USA), and allowed to
attach. Following a 24-h incubation, the medium was replaced and
the cells were treated with either the medium alone (control) or
the indicated concentrations of SFN and/or 3-MA for the indicated
periods of time. The samples were tested in duplicate, the
fluorescent signals were measured using the Infinite®
200 PRO multimode reader (Tecan Group Ltd, Maennedorf,
Switzerland).
Immunoblotting
The BE(2)-C cells
(1×106) were grown on 55-mm diameter plates and treated
with SFN and/or 3-MA, as described above. Whole cell extracts were
obtained according to the methods of Sadowski et al
(21). Briefly, following the
indicated period of incubation, the media was removed and the cells
were collected in cold phosphate-buffered saline (PBS), then washed
with PBS and centrifuged at 140 × g for 5 min at 4°C. The samples
were then resuspended in 70-100 μl lysis buffer (50 mM
Tris-HCl, pH 8.0, 10 mM 3-[(3-cholami-dopropyl)
dimethylammonio]-1-propanesulfonate, 2 mM EDTA, 1 mM
Na3VO4, 5 mM NaF, 1 mM dithiothreitol, 1 mM
phenylmethanesulfonylfluoride; 10% glycerol) and incubated on ice
for 20 min. All reagents were purchased from BioShop Canada, Inc.,
(Burlington, ON, Canada). The lysates were aspirated through a
syringe (0.6-mm diameter) and centrifuged at 11,400 × g for 5 min
at 4°C. The supernatants were collected and stored at −20°C until
further use. The protein lysates were separated by SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (Hybond P;
Merck Millipore, Warsaw, Poland). The membranes were treated with a
solution containing 10 mM Tris (pH 7.4; BioShop Canada), 150 mM
NaCl (POCH), 0.05% Tween® 20 (Sigma-Aldrich) and 5%
nonfat dry milk (Cell Signalling Technology, Inc.) and incubated
with the desired primary antibody for 1 h at room temperature.
Antibodies, raised against human antigens, included rabbit
monoclonal anti-beclin-1 antibody (1:1,000) and rabbit monoclonal
anti-LC3B antibody (1:1,000), from Cell Signaling Technology, Inc.
Rabbit polyclonal anti-Bcl-2 antibody (DC 21, 1:1,000; sc-783) and
mouse monoclonal anti-Bax antibody (B-9, 1:1,000; sc-7480) from
Santa Cruz Biotechnology, Inc. and mouse monoclonal anti-GAPDH
antibody G8795 (1:40,000) from Sigma-Aldrich. Following incubation
with these primary antibodies, the membranes were washed three
times for 5 min with the solution containing 10 mM Tris (pH 7.4),
150 mM NaCl with 0.1 % Tween® 20. After the washing steps, the
membranes were treated with the appropriate HRP-conjugated
secondary antibodies for 1 h at room temperature. The secondary
antibodies with indicated dilutions were as follows: rabbit
anti-mouse IgG-HRP, A9044, 1:50,000 (Sigma-Aldrich); goat
anti-rabbit IgG-HRP, A0545, 1:20,000 (Sigma-Aldrich); goat
anti-rabbit IgG-HRP, #7074, 1:2,000 (Cell Signaling Technology).
After secondary antibodies incubation, membranes were washed as
described above. The immunoreactive bands were visualized using an
Enhanced Chemiluminescence substrate (Immobilon Western HRP
substrate; Merck Millipore), according to the manufacturer’s
instructions. The intensity of the immunoreactive bands was
determined by densitometric scanning in order to quantify the
changes to the protein expression levels (Quest Spot Cutter,
Quantity One, version 4.6.9 software; Bio-Rad Laboratories,
Hercules, CA, USA). The experiment was repeated between two and
four times and representative results are shown.
Flow cytometric analysis of the
mitochondrial membrane potential
To measure the mitochondrial membrane potential
(∆ψm) in the neuroblastoma cells, flow cytometric
analyses were performed using the MitoTracker® Red
CMXRos dye (Invitrogen Life Technologies, Carlsbad, CA, USA). The
cells were plated and treated as described above. Cells treated
with 60 μM STS, an unspecific protein kinase inhibitor, were
used as a positive control. The CMXRos staining protocol was
performed as follows. The BE(2)-C
cells were collected, centrifuged at 250 × g for 10 min at 4°C and
stained with 1 ml 500 nM CMXRos solution per 1 ml cell suspension
[in medium with 10% fetal bovine serum (FBS), 15 min, 37°C]. The
cells were then washed twice with media supplemented with 2% FBS
and resuspended in 0.5 ml media. The cells were analyzed using a
FACScan (BD Biosciences, Franklin Lakes, NJ, USA). The experiment
was repeated three times.
Statistical analysis
Values are presented as the means. Error bars of the
means in Figs. 1B and C and
2A were calculated according to
the law of propagation of uncertainty. Otherwise, the error bars
denote the standard error of the mean. Experiments were repeated
between two and four times. A series of pairwise tests (t-test)
comparing the differences between the means of the groups, were
also performed. This was calculated using Microsoft Office Excel
2003; Microsoft Corporation, Redmond, WA, USA). P<0.05 was
considered to indicate a statistically significant difference
(*P<0.05; **P<0.01;
