Introduction
Diabetes mellitus is a group of metabolic diseases
characterized by hyperglycemia as a result of insulin secretion
and/or activity impairment. Diabetic neuropathy is a common
complication that affects sensory neurons, motor neurons and the
autonomic nervous system. Diabetic nerve pain is one of the most
common symptoms of diabetic neuropathy and is characterized by
spontaneous pain, hyperalgesia and paresthesias (1). The mechanisms underlying diabetic
pain are complex and involve multiple mechanisms, including
oxidative and nitrosative stress, immune system activation and
mitochondrial dysfunction (2,3).
Several lines of clinical and experimental evidence have indicated
that neuroinflammation is an important factor in the process of
peripheral and central diabetic neuropathy, which has been
associated with the elevation of pro-inflammatory cytokines,
including tumor necrosis factor-α (TNF-α) and interleukin-1β
(IL-1β) (4–6).
Microglia, which are the predominant resident types
of immune cells involved in the nervous system, mediate and
regulate multiple inflammatory processes and are associated with
the inflammatory changes underlying diabetic neuropathy (7). Minocycline is a tetracycline-derived
antibiotic that exhibits anti-inflammatory properties in the
central nervous system (8). It has
been used in a number of animal models of neuroinflammation in
which microglia are implicated. Studies using rat models of pain
facilitation have demonstrated that minocycline treatment inhibits
microglial activation and cytokine expression (9,10).
Activated microglia have been shown to cause or
exacerbate diabetic pain. The effects of minocycline on pain in
rats with streptozotocin (STZ)-induced diabetes were investigated
in the present study.
Materials and methods
Animals
A total of 42 10 week-old male Sprague-Dawley rats
(200–250 g) were provided by the Experimental Animal Center of the
Fourth Military Medical University (license no. RD-2010-06).
Animals were housed under standard laboratory conditions,
maintained on a 12 h light-dark cycle with food and water ad
libitum. The experimental protocols were approved by the
Institutional Animal Ethics Committee of the Fourth Military
Medical University (Xi’an, China).
Experimental diabetic model
Diabetes was induced in 30 rats by administering one
dose of STZ (Sigma-Aldrich, St. Louis, MO, USA) prepared in citrate
buffer (pH 4.4, 0.1 M; Abcam, Cambridge, MA, USA). Six rats were
removed from the study, due to failure of induction. STZ (65-mg/kg)
was injected intraperitoneally. Twelve age-matched control rats
were administered an equal volume of citrate buffer. Forty-eight
hours following the injection, diabetes was confirmed by collecting
blood samples from the tail vein. Plasma glucose levels were
estimated using a commercial blood glucose analyzer (Accusoft,
Roche Diagnostics, Laval, QC, Canada). Rats with plasma glucose
levels >300 mg/dl during fasting (12–16 h) were considered to be
diabetic and included in the study. Body weight and plasma glucose
levels were recorded twice per week for the duration of the
study.
Intrathecal (IT) catheter implantation
and drug administration
IT catheters were implanted as described by LoPachin
(11). Rats were anesthetized
using isoflurane (2% in oxygen; RWD Life Science, Shenzhen, China).
The occipital muscles were separated, and the cisternal membrane
was exposed. IT polyethylene catheters (catalogue no. PE-10; outer
diameter 0.5 mm, inner diameter 0.25-mm, BD Intramedic™
Polyethylene Tubing; BD Sciences, Franklin Lakes, NJ, USA) were
inserted via an incision in the cisterna magna and advanced
caudally 7–8 cm deep to the lumbar enlargement of the spinal cord.
The incision site was closed in layers, and the catheter was fixed
firmly under the skin and heat-sealed. Animals recovered for 3 days
until further treatment. Rats that exhibited no signs of motor
deficiency (hind limb paralysis and stiffness) were used in the
study. Drugs were injected via the IT catheter with an initial
volume of 10 μl STZ followed by 10 μl of saline
solution.
The rats with STZ-induced diabetes and control rats
were treated with 10, 50 or 100 μg/kg minocycline
(Sigma-Aldrich) dissolved in 10 μl saline, or saline vehicle
injection. IT administration of the drug or vehicle was initiated 4
days following STZ injection and was repeated twice daily at
consistent times for the following 31 days. Paw withdrawal
threshold (PWT) and paw withdrawal latency (PWL) were measured 1 h
prior to minocycline or saline administration.
Assessment of mechanical allodynia
PWT was measured using an up-down testing paradigm.
Rats were placed in cages with mesh floors and covered with
transparent plastic boxes. They acclimatized to their surroundings
for a minimum of 30 min in a temperature-controlled room (25°C)
prior to being tested. Von Frey hairs (Stoelting, Kiel, WI, USA) in
log increments of force (0.38, 0.57, 1.23, 1.83, 3.66, 5.93, 9.13,
and 13.1 g) were applied for 4–6 sec to the region between the foot
pads in the plantar surface of the hind paw. Abrupt paw withdrawal,
licking and shaking were interpreted as positive responses.
