Introduction
The incidence of severe sepsis is rising annually,
with a mortality rate approaching 50% worldwide (1). Sepsis is predominantly a consequence
of multiple organ failure, of which myocardial dysfunction is
recognized manifestation (2–4).
Endotoxin-induced cardiomyocyte apoptosis and the inflammatory
response in the cardiovascular system leads to a series of
pathophysiological injuries, which significantly increase the
mortality rate in patients with sepsis (5–7).
Previous studies have demonstrated that the lipopolysaccharide
(LPS) bacterial endotoxin reduces contractility and significantly
induces the expression of tumor necrosis factor (TNF)-α in
cardiomyocytes by binding to toll-like receptor-4 (TLR-4) (8–10).
This process activates nuclear factor-κB (NF-κB), which is an
important signal integrator controlling the production of
pro-inflammatory mediators (11–13).
The increased production of numerous inflammatory cytokines,
including TNF-α, interleukin (IL)-6, IL-1β, interferon (IFN)γ and
intercellular adhesion molecule (ICAM)-1, represses cardiac
function directly and indirectly (14). TNF-α is a major pro-inflammatory
cytokine, which mediates the signs and symptoms of sepsis and shock
(15). In addition, TNF-α-induced
apoptotic responses are triggered by the binding of death-receptor
ligands to TNF-α receptor 1 (TNF-R1), which is involved in the
pathogenesis of cardiac diseases (16).
Estrogen receptors (ERs) are important in preventing
endotoxin-induced cardiac dysfunction (16). Notably, clinical studies of
patients with sepsis indicate that the mortality rates and
expression levels of TNF-α are lower in females compared with males
(7,18,19).
Estrogen replacement therapy reduces the incidence of heart disease
following the menopause (20). In
previous years, considerable attention has been paid to identifying
natural phytoestrogens, which are plant-derived, polyphenolic,
non-steroidal compounds used in preventing and treating
cardiovascular diseases (21).
Panax notoginseng, termed as ‘sanchi’ or ‘san qi’ in
Chinese, has been used to prevent and manage cardiovascular disease
in China for several years (22).
Notoginsenoside R1 (NG-R1), a phytoestrogen, is beleived to be the
predominant ingredient of Panax notoginseng, which promotes
cardiovascular activity (23,24).
However, the effects of NG-R1 on cardiomyocytes, and the precise
cellular/molecular mechanisms, remain to be elucidated. The present
study demonstrated that NG-R1 inhibited the LPS-induced expression
of inflammatory cytokines and cell apoptosis in H9c2
cardiomyocytes. As the molecular structure of NG-R1 aglycone is
similar to that of estradiol (22), the present study further evaluated
whether its cardioprotective effects were dependent on ERs.
Materials and methods
Materials
NG-R1 was purchased from Shanghai Winherb Medical
S&T Development Co., Ltd. (Shanghai, China). All the tissue
culture materials were purchased from Gibco Life Technologies
(Grand Island, NY, USA). All antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and all other
chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The endotoxin-free materials used included Dulbecco’s modified
Eagle’s medium (DMEM; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco Life
Technologies) and 1% (v/v) penicillin/streptomycin (Gibco Life
Technologies). All the investigations performed in the present
study were approved by the Ethics Committee of The Second
Affiliated Hospital of Harbin Medical University (Heilongjiang,
China).
Cell culture and treatments
H9c2 cardiomyocytes were obtained from the Cell Bank
of the Chinese Academy of Sciences (Shanghai, China) and were
maintained in DMEM with 4.5 mg/l glucose, supplemented with 10%
(v/v) FBS and 1% penicillin/streptomycin (v/v), at 37°C in a
humidified atmosphere containing 5% CO2. The H9c2
cardiomyocytes (1−10×105) were treated with either the
vehicle (0.1% dimethyl sulfoxide; DMSO) or NG-R1 (0, 5, 10, 25 and
50 μM), in the presence or absence of 20 μg/ml LPS.
Following pretreatment for 1 h at 37°C with or without NG-R1, the
cells were exposed to 20 μg/ml LPS for 24 h at 37°C. In
separate experiments, the cells were pretreated with the ICI 182780
(ICI) non-selective ER antagonist, the methyl-piperidino-pyrazole
(MPP) selective ERα antagonist, or the selective NF-κB antagonist,
PDTC, for 30 min, prior to treatment with either the vehicle (0.1%
DMSO) or NG-R1 (25 μM) to examine the effects of ERα and
NF-κB in mediating the anti-inflammatory and anti-apoptotic effects
of NG-R1.
Assessment of cell viability and
apoptosis
The cell viability was determined using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
assay, as described previously (25). Cell apoptosis was determined by
terminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL) using an in situ cell death detection kit and
fluorescein (Roche Applied Science, Quebec, Canada), as described
previously (25). Samples were
visualized using a BZ-900 fluorescence microscope (Keyence, Osaka,
Japan) and Image-Pro Plus software, version 5.0 (Media Cybernetics,
Inc., Rockville, MD, USA). A total of 10–20 randomly selected
fields were visualized.
Caspase-3 activity assay
The activity of caspase-3 was measured using the
Caspase-3 Fluorometric Assay kit [containing glucose assay buffer,
glucose probe (in DMSO), glucose enzyme mix (lyophilized) and
glucose standard (100 nmol/ml); catalog no. K105–200; BioVision,
Mountain View, CA, USA), according to the manufacturer’s
instructions. Each sample in each well of the 96-well plate was
filled with 100 μl mixture, including 50 μl
resuspended cells in Cell Lysis Buffer, 50 μl 2X Reaction
buffer (containing 10 mM final concentration DTT) and 5 μl 1
mM DEVD-AFC substrate (50 μM final concentration). The
samples were read using a Fluoroskan Ascent FL fluorometer (Thermo
Fisher Scientific, Waltham, MA, USA) with an excitation wavelength
of 400 nm and an emission wavelength of 505 nm. The results were
expressed as the fold-change compared with the control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA). An aliquot of
the total RNA (~2 μg) was reverse transcribed using a
SuperScript First-Strand Synthesis system (Invitrogen Life
Technologies.). The resulting cDNA was synthesized from the
isolated RNA, and the cycle time values were obtained by RT-qPCR
using Power SYBR Green PCR Master mix (Applied Biosystems, Foster
City, California, USA) and an iQ5 Real-Time PCR Detection system
and analytical software (CFX Manager 2.1; Bio-Rad Laboratories,
Inc., Hercules, CA, USA), as described previously (25). The PCR cycling conditions were as
follows: Amplification at 95°C for 10 min, followed by 40 cycles of
95°C for 30 sec, 59°C for 30 sec and 72°C for 30 sec. Thermal
cycling started with 10 min denaturation at 95°C, 40 cycles of
denaturation at 95° C for 15 sec and combined primer
annealing/elongation at 60° for 1 min. Each sample was run in
triplicate. The primers (BBI Life Sciences Corp., Shanghai, China)
were designed using Applied Biosystems Primer Express software
(version 2.0) and are shown in Table
I. The mRNA expression levels were normalized against GAPDH,
and the relative mRNA expression levels are expressed using
arbitrary units, with the value of the control group defined as
one.