***P<0.001).
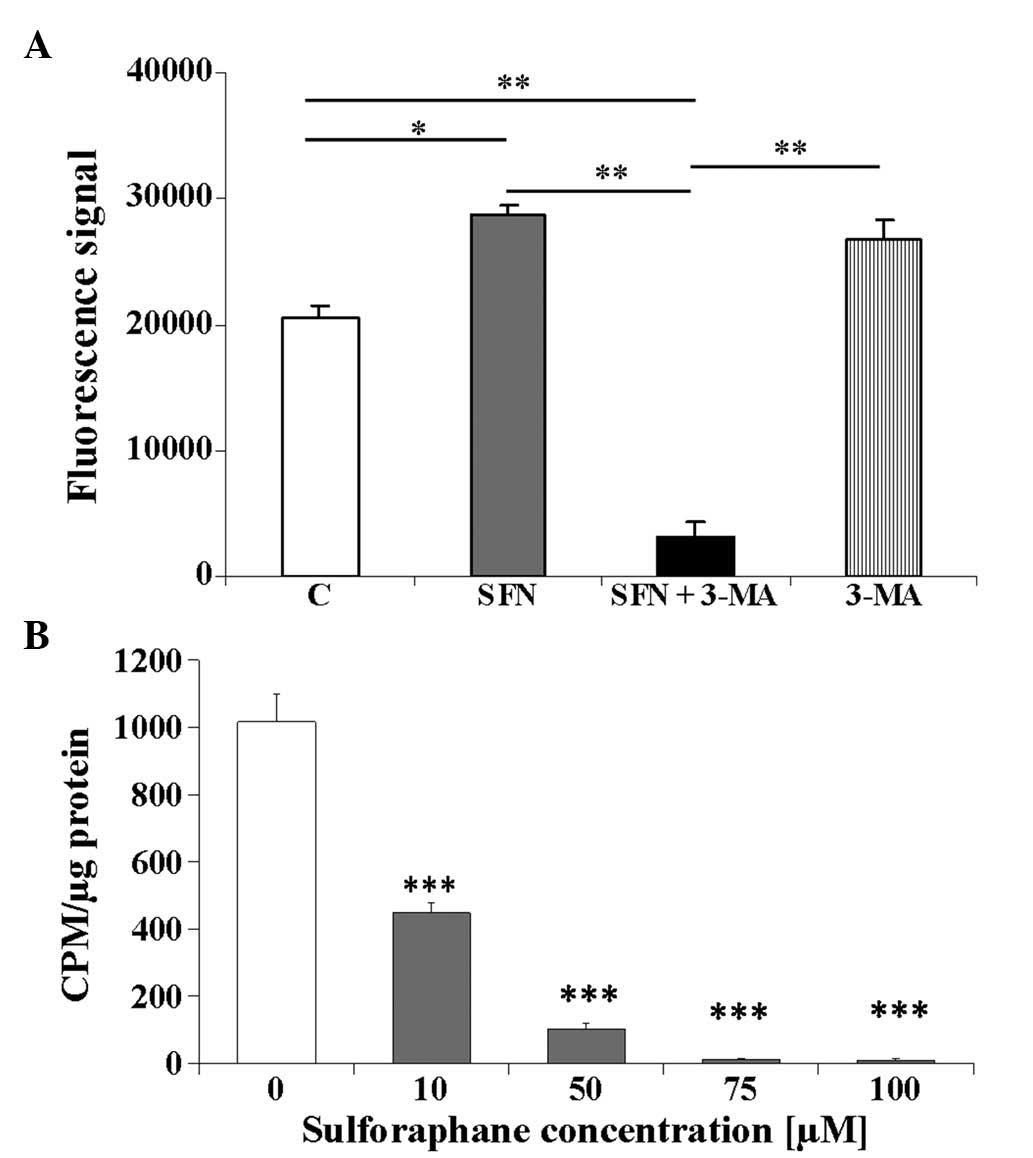 | Figure 2Caspase activity and DNA synthesis in
the BE(2)-C neuroblastoma cells
treated with SFN and 3-MA. The BE(2)-C cell cultures were treated with 40
μM SFN, 10 mM 3-MA or 40 μM SFN in combination with
10 mM 3-MA. (A) After treatment for 48 h, caspase activity was
measured in the cellular extracts. Values are presented as the
means from duplicated experiments, with error bars calculated
according to the law of propagation of uncertainty. One
representative experiment of three independent experiments is
shown. A series of pairwise t-tests were performed to compare the
mean values for the treated, vs. control cells; and the single, vs.
combined treatment groups. (B) Results of 3H-thymidyne
incorporation assay. A total of 1 μCurie/ml
3H-thymidine was added for 24 h following SFN treatment
for 24 h. The results are shown as CPMs per μg of total
cellular protein. Values are expressed as the mean ± standard error
of the mean. A series of pairwise t-tests, comparing the means of
the treated, vs. control cells was performed.
*P<0.05; **P<0.01;
***P<0.001. SFN, sulforaphane; CPM, counts per
minute; 3-MA, 3-methyladenine; C, untreated control. |
Results
SFN-induced inhibition of the growth of
BE(2)-C cells
In order to determine the inhibitory influence of
SFN on the BE(2)-C neuroblastoma
cell line, the cells were cultured with increasing concentrations
of the synthetic isothiocyanate (5-100 μM) for 24 and 48 h.
Untreated cells were also included. The IC50-value after
24 h was calculated as 38 μM (data not shown). Therefore,
based on earlier assays and previously published results (8,22),
40 μM SFN was subsequently selected for further experiments.