Assessment of thermal hyperalgesia
In order to assess nociceptive responses to thermal
stimuli, the rats were placed in plexiglass chambers (18 × 8 × 8
cm; RWD Life Science) and radiant heat (49°C), produced by an
analgesia meter (cat. no. 390G; IITC Life Science, Woodland Hills,
CA, USA), was applied to the plantar surface of the test paw. A
cut-off time of 20 sec was used in order to prevent tissue damage.
PWL from the radiant heat was recorded using a plantar test
(Hargreaves’ method) analgesia meter. Abrupt paw withdrawal,
licking and shaking were interpreted as positive responses.
Western blot analysis
Rats were anesthetized using 5% isoflurane and then
decapitated. Lumbar spinal cords were dissected at L4-L6. Samples
were homogenized in modified radioimmunopreciptation assay buffer
(50 mM Tris-HCl, pH 7.4; 1% nonyl phenoxypolyethoxylethanol, 1 mM
ethylenediaminetetraacetic acid and 150 mM NaCl) supplemented with
protease inhibitor cocktail (Sigma-Aldrich) diluted 1:10 and 2 mM
phenylmethanesulfonylfluoride, 2 mM NaF and phosphatase inhibitor
cocktail I and II were added (Sigma-Aldrich). Lysates were then
centrifuged at 4°C for 20 min at 12,000 × g. The supernatants were
collected and total protein concentration was measured using a
bicinchoninic acid kit (Pierce Biotechnology, Inc., Rockford, IL,
USA). Protein extracts were separated using SDS-PAGE in 7% Tris-HCl
gels (Bio-Rad Laboratories, Hercules, CA, USA) and transferred to
polyvinylidene difluoridemembranes (EMD Millipore, Billerica, MA,
USA), which were subsequently blocked using blocking solution [5%
dry non-fat milk in tris-buffered saline with 0.1% Tween
20® (TBST)] for 1 h. Membranes were incubated with
rabbit anti-Iba-1 antibody (1:1,000; cat. no. 016-20001; Wako
Chemicals USA, Inc., Richmond, VA, USA), rabbit anti-OX-42 antibody
(1:500; cat. no. RA25012; Neuromics, Edina, MN, USA), rabbit
anti-phospho-p38 MAPK antibody (1:500; cat. no. 9212; Cell
Signaling Technology, Inc., Danvers, MA, USA), mouse anti-p38
mitogen-activated protein kinase (MAPK) antibody (1:500; cat. no.
ab31828; Abcam), rabbit anti-TNF-α antibody (1:500; cat. no. 3707;
Cell Signaling Technology, Inc.), rabbit anti-IL-1β antibody
(1:5,000; cat. no. ab200478; Abcam), or rabbit anti-inducible
nitric oxide synthase (iNOS) antibody (1:2,000; cat. no. PA3-030A;
Thermo Fisher, Rockford, IL, USA) in blocking solution overnight at
4°C, washed in TBST and then incubated with horseradish
peroxidase-conjugated anti-rabbit (cat. no. A0545) or anti-mouse
(cat. no. A9044) immunoglobulin G (1:1,000; Sigma-Aldrich, St.
Louis, MO, USA) for 1 h at room temperature. Membranes were then
washed with TBST followed by TBS and developed using enhanced
chemiluminescence detection reagent (GE Healthcare, Bio-Sciences,
Pittsburgh, PA, USA) prior to film exposure (Kodak, Rochester, NY,
USA) a number of times. Membranes were reprobed with an antibody to
β-actin for use as an internal loading control.
Statistical analysis
Quantitative data are expressed as the mean ±
standard error of the mean. Statistical significance among
experimental groups was determined using one-way or two-way
analysis of variance with Bonferroni multiple-comparison post-hoc
analysis. P<0.05 was considered to indicate a statistically
significant difference. Statistical comparisons were computed using
SigmaPlot 12.0 software (Systat Software, Inc., Chicago, IL,
USA).
Results
Rats
Within the first week following STZ injection, rats
demonstrated signs of diabetes, including weight loss, polydipsia
and polyuria. Two weeks following STZ injection, rats exhibiting a
glucose level >300 mg/dl were included in the diabetic group.
Rats with STZ-induced diabetes exhibited significantly increased
blood glucose levels (473±28.6 mg/dl) compared with the control
group (121±12.7 mg/dl) and reduced body weight (195±12.2 g)
compared with the control group (293±12.8 g; Table I).
 | Table IBody weight and blood glucose levels
in rats of different groups. |
Table I
Body weight and blood glucose levels
in rats of different groups.