 | Table IPrimers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Target gene | Forward sequence
(5′–3′) | Base pairs | Reverse sequence
(5′–3′) | Base pairs |
|---|
| ER-α |
TCCCCAACACCATCTGAGAACT | 22 |
CGTTTCAGGGATTCGCAGAA | 20 |
| ER-β |
TCAGGAAAAGGAATATGGCATGT | 23 |
TTTTATGGCCACACAGTCCTACA | 23 |
| TNF-α |
CATCTTCTCAAAATTCGAGTGACAA | 25 |
TGGGAGTAGACAAGGTACAACCC | 23 |
| IL-1β |
CAACCAACAAGTGATATTCTCCATG | 25 |
GATCCACACTCTCCAGCTGCA | 21 |
| IL-6 |
GAGGATACCACTCCCAACAGACC | 23 |
AAGTGCATCATCGTTGTTCATACA | 24 |
| INFγ |
CGCCGCGTCTTGGTTTT | 27 |
GAGTGTGCCTTGGCAGTAACAG | 22 |
| GAPDH |
AACGACCCCTTCATTGAC | 22 |
TCCACGACATACTCAGCAC | 19 |
Western blot analysis
The cell lysate preparation and western blot
analysis were performed, as described previously (25), using a western blot kit (BBI Life
Science Corp.) according to the manufacturer’s instructions. Cell
lysates were subjected to SDS-PAGE (including a 5% stacking gel and
a 10% separating gel; Sigma-Aldrich) and transferred to
nitrocellulose membranes Beyotime Institute of Biotechnology,
Haimen, China). Subsequent to transferring, blots were blocked with
5% milk for 1 h at 37°C. The membranes were probed with the
following antibodies: Primary rabbit polyclonal anti-GAPDH (1:200;
sc-25778) at 4°C for 72 h as a loading control, mouse monoclonal
ERα (1:500; sc-73479), mouse monoclonal ERβ (1:500; sc-390243),
mouse monoclonal p-p65 (1:200; sc-166748), rabbit polyclonal total
p65 (1:200; sc-372) and mouse monoclonal I-κBα (1:200; sc-373893)
at 4°C for 24 h, and horseradish peroxidase-conjugated secondary
antibodies (1:5,000; goat anti-mouse IgG-HRP, sc-2005, and goat
anti-rabbit IgG-HRP, sc-2004) at room temperature for 30 min. The
membranes were washed with Tris-buffered saline with Tween 20 for
10 min three times following incubation with the antibodies. The
protein concentration was determined using a Bio-Rad DC Protein
Determination kit (Bio-Rad Laboratories, Inc.), with bovine serum
albumin as the standard. The immunoblots were developed using an
enhanced chemilluminescence kit (GE Healthcare, Little Chalfont,
UK). The signals were quantified by Quantity-One software (version
4.62; Bio-Rad Laboratories, Inc.) and the results from each
experimental group are expressed as the relative integrated
intensity compared with the control.
Indirect immunofluorescence assays
The H9c2 cardiomyocytes were cultured on Lab-Tek
chamber slides (Nalge Nunc International, Naperville, IL, USA) and
were fixed using cold 4% methanol at −20°C for 3 min. Indirect
immunofluorescence assays were performed, as described previously
(26). Briefly, the cells
(1−10×105) were treated with 0.3% Triton X-100 in
phosphate-buffered saline (PBS) for 15 min at room temperature, to
increase permeability. Following blocking with 10% normal goat
serum in PBS at room temperature for 1 h, the cell monolayers were
screened using a standard indirect immunofluorescence staining
procedure, with polyclonal antibodies against the p65 subunit of
NF-κB (1:200) and a fluorescein isothiocyanate-labeled anti-rabbit
antibody (1:200). The nuclei were stained using 10 μg/ml
4′,6-diamidino-2-phenylindole (Sigma-Aldrich). The negative
controls were incubated with preimmune rabbit sera rather than
primary antibodies.
Statistical analysis
The data are expressed as the mean ± standard error
of the mean. The significance of the differences between means were
assessed using Student’s t-test. A one-way analysis of variance
with Bonferroni corrections was used to determine the significance
for multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference. Statistical calculations were
performed using SPSS 11.0 software (SPSS, Inc., Chicago, IL,
USA).
Results
Inhibition of LPS-induced H9c2 cell death
by NG-R1 is mediated by ERα
Following incubation with various concentrations of
LPS (0–20 μg/ml) for 24 h, a significant, dose-dependent
reduction in cell viability was observed (Fig. 1). Therefore, a dose of 20
μg/ml was selected for subsequent experiments. As shown in
Fig. 1A, LPS (20 μg/ml)
significantly reduced cell viability by ~35%, whereas pretreatment
with 5, 10, and 25 μM NG-R1 maintained cell viability at
~73, 81 and 92%, respectively. By contrast, the viability of the
H9c2 cells remained unaltered following treatment with NG-R1 alone
(Fig. 1B). These results suggested
that NG-R1 inhibited LPS-induced cell death in a dose-dependent
manner. Since a higher concentration of NG-R1 (50 μM)
demonstrated no additional benefit on cell viability, a dose of 25
μM was selected for subsequent experiments.