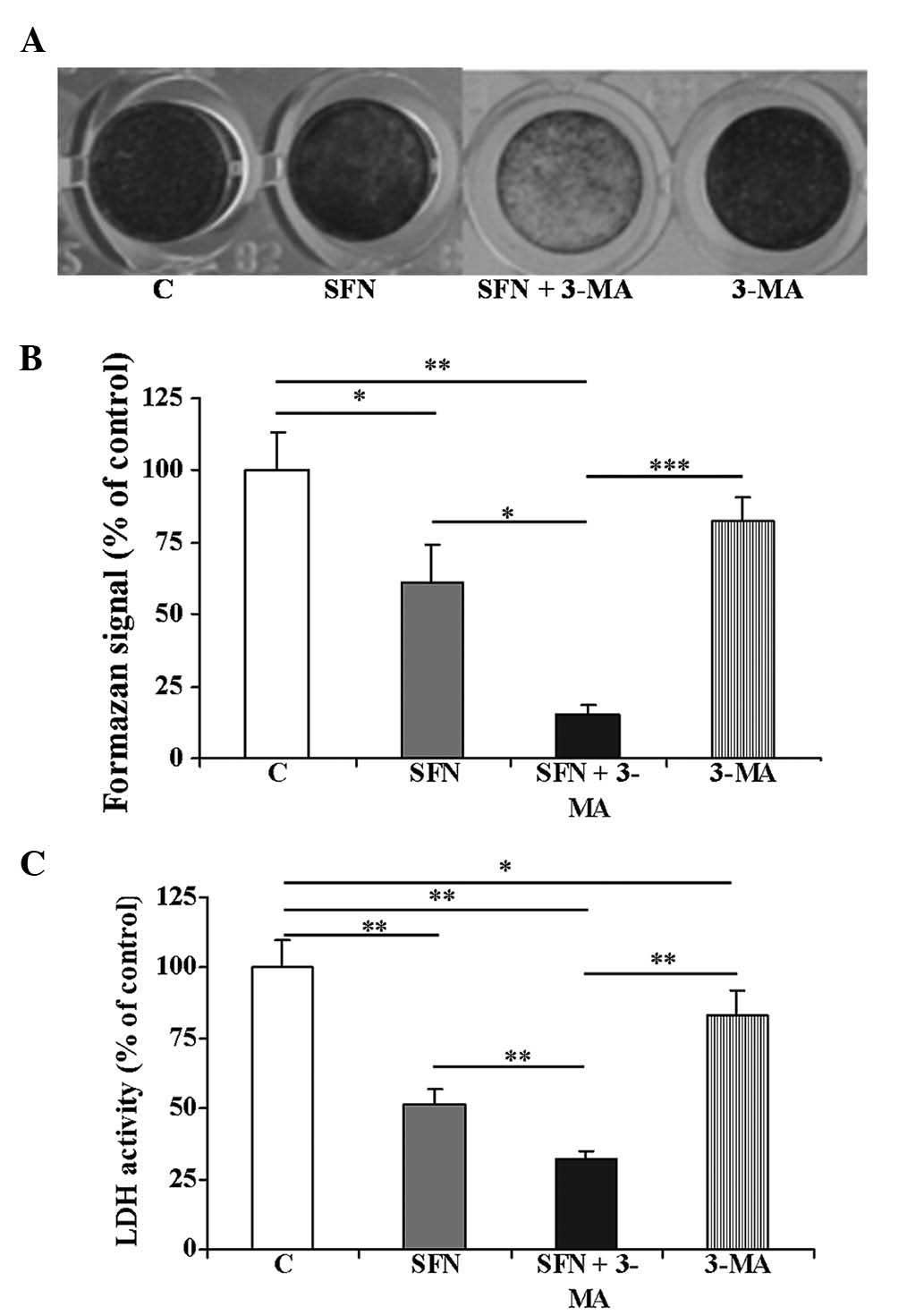
Microscopic evaluation (data not shown) and crystal violet staining
of the SFN-treated cells suggested that SFN treatment caused cell
death and after 48 h, the cells began to detach from the culture
dish (Fig. 1A). Combinatorial
treatment of the cells with SFN with 3-MA resulted in a marked
detachment of cultured cells. Treatment with 3-MA alone did not
alter the morphology of the cells. To assess the cytotoxicity of
SFN on the cells, the cell viability was analyzed using an MTT
assay (Fig. 1B). The inhibitory
effects of SFN were clearly visible at 48 h. Treatment of the
BE(2)-C cells with 40 μM
SFN also resulted in decreased cellular levels of LDH, a cytosolic
enzyme that leaks from damaged cells (Fig. 1C). Treatment with 3-MA slightly
decreased the viability of the cells, as estimated by MTT assay,
crystal violet staining and analysis of LDH activity (Fig. 1). However, the combined treatment
with SFN and 3-MA resulted in a statistically significant decrease
in the cellular parameters measured as compared with SFN treatment
alone (Fig. 1B and C; P<0.05
and P<0.01, respectively).
Purified broccoli sprout extracts containing ~90%
SFN, used in our preliminary studies, have also shown potent
inhibitory effects on the growth of numerous human cancer cell
lines, including BE(2)-C
neuroblastoma, HepG2 hepatoma and SK-MEL-28 melanoma (data not
shown).
SFN induces caspase activation and
decreases DNA content in neuroblastoma cells
To investigate whether apoptosis may be responsible
for the decreased survival of the BE(2)-C cells treated with SFN, total
activity of caspases 2, 3, 6, 7, 8, 9 and 10 was assayed. The cells
(4×104 per well) were cultured in the presence of 40
μM SFN for 48 h. Treatment with SFN resulted in a
significant ~1.5-fold increase in the total activity of caspases 2,
3, 6, 7, 8, 9, and 10 (Fig. 2A;
P<0.05) compared with the control cells. Furthermore, treatment
with autophagy inhibitor 3-MA (10 mM) increased caspase activity
when used alone (Fig. 2A). This
effect was expected, as the inhibition of autophagy may be
associated with increased apoptotic cell death (23). However, treatment with a
combination of 3-MA and 40 μM SFN resulted in a significant
decrease in caspase activity, possibly due to prolonged incubation
with high doses of SFN and 3-MA (Fig.