| Group | Body weight (g)
| Blood glucose (mg/dl)
|
|---|
| Day 0 | Day 14 | Day 0 | Day 14 |
|---|
| Control | 220±13.1 | 293±12.8 | 116±13.1 | 121±12.7 |
| MC | 226±13.6 | 285±14.1 | 116±17.5 | 123±16.4 |
| DM | 240±15.7 | 195±12.2a | 114±14.8 | 473±28.6b |
| DM+MC | 228±15.2 | 197±16.9 | 118±15.1 | 425±24.2 |
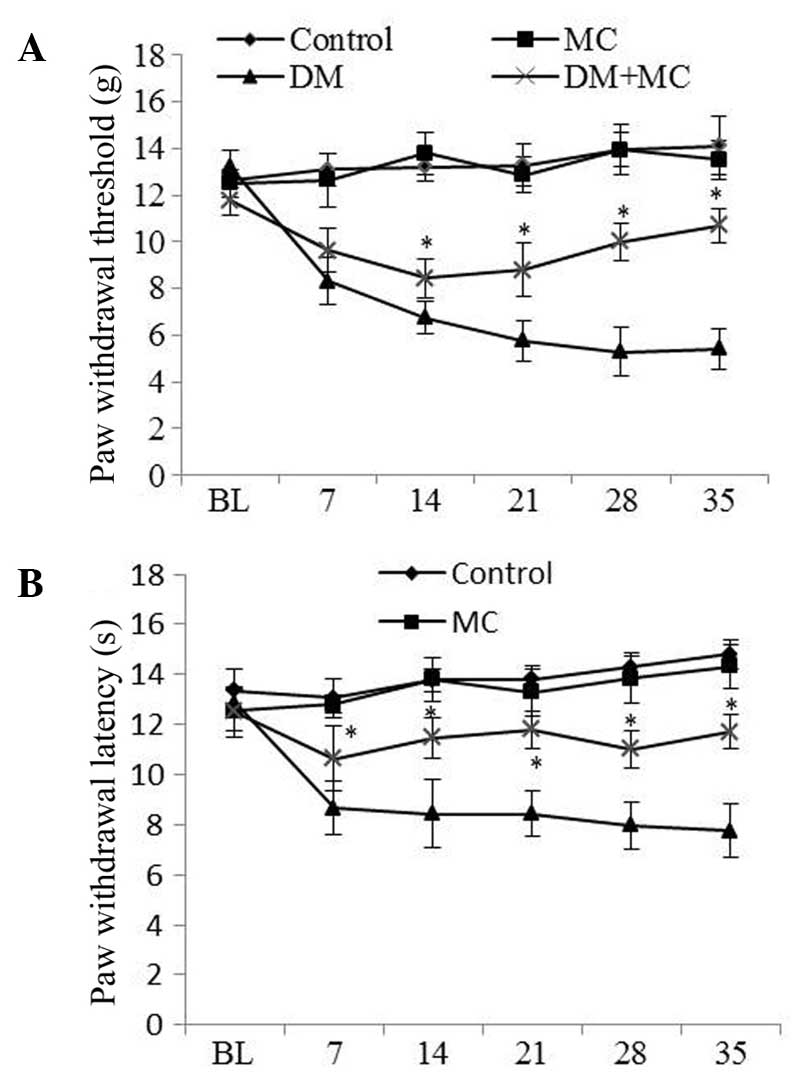
PWT values were significantly lower in diabetic rats
than that in control rats, suggesting the presence of mechanical
allodynia in diabetic rats. However, allodynia was reduced in
diabetic rats treated with minocycline compared with those treated
with vehicle (Fig. 1A). The
threshold for thermal hyperalgesia was significantly decreased
following STZ-injection compared with that in control rats. Similar
to mechanical allodynia, hyperalgesia was significantly lower in
diabetic rats treated with minocycline than in those treated with
saline vehicle (Fig. 1B).