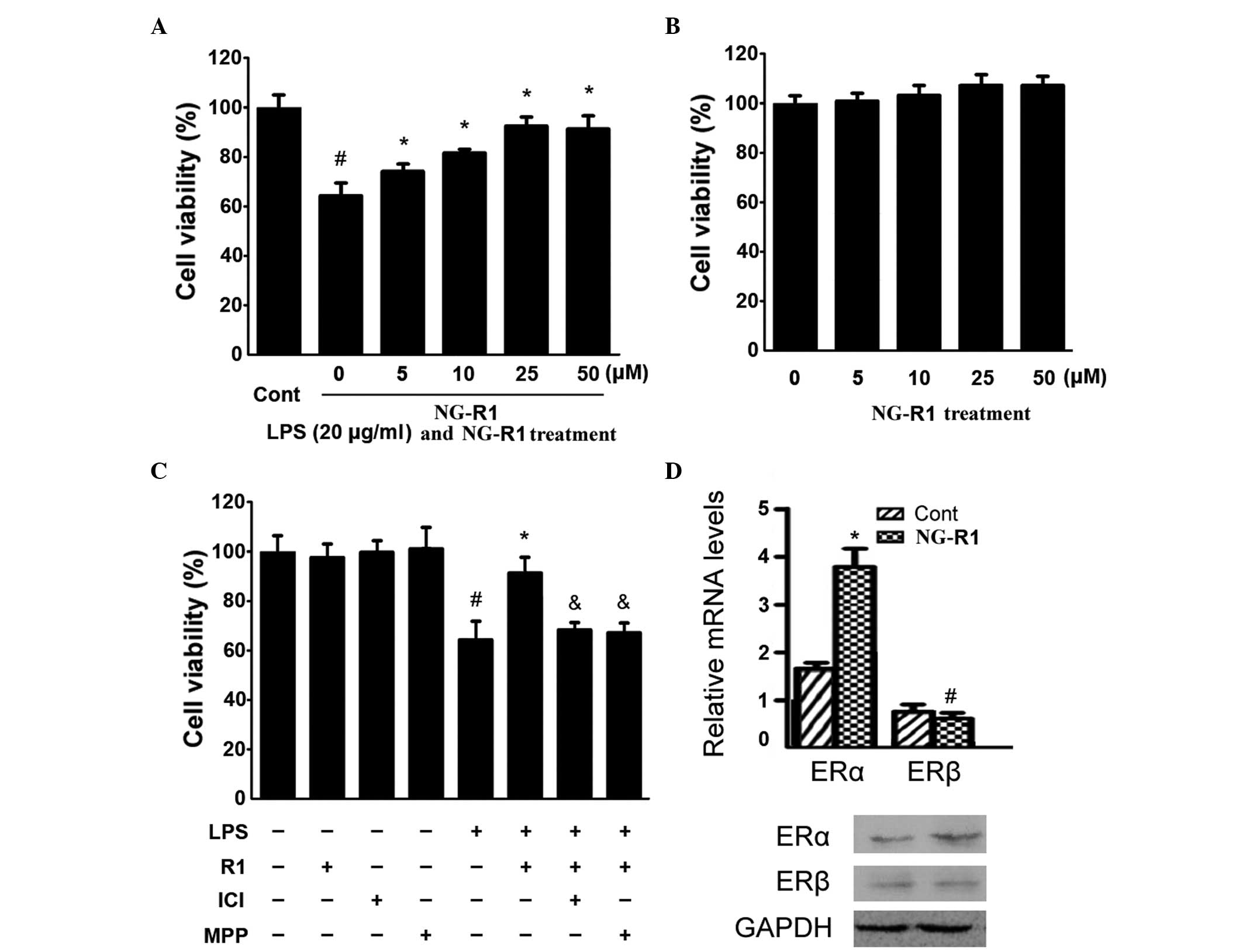 | Figure 1Effects of NG-R1 on the viability of
H9c2 cardiomyocytes and ER isoforms. (A) H9c2 cells were treated
with the indicated concentrations of NG-R1 (0–50 μM) for 1
h, followed by treatment with LPS (20 μg/ml) for 24 h or (B)
with the indicated concentrations (0–50 μM) of NG-R1 for 24
h, and cell viability was determined using an MTT assay expressed
as a percentage of the control (n=8 per group;
#P<0.05, vs. cells treated with NG-R1 only). (C) Cell
viability was determined using an MTT assay and the effects of the
ICI and MPP ERα antagonists on H9c2 cardiomyocyte viability were
assessed (#P<0.05, vs. cells treated without LPS).
(D) Reverse transcription-quantitative polymerase chain reaction
and immunoblotting revealed that NG-R1 selectively increased the
expression of ERα. The results are expressed as the mean ± standard
error of the mean. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide; NG-R1, notoginsenoside R1; Cont, vehicle (0.1%
dimethyl sulfoxide); LPS, lipopolysaccharide; R1, NG-R1; ICI, ICI
182780; MPP, methyl-piperidino-pyrazole; ER, estrogen receptor. |
To detect whether the inhibitory effect of NG-R1 on
LPS-induced H9c2 cell death is mediated by ER, an ERα antagonists
was used to pretreat the H9c2 cells prior to treatment with LPS and
NG-R1. As shown in Fig. 1C, the
effects of NG-R1 on H9c2 cell viability were attenuated by 30 min
pretreatment with ICI, a non-selective ERα antagonist, or MPP, a
selective ERα antagonist, prior to treatment with NG-R1 (25
μM) followed by LPS (20 μg/ml). Notably, NG-R1, ICI
or MPP alone exerted no effects on cell viability. These results
suggested that NG-R1 inhibited LPS-induced cell death in an
ER-dependent manner. The effect of NG-R1 on the expression of ER
was also determined. As shown in Fig.
1D, increases in the mRNA and protein expression levels of ERα
in NG-R1-treated cardiomyocytes were observed. However, the
expression of ERβ remained unaltered. Taken together, these results
demonstrated that NG-R1 acted through ERα.
Inhibition of LPS-induced H9c2 cell
apoptosis by NG-R1 is mediated by ERα
The apoptotic index and the activity of caspase-3
were examined in the H9c2 cardiomyocytes (Fig. 2). In cells treated with LPS (20
μg/ml), DNA fragmentation was observed following treatment
for 24 h (Fig. 2A and B). This
finding confirmed the data shown in Fig. 2C, demonstrating that caspase-3 was
activated following treatment with LPS. By contrast, treatment with
NG-R1 (25 μM) effectively ameliorated the LPS-induced DNA
fragmentation and activation of caspase-3. In addition, the effects
of NG-R1 on the apoptotic index and the activity of caspase-3 were
attenuated following 30 min pretreatment with ICI or MPP, prior to
treatment with NG-R1 (25 μM) and subsequently LPS (20
μg/ml). NG-R1, ICI or MPP alone exerted no effects on these
processes.