2A; P<0.01), compared with the control cells and to single
agents treatments. As the exact types of caspases involved in the
process cannot be distinguished by the test used in the present
study, it is possible that caspases of the receptor and
mitochondrial pathways are involved in SFN-induced apoptosis
(7).
The higher concentrations of SFN also inhibited DNA
synthesis. The effects of SFN on its ability to repress
3H-thymidine incorporation into DNA were studied at four
different concentrations (10, 50, 75 and 100 μM). The rate
of 3H-thymidine incorporation decreased significantly
with increasing SFN concentration (Fig. 2B; P<0.001) compared with the
control cells. This result is concordant with previous findings
reported by Rajendran et al (10), which showed that SFN may cause a
severe decrease in DNA content in HCT116 cells treated with 15
μM SFN for 24 h.
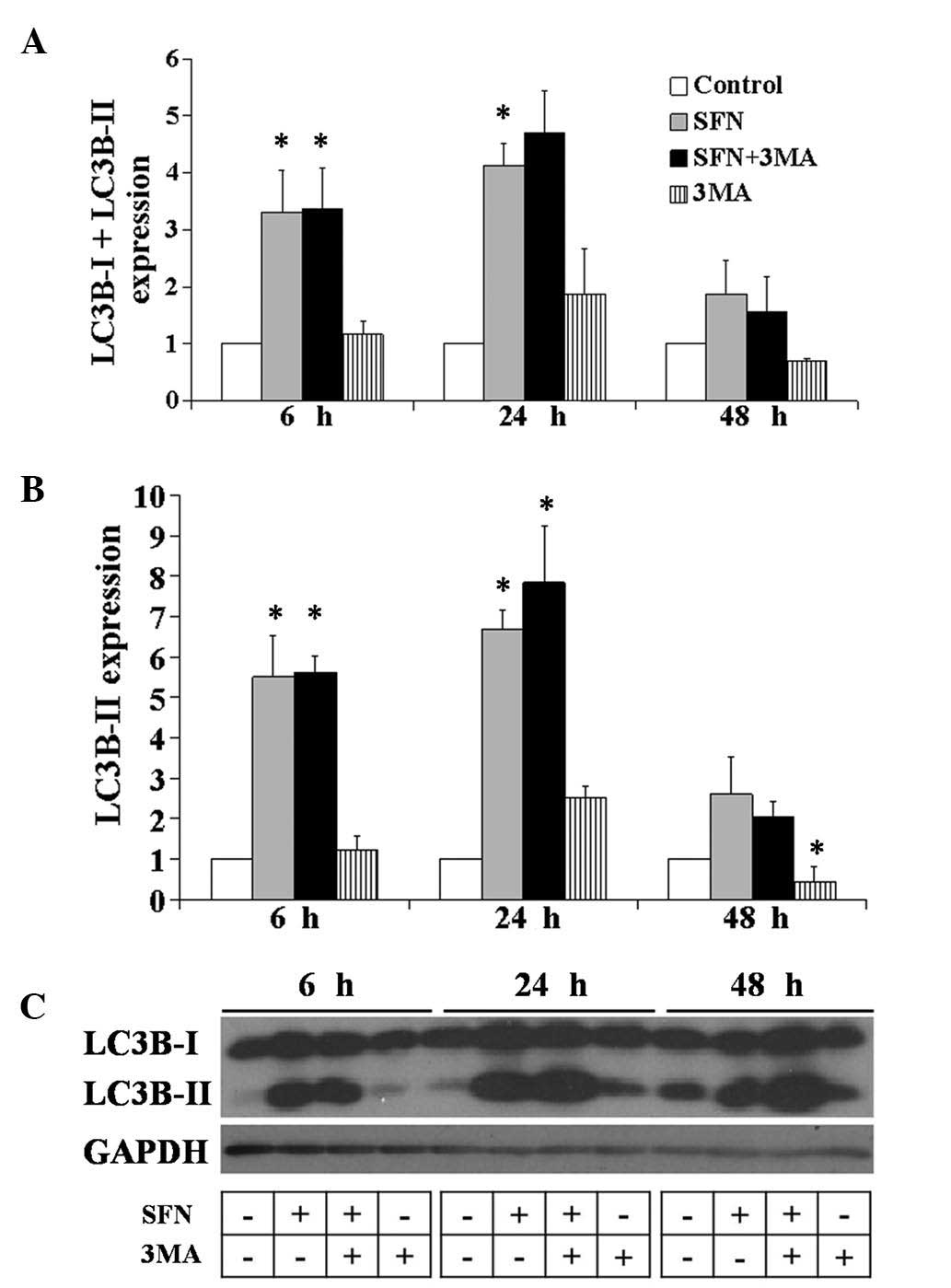
Analysis of LC3 protein modification
To investigate whether autophagy may be induced by
SFN treatment in the BE(2)-C
cells, the proteolytic process of MAP LC3 (in short LC3) was
assessed. The conversion from the endogenous, cytosolic LC3-I
protein to the LC3-II protein, which binds to autophagosomal
membranes, was detected by immunoblotting with antibodies against
LC3 (13,15). These results may provide evidence
of early autophagy induction. The cells were treated with 40
μM SFN, 10 mM 3-MA or the two agents simultaneously for 6,
24 and 48 h, and the isolated whole cells extracts were subjected
to western blot analysis (Fig. 3).