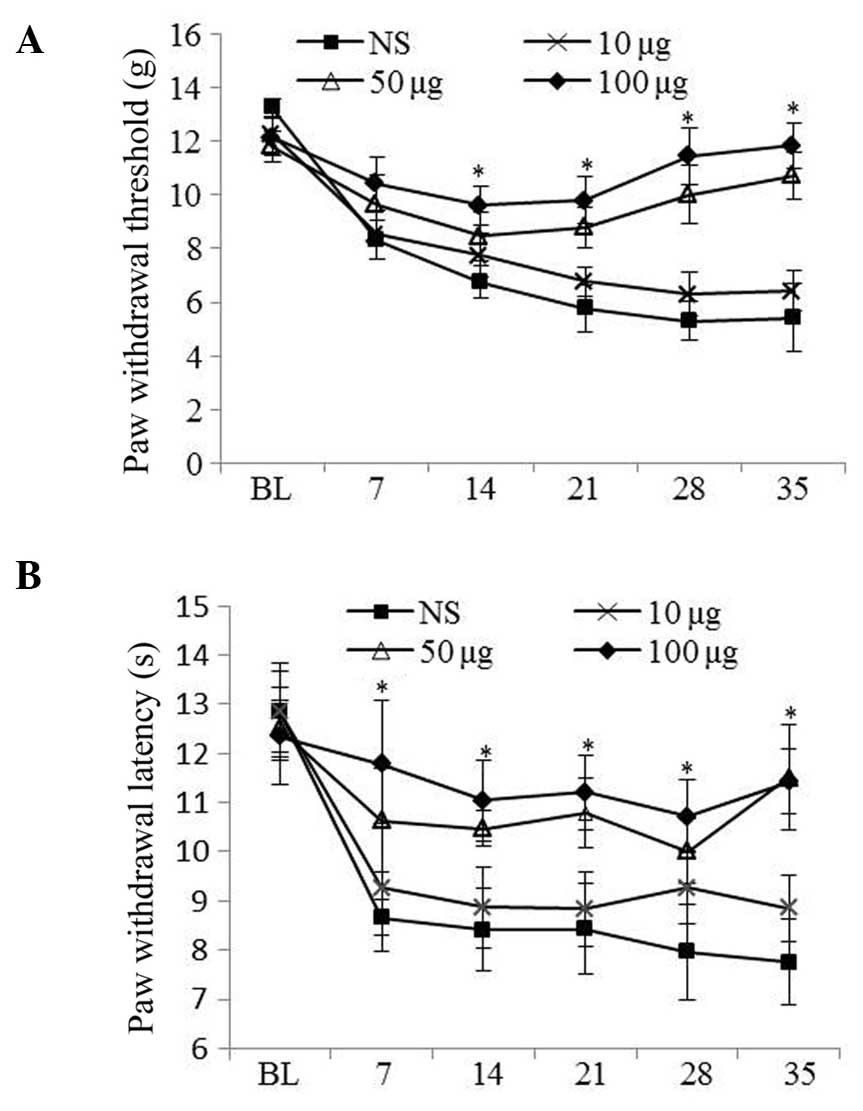
Minocycline treatment attenuated mechanical allodynia and
hyperalgesia in a dose-dependent manner. Minocycline treatment (50
and 100 μg) was shown to reduce diabetic pain in diabetic
rats compared with those treated with vehicle (P<0.05; Fig. 2A and 2B).
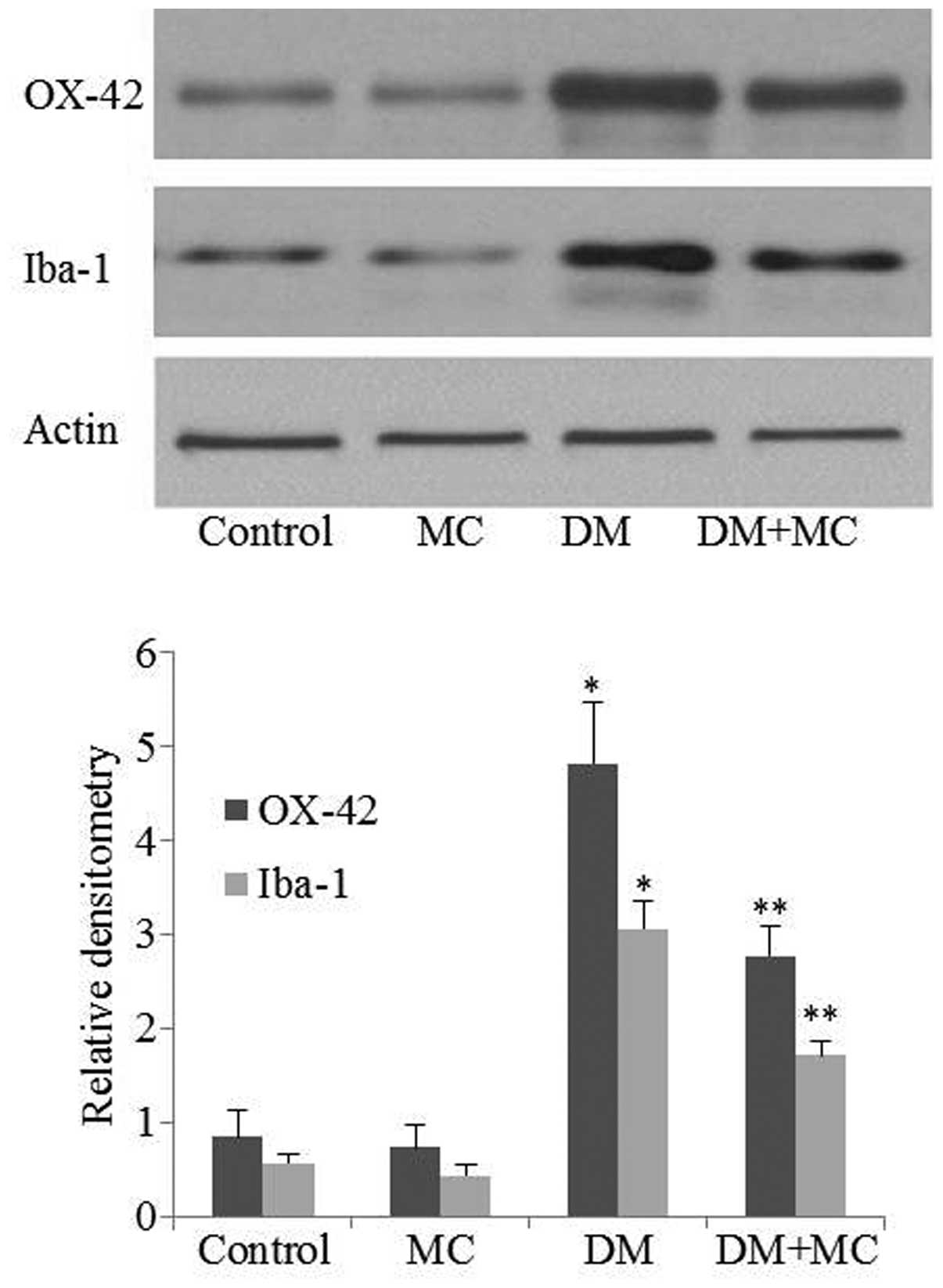
OX-42 and Iba-1 expression is upregulated
in activated microglia
In order to assess the effects of minocycline on
microglial activation in diabetic rats, OX-42 and Iba-1 expression
was measured using western blotting. Western blotting suggested
that OX-42 and Iba-1 expression levels were upregulated in diabetic
rats compared with control rats. However, their expression levels