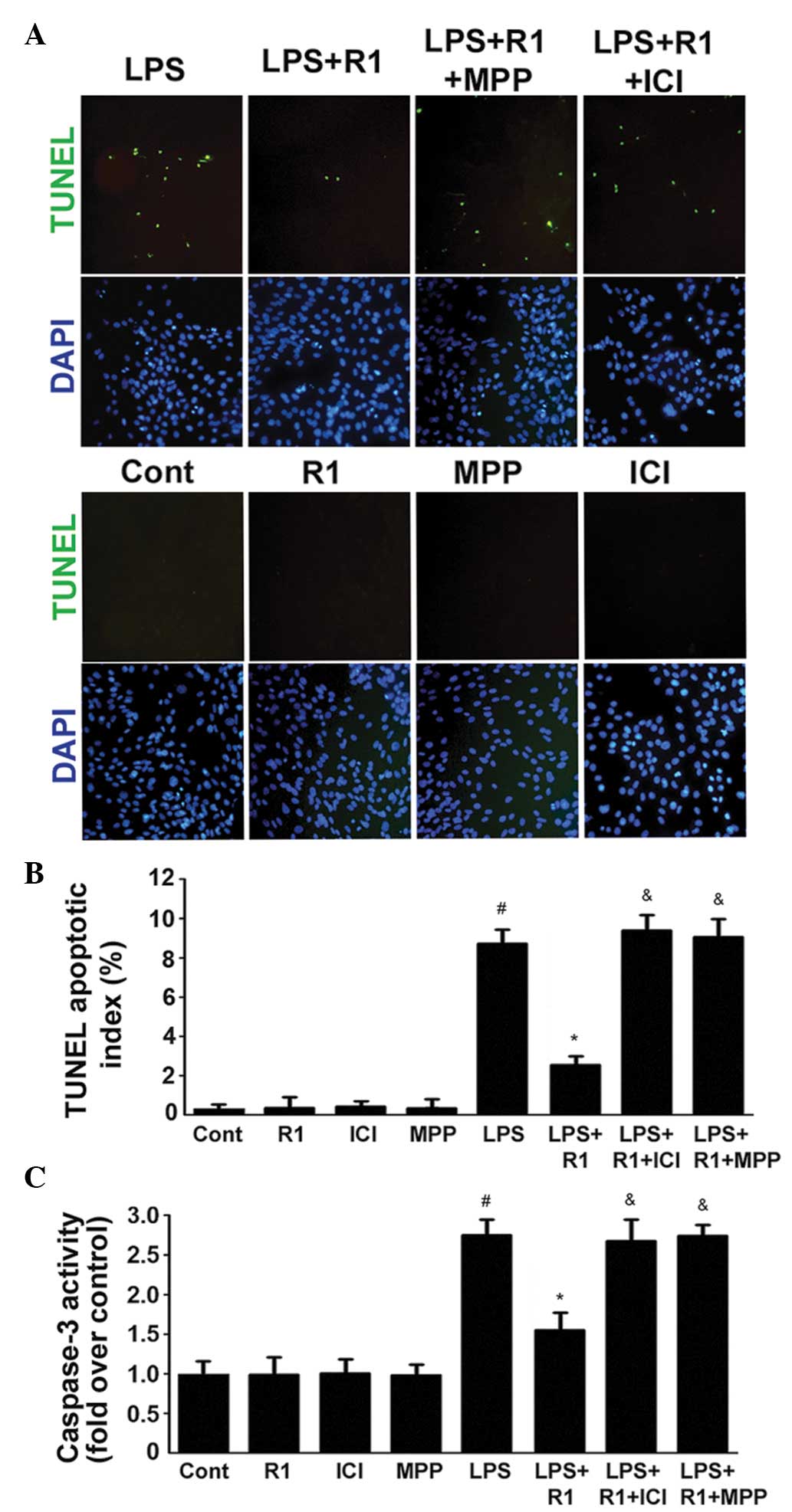 | Figure 2Effects of LPS, NG-R1 and/or ER
antagonists on the apoptosis of H9c2 cardiomyocytes. (A) ERα
mediates the effects of NG-R1 on endotoxin-induced inflammatory and
apoptotic responses in H9c2 cardiomyocytes. Cells were
pre-incubated with ICI or MPP for 30 min prior to treatment with or
without NG-R1 (25 μM) for 1 h, followed by LPS (20
μg/ml) for 24 h. The cells were subsequently fixed and
subjected to TUNEL and DAPI staining (magnification, ×200). (B)
TUNEL apoptotic index was determined by calculating the ratio of
TUNEL-positive cells to total cells. (C) Caspase-3 activity was
measured using a fluorometric assay, and expressed as the
fold-change compared with the control. The data are presented as
the mean ± standard error of the mean (n=8 per group;
#P<0.05, vs. Cont; *P<0.05, vs. LPS
treatment; &P<0.05, vs. NG-R1 and LPS
co-treatment). TUNEL, terminal deoxynucleotidyl transferase dUTP
nick end labeling; DAPI, 4′,6-diamidino-2-phenylindole; NG-R1,
notoginsenoside R1; Cont, vehicle (0.1% dimethyl sulfoxide); LPS,
lipopolysaccharide; R1, NG-R1; ICI, ICI 182780; MPP,
methyl-piperidino-pyrazole; ER, estrogen receptor. |
ERa-mediated inhibition of NF-κB
contributes to the inhibitory effect of NG-R1 on LPS-induced
apoptosis of H9c2 cells
Treatment with LPS resulted in the activation of
NF-κB in several types of cells, which was characterized by the
nuclear translocation of NF-κB following the phosphorylation of
NF-κB p65 and degradation of the NF-κB inhibitor α (I-κBα)
(27–29). The present study used
immunofluorescence staining of NF-κB p65 and demonstrated that
treatment with LPS led to nuclear accumulation of NF-κB in the H9c2
cells (Fig. 3A). Western blotting
revealed that treatment with LPS led to the phosphorylation of p65
and degradation of I-κBα (Fig.
3B). By contrast, treatment with NG-R1 (25 μM) reduced
the LPS-induced phosphorylation of NF-κB p65, degradation of I-κBα
and nuclear localization of NF-κB. These effects were attenuated by
pretreatment with MPP 30 min prior to treatment with NG-R1 (25
μM) and subsequently LPS (20 μg/ml). Treatment with
either NG-R1 or MPP alone exerted no effects on the phosphorylation
of NF-κB p65, degradation of I-κBα or nuclear localization of
NF-κB. as shown in Fig. 3C and D,
exposure of the H9c2 cells to LPS increased the production of NF-κB
target genes, including IL-6 and IL-1β. The expression levels of
these genes increased following treatment with LPS, however they
were significantly inhibited by additional treatment with NG-R1.