Treatment with SFN led to increased protein expression levels of
LC3-I and II at 6, 24 and 48 h (P<0.05) compared with the
SFN-treated cells compared with the control cells. These results
suggested the involvement of the autophagic process, since the
amount of LC3-II usually correlates well with the number of
autophagosomes (16). Simultaneous
treatment of SFN with 3-MA also resulted in increased expression
levels of the two forms of LC3, compared with the control cells,
from as early as 6 h and the tendency remained unchanged up to 48 h
(Fig. 3A and C; P<0.05). 3-MA
treatment alone was used as a control and caused a moderate change
in the LC3 protein expression pattern, suggesting that autophagy
induction was almost completely blocked. In addition, microscopic
observation of the SFN and 3-MA treated cells suggested that the
treatment caused cell death and after 48 h the cells began to
detach from the culture dish (Fig.
1A).
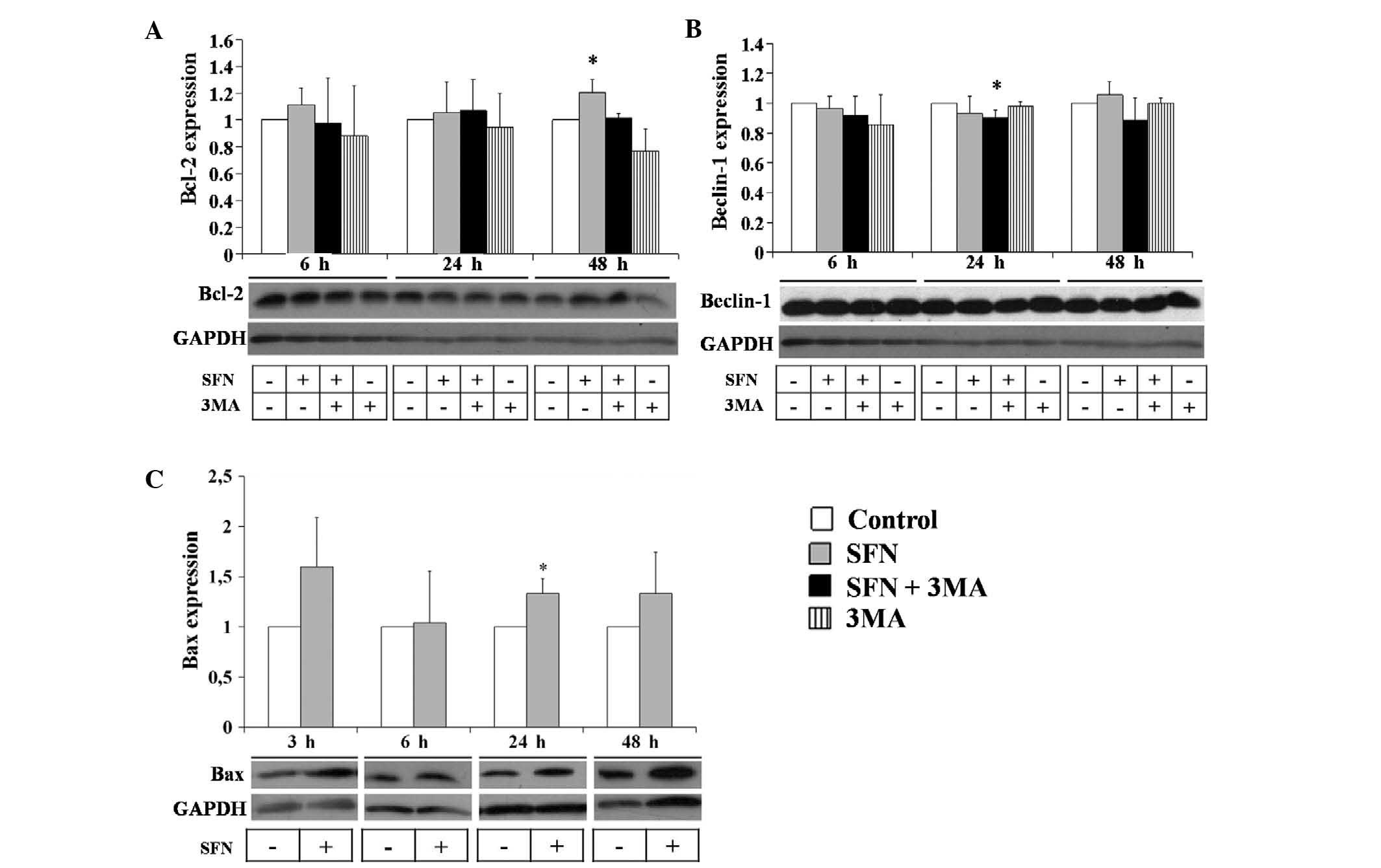
SFN elevates Bax protein expression
levels in neuroblastoma cells
In order to further investigate the mechanisms of
the observed cell death, the BE(2)-C cells were treated with SFN and 3-MA
and the protein expression levels of the antiapoptotic gene Bcl-2
were measured (Fig. 4). After 6,
24 and 48 h of SFN treatment, the protein expression levels of
Bcl-2 remained unchanged. Combined treatment of SFN with 3-MA
decreased the protein expression levels of Bcl-2 as compared with
SFN treatment alone (Fig. 4A).