in the spinal cord decreased significantly following minocycline
treatment of diabetic rats compared with those treated with saline
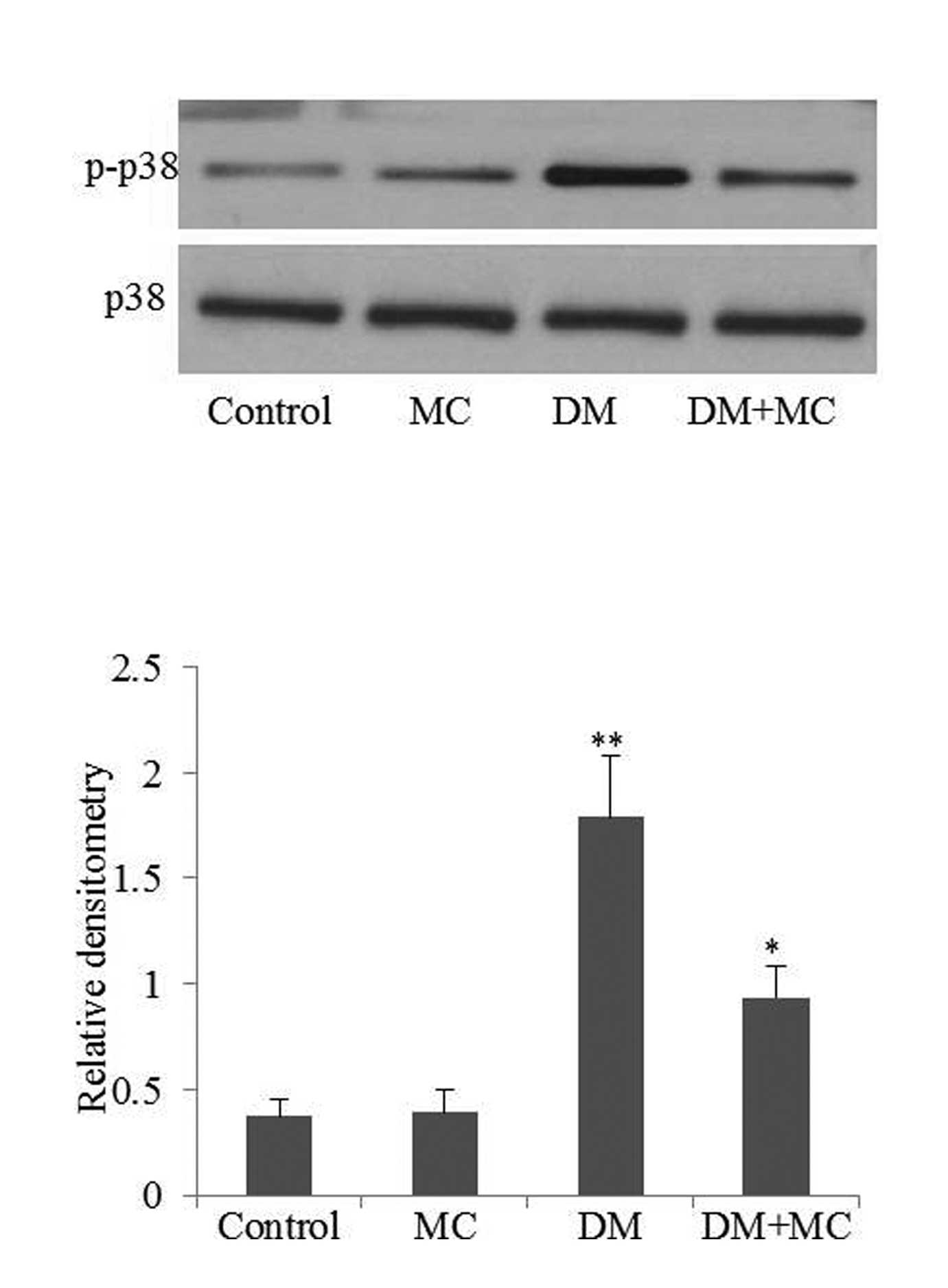
vehicle (Fig. 3; P<0.05). The
p38 MAPK signaling pathway is involved in microglial activation and
cytokine production. Therefore, phospho-p38 MAPK levels were
analyzed in the present study. The results demonstrated that
phospho-p38 MAPK expression in diabetic rats was significantly
higher compared with the control group (Fig. 4; P<0.05). However, the level of
phospho-p38 MAPK expression decreased following minocycline
treatment. The results suggested that microglia were activated in
diabetic rats, while minocycline treatment of diabetic rats
inhibited microglial activation.