The effects of NG-R1 on the expression levels of these NF-κB target
genes were attenuated by pretreatment with ICI or MPP 30 min prior
to treatment with NG-R1 (25 μM) and subsequently LPS (20
μg/ml). NG-R1, ICI or MPP alone exerted no effects on the
expression levels of the NF-κB target genes. These data indicated
that NG-R1 inhibited the LPS-induced activation of NF-κB in an
ERα-dependent manner. TNF-α (0–16 ng/ml) significantly activated
caspase-3 in a dose-dependent manner (Fig. 4A) and TUNEL staining revealed
TNF-α-induced myocardial cell apoptosis (Fig. 4B). In the H9c2 cells, NG-R1
inhibited the LPS-induced expression of TNF-α, and this effect was
attenuated by treatment with ICI or MMP (Fig. 4C). Similar to the effects of the
NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), NG-R1
significantly inhibited the LPS-induced expression of TNF-α and the
activation of caspase-3 (Fig. 4D and
E). NG-R1 alone exerted no effects on the expression of TNF-α
or the activation of caspase-3. These data confirmed that the
inhibitory effects of NG-R1 on the TNF-α-mediated activation of
caspase-3 and apoptosis in H9c2 cells were closely associated with
the inactivation of NF-κB.
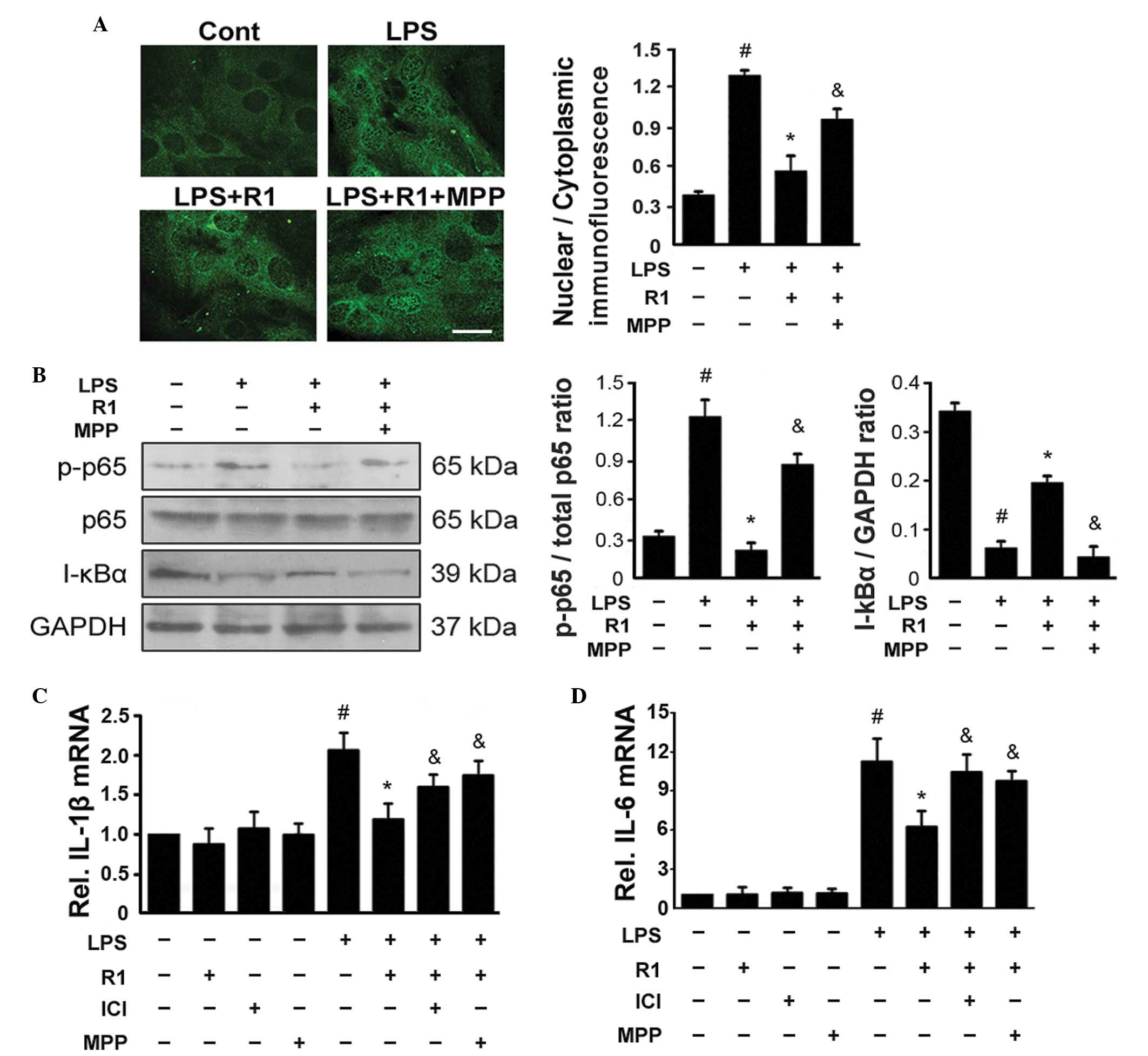 | Figure 3NG-R1-mediated inhibition of
LPS-mediated activation of NF-κB is dependent on ERα. (A) Cells
were pre-incubated with MPP, a selective ERα antagonist, for 30 min
prior to treatment with or without NG-R1 (25 μM) for 1 h,
followed by LPS (20 μg/ml) for 24 h. Representative images
of indirect immunofluorescence for the p65 subunit of NF-κB in H9c2
cells are shown (scale bar=10 μm). The mean density ratios
of nuclear/cytoplasmic immunofluorescence were analyzed. (B)
Lysates were prepared from the H9c2 cells and the immunoblots were
probed for p-p65, total p65 and I-κBα, and quantitative analyses of
the phosphorylation of NF-κB and degradation of I-κBα in the H9c2
cells was performed. GAPDH was used as a loading control in all
western blotting experiments. (C and D) Expression levels of IL-1β
and IL-6 were measured by reverse transcription-quantitative
polymerase chain reaction (n=6 per group; #P<0.05,
vs. Cont; *P<0.05, vs. LPS-treatment; &P<0.05,
vs. NG-R1 and LPS co-treatment. NG-R1, notoginsenoside R1; Cont,
vehicle (0.1% dimethyl sulfoxide); LPS, lipopolysaccharide; R1,