Treatment with 3-MA alone decreased the Bcl-2 protein expression
levels, particularly at 48 h; however this change was not
statistically significant. Furthermore, the protein expression
levels of another autophagic factor, beclin-1, were measured and
shown to remain unchanged following treatment with the agents
tested (Fig. 4B). In addition, Bax
protein expression levels were evaluated by immunoblotting and
increased levels were observed at 6, 24 and 48 h of SFN treatment
(Fig. 4C; P<0.05). Changes in
the Bax protein levels were statistically significant at 24 h,
suggesting the induction of apoptosis.
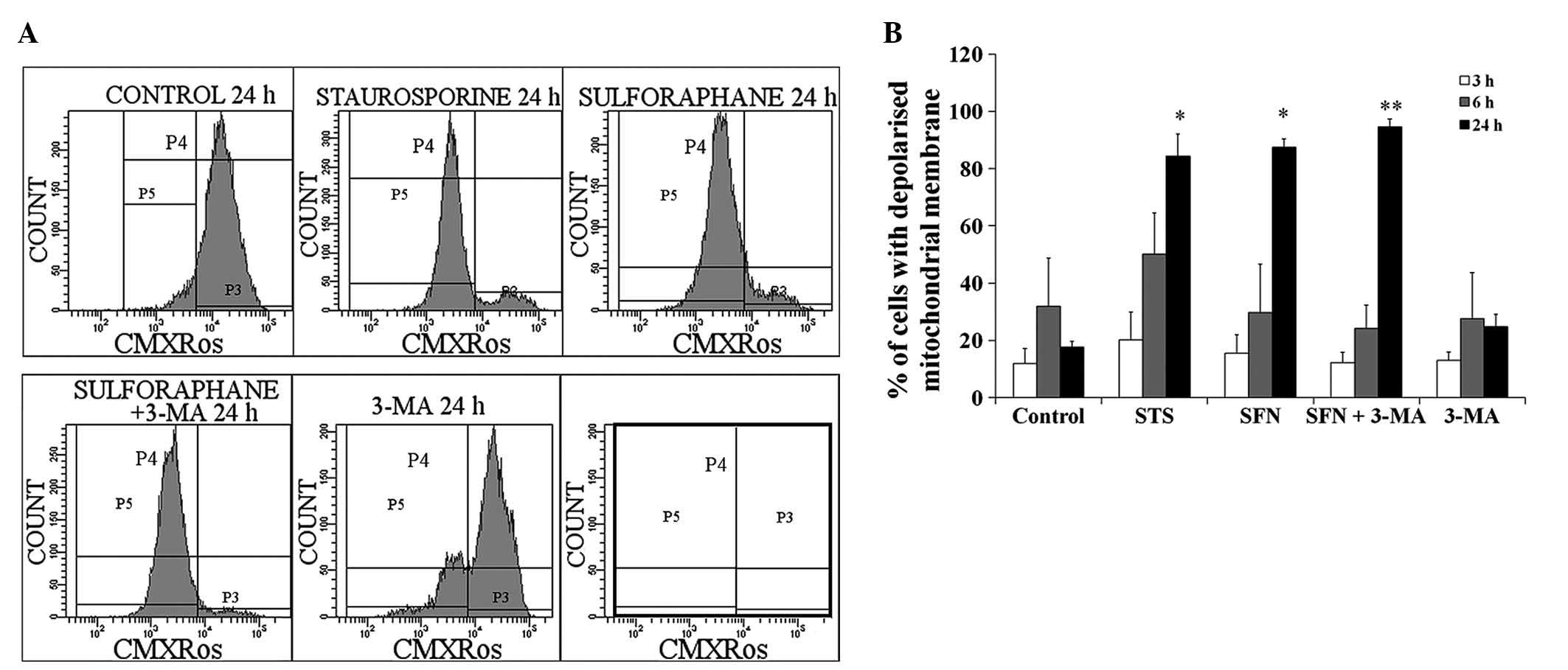
Analysis of mitochondrial membrane
potential in SFN-treated neuroblastoma cells
Flow cytometry was used to measure changes to the
mitochondrial membrane potential upon treatment with SFN. CMXRos
served as a dye, which actively accumulates in the mitochondria of
live cells. Control cells with functional mitochondrial membranes
had a high accumulation of CMXRos (Fig. 5A; channel P3). The positive control
apoptotic cells had a significant loss in mitochondrial membrane
potential and were obtained following treatment with 60 μM
STS, a known proapoptotic compound, for 24 h (channel P5). As
compared with STS treatment, treatment with 40 μM SFN also
caused a significant decrease in the mitochondrial membrane
potential (Fig. 5A). Treatment
with 3-MA alone caused only a minimal decrease in mitochondrial
membrane potential, but did not alter the effects of SFN.
In addition, the cells were cultured for 3, 6 and 24
h in the presence of 40 μM SFN, 10 mM 3-MA or 60 μM
STS. No effect was observed on the mitochondrial membrane
depolarization in response to treatment with any of the compounds
at 3 h (Fig. 5B), whereas an
increase in depolarization was observed in response to STS
treatment for 6 h. A significant increase in the number of cells
with depolarized mitochondrial membranes was observed in response
to treatment with SFN for 24 h and was comparable with the increase
caused by STS and combined treatment of SFN with 3-MA. Treatment
with 3-MA alone did not affect mitochondrial membrane
depolarization.