Activated microglia secrete
proinflammatory cytokines and iNOS, which are involved in pain
hypersensitivity
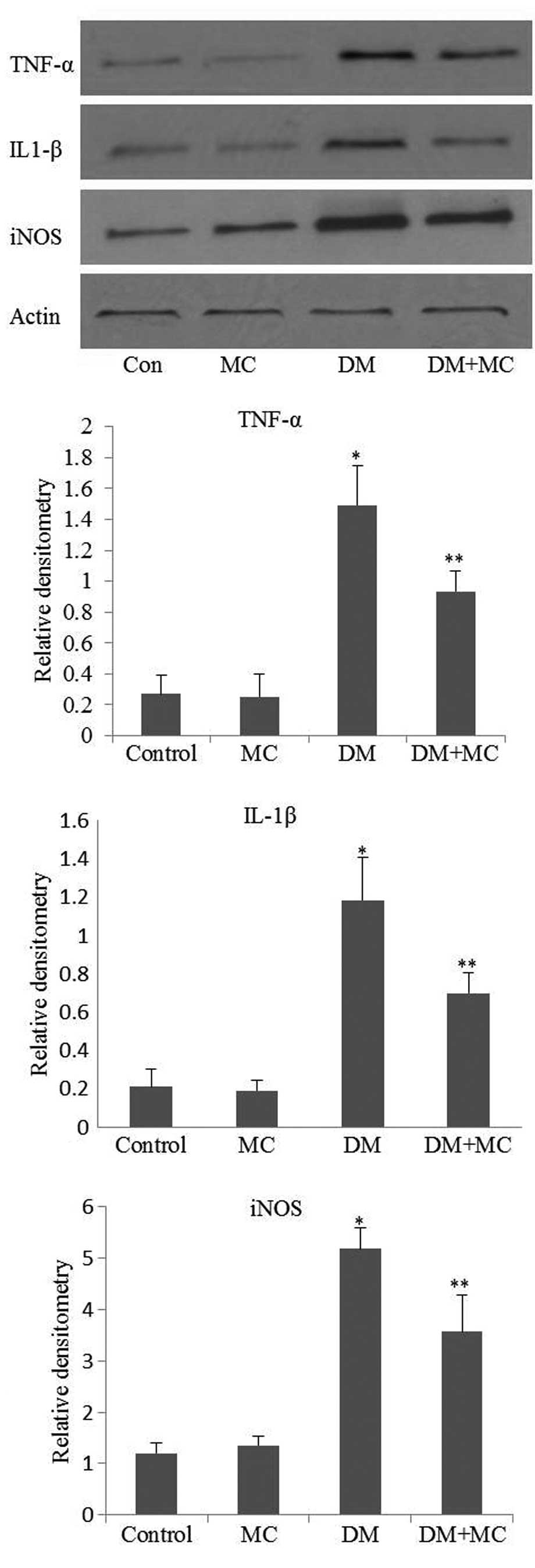
The expression of TNF-α, IL-1β and iNOS in the
spinal cords of diabetic rats was significantly higher compared
with that in the control group. However, following minocycline
treatment, the expression levels of TNF-α, IL-1β and iNOS were
significantly lower in diabetic rats compared with those of the
saline vehicle-treated group (Fig.
5; P<0.05). Proinflammatory cytokines and iNOS expression
was not completely inhibited in minocycline-treated diabetic rats
compared with saline vehicle-treated diabetic rats. This may be
explained by the presence of astrocytes, which are not inhibited by
minocycline and may contribute to the expression of proinflammatory
cytokines and iNOS (12).
Discussion
Inflammation is involved in the progression of
diabetic neuropathic complications. Proinflammatory cytokines, such
as TNF-α and IL-1β have been found to exhibit increased expression
in diabetic patients (13). In
chronic hyperglycemia, proinflammatory cytokines and reactive
oxygen species (ROS) infiltrate vascular tissues and activate
microglia. Recent studies have shown that microglia activation is
associated with the initiation and maintenance of neuropathic pain
(14,15). Activated microglia release a number
of neurotoxins that further enhance microglial proliferation and
activation (10). Activated
microglia excrete TNF-α and IL-1β. In addition, via the expression
of iNOS, activated microglia produce toxic mediators, such as ROS
and nitric oxide (NO). Proinflammatory cytokines and chemokines are
associated with hyperalgesia (5).