NG-R1; ICI, ICI 182780; MPP, methyl-piperidino-pyrazole; ER,
estrogen receptor; p-, phosphorylated; IL, interleukin. |
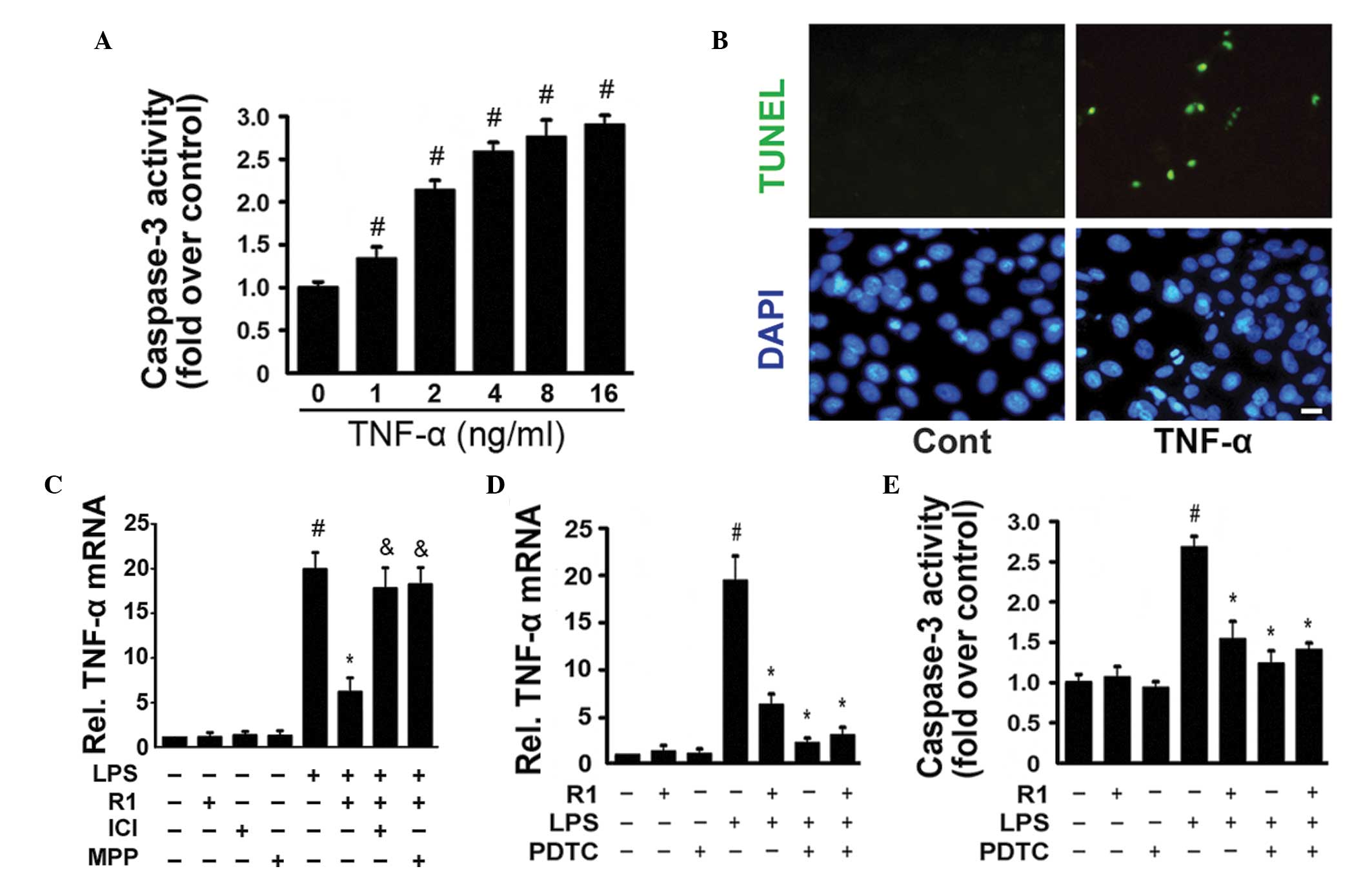 | Figure 4TNFα stimulates the activation of
caspase-3 and apoptosis in H9c2 cardiomyocytes. (A) H9c2 cells were
cultured with the indicated concentrations (0–16 ng/ml) of TNF-α
for 24 h. The activity of caspase-3 was measured using a
fluorometric assay and expressed as the fold-change compared with
the control (n=6 per group; #P<0.05, vs. Cont). (B)
Cells were exposed to TNF-α (16 ng/ml) for 24 h and were stained
using TUNEL and DAPI staining (scale bar=10 μm). (C) mRNA
expression of TNF-α was measured by reverse
transcription-quantitative polymerase chain reaction (n=6 per
group; #P<0.05, vs. Cont; *P<0.05, vs.
LPS-treatment; &P<0.05, vs. NG-R1 and LPS
co-treatment. (D) Cells were pre-incubated with PDTC (a specific
inhibitor of NF-κB) for 30 min prior to treatment with or without
NG-R1 (25 μM) for 1 h, followed by LPS (20 μg/ml) for
24 h. The mRNA expression of TNF-α was determined byRT-qPCR (E)
Caspase-3 activity was measured using a fluorometric assay and
expressed as the fold-change compared with the control (n=6 per
group; #P<0.05, vs. Cont; *P<0.05, vs. LPS-treatment;
&P<0.05, vs. NG-R1 and LPS co-treatment). The
results are expressed as the mean ± standard error of the mean.
TUNEL, terminal deoxynucleotidyl transferase dUTP nick end
labeling; DAPI, 4′,6-diamidino-2-phenylindole; NG-R1,
notoginsenoside R1; Cont, vehicle (0.1% dimethyl sulfoxide); LPS,
lipopolysaccharide; R1, NG-R1; ICI, ICI 182780; MPP,
methyl-piperidino-pyrazole; TNF, tumor necrosis factor; PDTC,
pyrrolidine dithiocarbamate; RT-qPCR, reverse transcription
quantitative polymerase chain reaction. |
Discussion
NG-R1, a phytoestrogen, is believed to be the
predominant ingredient in Panax notoginseng responsible for
its cardiovascular activity. However, the effects of NG-R1 on
cardiomyocytes, and its precise cellular/molecular mechanisms,
remain to be elucidated. The present study observed for the first
time, to the best of our knowledge, that NG-R1 significantly
attenuated endotoxin-induced inflammatory and apoptotic responses
in H9c2 cardiomyocytes. Furthermore, the cardioprotective effects
of NG-R1 were dependent on the activation of ERα and the
inactivation of NF-κB in these cells.