Discussion
The present study aimed to determine how
neuroblastoma cells respond to the potential anticancer agent SFN,
as well as 3-MA, an inhibitor of autophagy. Naturally occurring
isothiocyanates of cruciferous origin have previously been shown to
inhibit carcinogenesis (2,4). They may therefore be considered as
potential protective and therapeutic agents. Despite intensive
treatment, neuroblastoma tumors are difficult to eradicate and
>60% of children above the age of one year with neuroblastoma
have poor prognosis (24,25). Naturally occurring compounds, such
as SFN, may be used as a supplementary treatment for cancer
therapy, or may be used in combination with conventional
therapeutics. It was previously shown that administration of SFN to
female rats, through gavage, resulted in a maximum concentration of
the compound in the blood and mammary glands as early as at 1 h,
reducing the incidence, multiplicity and weight of mammary tumors
(1). Therefore, it is likely that
the concentration of SFN required to induce cell death may be
achievable in humans (7).
In the present study, 40 μM SFN induced
apoptosis of BE(2)-C cells after
24 and 48 h of treatment, as determined by a decrease in the
mitochondrial membrane potential, an increase in caspase activity
and inhibition of DNA synthesis. Flow cytometry showed that
treatment with SFN decreased the mitochondrial membrane potential
in ~40% of cells at 24 h, and ~62% of cells at 48 h. Due to its
electrophilic nature, SFN may directly modify nucleophilic sites,
such as sulfhydryl groups, in mitochondrial respiratory chain
enzymes or mitochondrial membranes. Conversely, a previous study
reported that apoptosis was inhibited in the PC-3 and LNCaP human
prostate cancer cells lines through autophagy induced by treatment
with SFN (8). SFN was shown to
induce the formation of acidic vesicle organelles, which enclosed
whole mitochondria and inhibited apoptosis in the prostate cell
lines. Mitochondrial digestion within autophagosomes prevents the
leakage of cytochrome c into the cytoplasm and consequently
blocks the induction of apoptosis (8). In the present study, SFN induced
autophagy and apoptosis, as documented by an increase in LC3II
expression levels, as well as caspase activation, an increase in
Bax expression levels and loss of mitochondrial membrane potential,
respectively. The two forms of LC3, I and II, were increased upon
treatment with SFN, and combined treatment with 3-MA resulted in
even higher levels at 24 h. Therefore, it may be concluded that SFN
causes autophagy by a mechanism independent of autophagy inhibition
by 3-MA. However, in the present study, fluorescent fusion methods
were not used to determine the protein fate of LC3 within
neuroblastoma cells. This method is expected to provide clear
evidence of autophagosomal formation.
In the PC-3 prostate cancer cell line, SFN was
previously shown to increase the Bax/Bcl-2 ratio, favoring
apoptosis (26). Similar
conclusions may be drawn from the results of the present study, as
Bax protein expression levels were increased early upon treatment
with SFN and remained high at up to 48 h. In addition, in the
BE(2)-C cells, a negligible change
in the protein expression levels of Bcl-2 was observed, thus
increasing the Bax/Bcl-2 ratio. Similarly, the lack of stimulatory
effects of SFN on Bcl-2 expression levels were demonstrated in a
previous study on the Jurkat T-cell lymphoma cell line (6). Beclin-1 interacts with class III PI3K
complex and participates in autophagosomal formation (27,28).
In the present study, the protein expression levels of beclin-1
remained unchanged in response to treatment with SFN and 3-MA, or a
combination of the two, at 6 h. At later time-points, only slight
inhibitory effects on the expression levels of beclin-1 were
observed in response to combined treatment. These results indicated
that beclin-1 was constitutively expressed in the BE(2)-C neuro-blastoma cells.
Apoptosis and autophagy are precisely regulated and
have important roles in the maintenance of homeostasis and disease.
Therefore, it is important to distinguish the effects of any
potential anti-cancer agent on these two processes. An enhanced
understanding of the role of SFN in the induction of cell death may
have implications in the design of future clinical trials for
combination cancer treatments using SFN and conventional
anti-cancer drugs (24,25). The inhibition of autophagy is also
an attractive therapeutic approach, since inhibition of autophagy
in cancer cells is associated with increased apoptotic cell death
(8,23). It has previously been reported that
the combination of SFN with autophagy inhibition, by 3-MA or
bafilomycin A1, may be a promising strategy for breast cancer
(29), human colon cancer
(30) or human prostate cancer
(8) therapy. Furthermore, the
chemopreventive efficacy of SFN may be augmented by inhibition of
autophagy using chloroquine in a transgenic adenocarcinoma of mouse
prostate as shown in an in vivo study (31). The present study showed that the
inhibition of autophagy by 3-MA does not alter SFN-induced
autophagy and apoptosis in human neuroblastoma cells, although the
combined effect of SFN and 3-MA is evidenced by a significant
decline in cell viability.
Acknowledgments
The present study was supported by the Jagiellonian
University grant DS/8/WBBiB. The authors acknowledge the help of Dr
Aleksandra Kowalczyk in design of the studies and Dr Małgorzata
Bzowska for flow cytometric analysis.
References
|
1
|
Fahey JW, Zhang Y and Talalay P: Broccoli
sprouts: an exceptionally rich source of inducers of enzymes that
protect against chemical carcinogens. Proc Natl Acad Sci USA.
94:10367–10372. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barcelo S, Gardiner JM, Gescher A and
Chipman JK: CYP2E1-mediated mechanism of anti-genotoxicity of the
broccoli constituent sulforaphane. Carcinogenesis. 17:277–282.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brooks JD, Paton VG and Vidanes G: Potent
induction of phase 2 enzymes in human prostate cells by
sulforaphane. Cancer Epidemiol Biomarkers Prev. 10:949–954.