TNF-α expression induces the phosphorylation of
c-Jun N-terminal kinase 1 and activates nuclear factor κB (NF-κB),
leading to chemokine (C-C motif) ligand 2 (CCL2) release. CCL2 then
acts on C-C chemokine receptor type 2 (CCR2) receptors on neurons
and interacts positively with neuronal N-methyl-D-aspartate (NMDA)
and α-Amino-3-hydroxy-5 -methyl-4-isoxazolepropionic acid (AMPA)
receptors (16). In the rostral
ventromedial medulla, which is responsible for maintaining chronic
neuropathic pain, TNF-α is induced following nerve injury and
facilitates NMDA receptor phosphorylation (17). TNF-α also stimulates
phosphorylation of the glutamate A1 subunit of the AMPA receptor
and promotes its trafficking to the membrane in dorsal horn neurons
(9,18).
IL-1β is another proinflammatory cytokine involved
in pain hypersensitivity. The release of IL-1β is mediated by
chemokine (C-X3-C motif) ligand 1 signaling and p38 MAPK
activation, and is dependent on adenosine triphosphate. Following
spinal nerve injury or inflammation, pro-IL-1β is cleaved by matrix
metalloproteinase-9 in microglia. IL-1β is an important messenger
between glial cells and neurons. Activation of the IL-1 receptor
causes it to colocalize with NMDA receptors, facilitating NMDA
receptor phosphorylation, which induces changes in synaptic
strength and results in hyperalgesic behavior (17,19).
However, IL-1β may also function in an NMDA receptor-independent
manner (20).
ROS are associated with the development of
persistent pain that results from nerve injury or inflammatory
insult (21). Studies have shown
that ROS in the spinal cord may induce pain by reducing the
inhibitory effect of γ-aminobutyric acid on substantia gelatinosa
neurons that are involved in pain transmission (22). High NO is associated with diabetic
neuropathic pain (23). The
production of NO occurs via increased iNOS activity following
chronic inflammation in diabetic patients. There is evidence to
suggest that there is a reciprocal correlation between NO and
prostaglandin (PG) biosynthetic pathways (24). NO directly influences
cyclooxygenase expression and PG biosynthesis (23).
In the present study, the PWT and PWL in diabetic
rats were markedly lower compared with control rats. Spinal cord
expression levels of OX-42, Iba-1 and phospho-p38 MAPK were
elevated in diabetic rats compared with those in control rats.
Microglia activation produced additional proinflammatory cytokines
and toxic mediators, which are involved in diabetic pain. When
treated with minocycline, diabetic rats exhibited reduced
mechanical allodynia and thermal hyperalgesia compared with those
treated with saline vehicle. The inhibition of microglial
activation was confirmed by a decrease in the expression of
microglia markers and proinflammatory cytokines. The results of the
present study also demonstrated that the expression of
proinflammatory cytokines and iNOS could not be completely blocked
by treatment with minocycline. This was partly because astrocytes,
which cannot be inhibited by minocycline, also contribute to the
expression of proinflammatory cytokines and iNOS (12). These results suggested that
minocycline may attenuate diabetic pain by inhibiting microglial
activity.
The mechanisms underlying the influence of
minocycline on diabetic rats are currently unclear. However, recent
studies have suggested that minocycline inhibits MAPK- and
NF-κB-dependent signaling pathways in primary microglia and
microglial cell cultures compared with controls (13). Furthermore, minocycline suppresses
microglial expression of OX-42 and major histocompatibility complex
class II in rat brains via a protein kinase C-dependent mechanism
(15).
In conclusion, the results of the present study
indicated a potential effect of minocycline for the treatment of
diabetic hypersensitivity. Minocycline may inhibit spinal
microglial activation and attenuate diabetic pain in diabetic rats.
Therefore, further investigation into the use of microglial
inhibition in the treatment of incurable diabetic pain may be
beneficial.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81100816 and
81000563).
References
|
1
|
Yagihashi S, Yamagishi S and Wada R:
Pathology and pathogenetic mechanisms of diabetic neuropathy:
Correlation with clinical signs and symptoms. Diabetes Res Clin
Pract. 77(Suppl 1): S184–S189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Green CJ, Pedersen M, Pedersen BK and
Scheele C: Elevated NF-κB activation is conserved in human myocytes
cultured from obese type 2 diabetic patients and attenuated by
AMP-activated protein kinase. Diabetes. 60:2810–2819. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Newsholme P, Gaudel C and Krause M:
Mitochondria and diabetes. An intriguing pathogenetic role. Adv Exp
Med Biol. 942:235–247. 2012.PubMed/NCBI
|
|
4
|
Zhang YL, Xu JM, Zhou P, Zhong XL and Dai
RP: Distinct activation of tumor necrosis factor-α and
interleukin-6 in the spinal cord after surgical incision in rats.