Septic shock, resulting from host stimulation of
inflammatory cytokines, causes cardiac dysfunction by suppressing
myocardial contractility, which significantly increases mortality
rates in patients with sepsis (27). Bacterial LPS is a potent stimulator
of proinflammatory cytokines, including TNF-α, IL-6, IL-1β, IFNγ
and ICAM-1, in cardiomyocytes (27). The results of the present study
demonstrated that NG-R1 increased cell viability and reduced
apoptotic damage in cardiomyocytes via the inhibition of a series
of proinflammatory cytokines, including TNF-α, IL-6, IL-1β and IFNγ
(Figs. 1Figure 23). NG-R1 also inhibited the activation of
NF-κB signaling in cardiomyocytes, as demonstrated by
phosphorylation of the p65 subunit of NF-κB and degradation of
I-κBα (Fig. 4). In cardiomyocytes,
TLR4 specifically recognizes LPS, resulting in the activation of
NF-κB, which is an important signal integrator controlling the
production of pro-inflammatory mediators (27). Among these mediators, TNF-α, a
major proinflammatory cytokine, induces an apoptotic responses by
promoting the binding of death-receptor ligands to TNF-R1,
subsequently initiating the death-receptor-mediated apoptotic
pathway (29). The present study
suggested that activation of NF-κB caused the upregulation of TNF-α
in myocardial cells, which directly contributed to cardiac
apoptosis, as demonstrated by the increased quantities of
TUNEL-positive cells and the activation of caspase-3 in
cardiomyocytes following stimulation with TNF-α (Fig. 2). In addition, the NF-κB activation
inhibitor, PDTC, partially inhibited the production of TNF-α and
LPS-mediated activation of caspase-3 in myocardial cells (Figs. 4C and D). These results confirmed
those of previous studies demonstrating that the induction of
myocardial inflammatory cytokines, including TNF-α, IL-1β, and
IL-6, is critical for activation of caspase in endotoxemic models
(19,30). The data also confirmed previous
reports that LPS-induced TNF-α is responsible for myocardial cell
apoptosis via the NF-κB signaling pathway (29).
Estrogen and ERs are implicated in the cellular
survival of cardiomyocytes (31).
The 17β-estradiol ERα agonist reduces pathological cardiac
hypertrophy and heart failure (32). To investigate the direct effects of
LPS and NG-R1 on cardiomyocytes, and the role of ERα in this
process, the present study used pharmacological inhibitors of ERα,
ICI and MPP The results revealed that the ability of NG-R1 to
inhibit apoptotic and inflammatory responses was dependent on the
activation of ERα. These findings were supported by the observation
that pharmacologic inhibition of ERα, using ICI or MPP, eliminated
the protective effect of NG-R1 against LPS-induced cell death,
proinflammatory cytokine production and activation of NF-κB in
cardiomyocytes (Figs. 1Figure 2Figure 34). In addition, NG-R1 increased the mRNA
and protein expression levels of ERα in the NG-R1-treated H9c2
cardiomyocytes, but, it did not alter the expression of ERβ
(Fig. 1C and D). This finding was
in accordance with previous reports, which suggested that the
activation of ERα in cardiomyocytes attenuates the LPS-induced
expression of TNF-α and myocardial cell apoptosis (29).
In the present study, pretreatment with NG-R1 caused
the activation of ERα (Fig. 1).
There is a missing link between the NG-R1-mediated activation of
ERα and the NG-R1-mediated inhibition of cell apoptosis, decreased
caspase-3 activity, or NG-R1-mediated attenuation of the
inflammatory response (downregulated NF-κB activation and reduced
cytokine expression_. It is well-documented that ERα activates the
phosphoinositide 3-kinase (PI3K)/Akt and mitogen-activated protein
kinase (MAPK) signaling pathways, thereby negatively regulating
LPS-induced NF-κB-dependent inflammatory responses in several cell
types, including cardiomyocytes (32). Therefore, NG-R1 may also inhibit
apoptotic and inflammatory responses through the PI3K/Akt and/or
MAPK signaling pathways, although further investigation is
required.
Another issue to address is that, as an
estrogen-like compound, NG-R1 is a tetracyclic triterpenoid saponin
with a weak estrogenic effect, and the binding capacity of saponins
to ERs is poor in vivo (32). Therefore, the significant
protective effects of NG-R1 in the present study are not limited to
its estrogenic properties. Previous studies have demonstrated that
pretreatment with NG-R1 may also act on the PI3K/Akt and reactive
oxygen species (ROS)/extracellular signal-regulated kinase
signaling pathways and directly scavenge ROS (22). In addition, NG-R1 has exhibited
other multifunctional functions in cardioprotection, including
attenuating the LPS-induced activation of the coagulation system,
reducing fibrinolytic capacity and inhibiting neutrophil/leukocyte
infiltration and inflammatory reactions (24).
In conclusion, the present study revealed that
pretreatment with NG-R1 improved cell viability, inhibited
inflammatory cytokine production and attenuated the LPS-induced
activation of NF-κB in cardiomyocytes. The activation of ERα and
inhibition of the NF-κB signaling pathway in cardiomyocytes is,
therefore, important for the cardioprotective effects of NG-R1. In
addition to our previous studies demonstrating that NG-R1
attenuates cardiac dysfunction in the myocardium of endotoxemic
mice (33,34), the present study suggested that
NG-R1 exerts direct anti-inflammatory effects on cardiomyocytes.
Thus, NG-R1 represents a potent reagent for the treatment of
myocardial inflammation during septic shock.
Acknowledgments
This study was funded by a grant from the Department
of Health of Heilongjiang Province Foundation of China (no.
2011-059).
References
|
1
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crouser E, Exline M, Knoell D and Wewers
MD: Sepsis: links between pathogen sensing and organ damage. Curr
Pharm Des. 14:1840–1852. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin GS, Mannino DM, Eaton S and Moss M:
The epidemiology of sepsis in the United States from 1979 through
2000. N Engl J Med. 348:1546–1554. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baumgarten G, Knuefermann P, Schuhmacher
G, et al: Toll-like receptor 4, nitric oxide, and myocardial
depression in endotoxemia. Shock. 25:43–49. 2006. View Article : Google Scholar
|
|
6
|
Hickson-Bick DL, Jones C and Buja LM:
Stimulation of mitochondrial biogenesis and autophagy by
lipopolysaccharide in the neonatal rat cardiomyocyte protects
against programmed cell death. J Mol Cell Cardiol. 44:411–418.