2001.PubMed/NCBI
|
|
4
|
Tan XL and Spivack SD: Dietary
chemoprevention strategies for induction of phase II
xenobiotic-metabolizing enzymes in lung carcinogenesis: A review.
Lung Cancer. 65:129–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gamet-Payrastre L, Li P, Lumeau S, Cassar
G, Dupont MA, Chevolleau S, Gasc N, Tulliez J and Tercé F:
Sulforaphane, a naturally occurring isothiocyanate, induces cell
cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer
Res. 60:1426–1433. 2000.PubMed/NCBI
|
|
6
|
Fimognari C, Nüsse M, Cesari R, Iori R,
Cantelli-Forti G and Hrelia P: Growth inhibition, cell-cycle arrest
and apoptosis in human T-cell leukemia by the isothiocyanate
sulforaphane. Carcinogenesis. 23:581–586. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh SV, Srivastava SK, Choi S, Lew KL,
Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL,
Lee YJ, Xiao H and Herman-Antosiewicz A: Sulforaphane-induced cell
death in human prostate cancer cells is initiated by reactive
oxygen species. J Biol Chem. 280:19911–19924. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
C and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nishikawa T, Tsuno NH, Tsuchiya T,
Yoneyama S, Yamada J, Shuno Y, Okaji Y, Tanaka J, Kitayama J,
Takahashi K and Nagawa H: Sulforaphane stimulates activation of
proapoptotic protein bax leading to apoptosis of endothelial
progenitor cells. Ann Surg Oncol. 16:534–543. 2009. View Article : Google Scholar
|
|
10
|
Rajendran P, Kidane AI, Yu TW, Dashwood
WM, Bisson WH, Löhr CV, Ho E, Williams DE and Dashwood RH: HDAC
turnover, CtIP acetylation and dysregulated DNA damage signaling in
colon cancer cells treated with sulforaphane and related dietary
isothiocyanates. Epigenetics. 8:612–623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao D, Powolny AA, Antosiewicz J, Hahm
ER, Bommareddy A, Zeng Y, Desai D, Amin S, Herman-Antosiewicz A and
Singh SV: Cellular responses to cancer chemopreventive agent
D,L-sulforaphane in human prostate cancer cells are initiated by
mitochondrial reactive oxygen species. Pharm Res. 26:1729–1738.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Klionsky DJ: Autophagy: from phenomenology
to molecular understanding in less than a decade. Nat Rev Mol Cell
Biol. 8:931–937. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsuchihara K, Fujii S and Esumi H:
Autophagy and cancer: dynamism of the metabolism of tumor cells and
tissues. Cancer Lett. 278:130–138. 2009. View Article : Google Scholar
|
|
14
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giménez-Xavier P, Francisco R, Santidrián
AF, Gil J and Ambrosio S: Effects of dopamine on LC3-II activation
as a marker of autophagy in a neuroblastoma cell model.
Neurotoxicology. 30:658–665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmidt H and Karrer P: Synthese der
recemischen und der optisch aktiven formen des sulforaphans. Helv
Chim Acta. 31:1067–1074. 1948.In German. PubMed/NCBI
|
|
19
|
Denizot F and Lang R: Rapid colorimetric
assay for cell growth and survival. Modifications to the
tetrazolium dye procedure giving improved sensitivity and
reliability. J Immunol Methods. 89:271–277. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Riss TL and Sirbasku DA: Growth and
continuous passage of COMMA-D mouse mammary epithelial cells in
hormonally defined serum-free medium. Cancer Res. 47:3776–3782.
1987.PubMed/NCBI
|
|
21
|
Sadowski HB, Shuai K, Darnell JE Jr and
Gilman MZ: A common nuclear signal transduction pathway activated
by growth factor and cytokine receptors. Science. 261:1739–1744.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Myzak MC and Dashwood RH: Chemoprotection
by sulforaphane: keep one eye beyond Keap1. Cancer Lett.
233:208–218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livesey KM, Tang D, Zeh HJ and Lotze MT:
Autophagy inhibition in combination cancer teatment. Curr Opin
Investig Drugs. 10:1269–1279. 2009.PubMed/NCBI
|
|
24
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singh AV, Xiao D, Lew KL, Dhir R and Singh
SV: Sulforaphane induces caspase-mediated apoptosis in cultured
PC-3 human prostate cancer cells and retards growth of PC-3
xenografts in vivo. Carcinogenesis. 25:83–90. 2004. View Article : Google Scholar
|
|
27
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao L, Zhu Y, Wang D, Chen M, Gao P, Xiao
W, Rao G, Wang X, Jin H, Sui N and Chen Q: Morphine induces
Beclin-1 and ATG5-dependent autophagy in human neuroblastoma
SH-SY5Y cells and in the rat hippocampus. Autophagy. 6:386–394.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kanematsu S, Uehara N, Miki H, Yoshizawa
K, Kawanaka A, Yuri T and Tsubura A: Autophagy inhibition enhances
sulforaphane-induced apoptosis in human breast cancer cells.
Anticancer Res. 30:3381–3390. 2010.PubMed/NCBI
|
|
30
|
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y,
Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K and Nagawa H:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar
|
|
31
|
Vyas AR, Hahm E-R, Arlotti JA, Watkins S,
Stolz DB, Desai D, Amin S and Singh SV: Chemoprevention of prostate
cancer by d,l-sulforaphane is augmented by pharmacological
inhibition of autophagy. Cancer Res. 73:5985–5995. 2013. View Article : Google Scholar : PubMed/NCBI
|