Mol Med Rep. 5:1423–1427. 2012.PubMed/NCBI
|
|
5
|
Ren K and Dubner R: Interactions between
the immune and nervous systems in pain. Nat Med. 16:1267–1276.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung WW, Kim HS, Shon JR, Lee M, Lee SH,
Sul D, Na HS, Kim JH and Kim BJ: Intervertebral disc
degeneration-induced expression of pain-related molecules: Glial
cell-derived neurotropic factor as a key factor. J Neurosurg
Anesthesiol. 23:329–334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prinz M, Tay TL, Wolf Y and Jung S:
Microglia: Unique and common features with other tissue
macrophages. Acta Neuropathol. 128:319–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amin AR, Attur MG, Thakker GD, Patel PD,
Vyas PR, Patel RN, Patel IR and Abramson SB: A novel mechanism of
action of tetracyclines: Effects on nitric oxide synthases. Proc
Natl Acad Sci USA. 93:14014–14019. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leung L and Cahill CM: TNF-alpha and
neuropathic pain - a review. J Neuroinflammation. 7:272010.
View Article : Google Scholar
|
|
10
|
Naseri K, Saghaei E, Abbaszadeh F, Afhami
M, Haeri A, Rahimi F and Jorjani M: Role of microglia and astrocyte
in central pain syndrome following electrolytic lesion at the
spinothalamic tract in rats. J Mol Neurosci. 49:470–479. 2013.
View Article : Google Scholar
|
|
11
|
LoPachin RM, Rudy TA and Yaksh TL: An
improved method for chronic catheterization of the rat spinal
subarachnoid space. Physiol Behav. 27:559–561. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ihara H, Yamamoto H, Ida T, Tsutsuki H,
Sakamoto T, Fujita T, Okada T and Kozaki S: Inhibition of nitric
oxide production and inducible nitric oxide synthase expression by
a polymethoxyflavone from young fruits of Citrus unshiu in rat
primary astrocytes. Biosci Biotechnol Biochem. 76:1843–1848. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nikodemova M, Duncan ID and Watters JJ:
Minocycline exerts inhibitory effects on multiple mitogen-activated
protein kinases and IkappaBalpha degradation in a stimulus-specific
manner in microglia. J Neurochem. 96:314–323. 2006. View Article : Google Scholar
|
|
14
|
Lim H, Kim D and Lee SJ: Toll-like
receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2
expression in spinal cord microglia. J Biol Chem. 288:7572–7579.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nikodemova M, Watters JJ, Jackson SJ, Yang
SK and Duncan ID: Minocycline down-regulates MHC II expression in
microglia and macrophages through inhibition of IRF-1 and protein
kinase C (PKC)alpha/betaII. J Biol Chem. 282:15208–15216. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I
and Ji RR: The c-Jun N-terminal kinase 1 (JNK1) in spinal
astrocytes is required for the maintenance of bilateral mechanical
allodynia under a persistent inflammatory pain condition. Pain.
148:309–319. 2010. View Article : Google Scholar :
|
|
17
|
Wei F, Guo W, Zou S, Ren K and Dubner R:
Supraspinal glial-neuronal interactions contribute to descending
pain facilitation. J Neurosci. 28:10482–10495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi JI, Svensson CI, Koehrn FJ, Bhuskute
A and Sorkin LS: Peripheral inflammation induces tumor necrosis
factor dependent AMPA receptor trafficking and Akt phosphorylation
in spinal cord in addition to pain behavior. Pain. 149:243–253.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren K: Emerging role of astroglia in pain
hypersensitivity. Jpn Dent Sci Rev. 46:862010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weyerbacher AR, Xu Q, Tamasdan C, Shin SJ
and Inturrisi CE: N-Methyl-D-aspartate receptor (NMDAR) independent
maintenance of inflammatory pain. Pain. 148:237–246. 2010.
View Article : Google Scholar :
|
|
21
|
Gao X, Kim HK, Chung JM and Chung K:
Reactive oxygen species (ROS) are involved in enhancement of
NMDA-receptor phosphorylation in animal models of pain. Pain.
131:262–271. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yowtak J, Lee KY, Kim HY, Wang J, Kim HK,
Chung K and Chung JM: Reactive oxygen species contribute to
neuropathic pain by reducing spinal GABA release. Pain.
152:844–852. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mollace V, Muscoli C, Rotiroti D and
Nisticó G: Spontaneous induction of nitric oxide- and prostaglandin
E2-release by hypoxic astroglial cells is modulated by interleukin
1 beta. Biochem Biophys Res Commun. 238:916–919. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Purwata TE: High TNF-alpha plasma levels
and macrophages iNOS and TNF-alpha expression as risk factors for
painful diabetic neuropathy. J Pain Res. 4:169–175. 2011.
View Article : Google Scholar : PubMed/NCBI
|