2008. View Article : Google Scholar
|
|
7
|
Zanotti-Cavazzoni SL and Hollenberg SM:
Cardiac dysfunction in severe sepsis and septic shock. Curr Opin
Crit Care. 15:392–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davani EY, Boyd JH, Dorscheid DR, et al:
Cardiac ICAM-1 mediates leukocyte-dependent decreased ventricular
contractility in endotoxemic mice. Cardiovasc Res. 72:134–142.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu H, Shan L, Schiller PW, Mai A and Peng
T: Histone deacetylase-3 activation promotes tumor necrosis
factor-alpha (TNF-α) expression in cardiomyocytes during
lipopolysaccharide stimulation. J Biol Chem. 285:9429–9436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adamopoulos S, Parissis JT and Kremastinos
DT: A glossary of circulating cytokines in chronic heart failure.
Eur J Heart Fail. 3:517–526. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Collins T, Read MA, Neish AS, Whitley MZ,
Thanos D and Maniatis T: Transcriptional regulation of endothelial
cell adhesion molecules: NF-κB and cytokine-inducible enhancers.
FASEB J. 9:899–909. 1995.PubMed/NCBI
|
|
12
|
Fischer E, Van Zee KJ, Marano MA, et al:
Interleukin-1 receptor antagonist circulates in experimental
inflammation and in human disease. Blood. 79:2196–2200.
1992.PubMed/NCBI
|
|
13
|
Ceylan-Isik AF, Zhao P, Zhang B, Xiao X,
Su G and Ren J: Cardiac overexpression of metallothionein rescues
cardiac contractile dysfunction and endoplasmic reticulum stress
but not autophagy in sepsis. J Mol Cell Cardiol. 48:367–378. 2010.
View Article : Google Scholar :
|
|
14
|
Peng T, Lu X, Lei M, Moe GW and Feng Q:
Inhibition of p38 MAPK decreases myocardial TNF-α expression and
improves myocardial function and survival in endotoxemia.
Cardiovasc Res. 59:893–900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van Empel VP, Bertrand AT, Hofstra L,
Crijns HJ, Doevendans PA and De Windt LJ: Myocyte apoptosis in
heart failure. Cardiovasc Res. 67:21–29. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hale SL, Birnbaum Y and Kloner RA:
Estradiol, administered acutely, protects ischemic myocardium in
both female and male rabbits. J Cardiovasc Pharmacol Ther. 2:47–52.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baker L, Meldrum KK, Wang M, et al: The
role of estrogen in cardiovascular disease. J Surg Res.
115:325–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ylikorkala O: HRT as secondary prevention
of cardiovascular disease. Maturitas. 47:315–318. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schröder J, Kahlke V, Staubach KH, Zabel P
and Stuber F: Gender differences in human sepsis. Arch Surg.
133:1200–1205. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Usui T: Pharmaceutical prospects of
phytoestrogens. Endocr J. 53:7–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang HS and Wang SQ: Notoginsenoside R1
inhibits TNF-α-induced fibronectin production in smooth muscle
cells via the ROS/ERK pathway. Free Radic Biol Med. 40:1664–1674.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun K, Wang CS, Guo J, et al: Protective
effects of ginsenoside Rb1, ginsenoside Rg1, and notoginsenoside R1
on lipopolysaccharide-induced microcirculatory disturbance in rat
mesentery. Life Sci. 81:509–518. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang WJ, Wojta J and Binder BR:
Notoginsenoside R1 counteracts endotoxin-induced activation of
endothelial cells in vitro and endotoxin-induced lethality in mice
in vivo. Arterioscler Thromb Vasc Biol. 17:465–474. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baeuerle PA and Henkel T: Function and
activation of nf-κb in the immune system. Annu Rev Immunol.
12:141–179. 1994. View Article : Google Scholar
|
|
25
|
Lennikov A, Kitaichi N, Noda K, et al:
Amelioration of endotoxin-induced uveitis treated with an iκb
kinase β inhibitor in rats. Mol Vis. 18:2586–2597. 2012.
|
|
26
|
Zandi E, Chen Y and Karin M: Direct
phosphorylation of IkappaB by IKKα and IKKβ: discrimination between
free and NF-κB-bound substrate. Science. 281:1360–1363. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carlson DL, Willis MS, White DJ, Horton JW
and Giroir BP: Tumor necrosis factor-α-induced caspase activation
mediates endotoxin-related cardiac dysfunction. Crit Care Med.
33:1021–1028. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tatsumi T, Akashi K, Keira N, et al:
Cytokine-induced nitric oxide inhibits mitochondrial energy
production and induces myocardial dysfunction in endotoxin-treated
rat hearts. J Mol Cell Cardiol. 37:775–784. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brinckmann M, Kaschina E, Altarche-Xifró
W, et al: Estrogen receptor α supports cardiomyocytes indirectly
through post-infarct cardiac c-kit+ cells. J Mol Cell Cardiol.
47:66–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu CH, Liu JY, Wu JP, et al: et al
17β-estradiol reduces cardiac hypertrophy mediated through the
up-regulation of PI3K/Akt and the suppression of calcineurin/NF-AT3
signaling pathways in rats. Life Sci. 78:347–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murphy E: Estrogen signaling and
cardiovascular disease. Circ Res. 109:687–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xi YD, Yu HL, Ding J, et al: Flavonoids
protect cerebrovascular endothelial cells through Nrf2 and PI3K
from β-amyloid peptide-induced oxidative damage. Curr Neurovasc
Res. 9:32–41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun B, Xiao J, Sun XB and Wu Y:
Notoginsenoside R1 attenuates cardiac dysfunction in endotoxemic
mice: an insight into oestrogen receptor activation and PI3K/Akt
signalling. Br J Pharmacol. 168:1758–1770. 2013. View Article : Google Scholar :
|
|
34
|
Xiao J, Wang NL, Sun B and Cai GP:
Estrogen receptor mediates the effects of pseudoprotodiocsin on
adipogenesis in 3T3-L1 cells. Am J Physiol Cell Physiol.
299:C128–C138. 2010. View Article : Google Scholar : PubMed/NCBI
|














