Introduction
Myocardial hypertrophy is characterized by an
increased thickness of the heart wall, and is a common clinical
manifestation of a number of hereditary disorders. During
childhood, several diseases may lead to a phenotype mimicking
cardiac hypertrophy (1). The
mitochondrial syndromes MELAS, MERRF and Kearns-Sayre syndrome
affect multiple mitochondrial proteins. If gene defects occur in
respiratory chain enzymes, it usually takes the form of
hypertrophic cardiomyopathy (HCM) (2). The majority of instances of
myocardial hypertrophy are caused by HCM, which is an autosomal
dominant disease that is predominantly characterized by ventricular
hypertrophy, including hypertrophy of the interventricular septum,
and atrial enlargement. Mutations are detected in the sarcomere
protein in 60% of patients with HCM, which is a functional unit
required for the contraction of myocytes (3,4).
However, <10% of cases of cardiac hypertrophy are caused by
other diseases, including aortic valve disease, Fabry's disease
(5), Danon disease, Pompe disease,
Noonan syndrome (6) and
hypertension (1).
Although HCM and other diseases may all result in
myocardial hypertrophy, genetic studies have revealed that the
pathogenic genes and mutations differ among these conditions
(7). To date, >630 mutations in
10 HCM susceptibility genes have been identified. These genes
encode sarcomeric proteins, including β-myosin heavy chain (β MHC),
regulatory myosin light chain, essential myosin light chain,
β-cardiac myosin binding protein C, cardiac troponin T, cardiac
troponin I, α-tropomyosin and cardiac actin (8). The β MHC gene was the first to be
identified as a cause of familial hypertrophic cardiomyopathy
(9). Subsequently, ~100
disease-associated mutations have been defined, including myosin
heavy chain 7 (MYH7), myosin light chain 3 (MYL3), MYL2, actin α
cardiac muscle 1 (ACTC1), tropomyosin 1 (TPM1), troponin T type 2
(TNNT2), troponin I type 3 (TNNI3), troponin C type 1 (TNNC1),
cardiac myosin -binding protein C (MYBPC3), actinin α2 (ACTN2),
titin (TTN) and 5′-AMP-activated protein kinase subunit γ2 (PRKAG2)
(10). Furthermore, other
candidate genes, including acid α-glucosidase (GAA) (11), bone morphogenetic protein 2 (BMP2)
(12), NK2 homeobox 5 (NKX2.5)
(13), lysosome-associated
membrane protein 2 (LAMP-2) (14),
tyrosine-protein phosphatase non-receptor type 11 (PTPN11)
(15), son of sevenless homolog 1
(SOS1) (16) and V-Ki-ras2 Kirsten
rat sarcoma viral oncogene homolog (KRAS) (17), have a close association with
metabolic disease and malformation syndrome.
It is important to identify the causes of myocardial
hypertrophy, in order to provide a theoretical basis for improved
clinical diagnosis and assessment of prognosis. New generation
sequencing technologies have been employed to detect the genetic
causes of various human diseases. Exome sequencing is a high
throughput sequencing technology, which focuses on exonic regions,
which harbor 85% of mutations and exert large effects on
disease-associated traits in the human genome (18,19).
Therefore, exome sequencing may be a better choice with which to
identify mutations involved in human diseases than Genome wide
association studies and Sanger sequencing. Furthermore, exome
sequencing may be used to identify the cause of several
Mendelian-inherited diseases and to identify genes associated with
an increased risk of sudden cardiac death (SCD).
The present study aimed to identify causes of
cardiac hypertrophy. A 10-year-old boy exhibiting left ventricular
hypertrophy (LVH) and pre-excitation with a short PR interval, was
selected as the proband of a Chinese family. Exome sequencing was
used to identify the mutation responsible. The results suggested
that it is important to confirm the genetic causes of cardiac
hypertrophy in order to improve the diagnosis and treatment of this
disease.
Materials and methods
Clinical evaluation
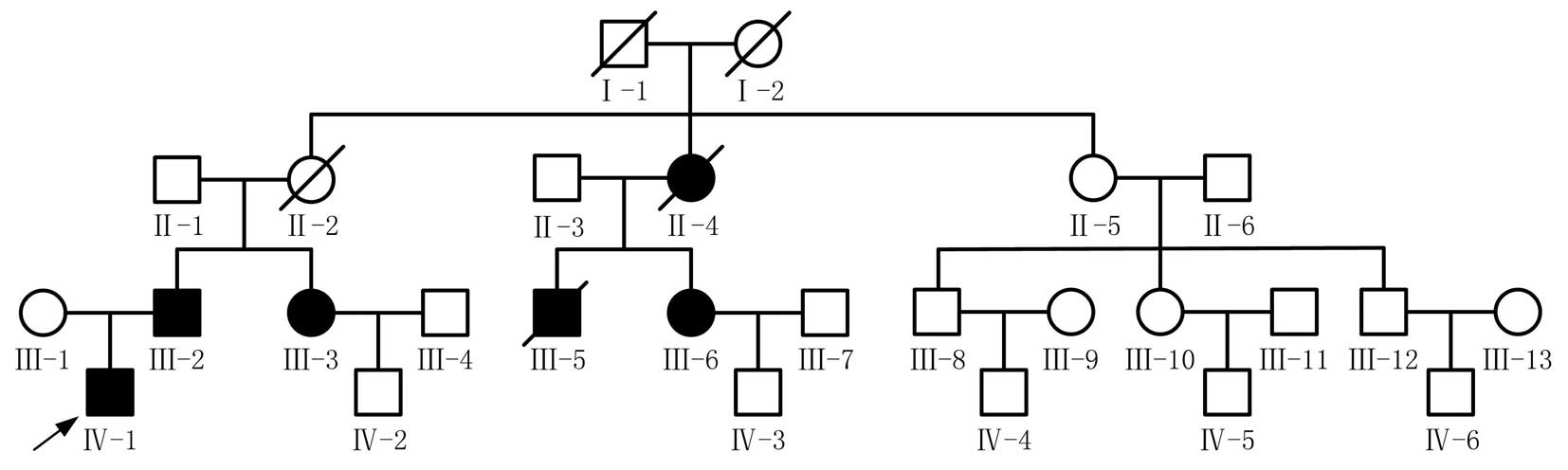
Surviving members of the family (n=22) were assessed
by a detailed analysis of their medical history, a physical
examination, 12-lead electrocardiography (ECG) and two-dimensional
echocardiography (Fig. 1).
Ventricular pre-excitation was diagnosed on the basis of a short PR
interval (<120 ms), a widened QRS interval (>110 ms) and an
abnormal initial QRS vector (δ wave). When the thickness of the
left ventricular free wall or the ventricular septum was >13 mm,
left ventricular hypertrophy was diagnosed. Written, informed
consent was obtained from all participants, according to the
guidelines of the Ethics committee of Xinhua Hospital (Shanghai,
China). Clinicians and nurses undertook the clinical assessments
for the proband and their family in the Department of Pediatric
Cardiology, XinHua Hospital.
Exome capture sequencing
Blood samples were obtained from each individual and
DNA was extracted from whole blood using the QIAamp DNA blood mini
kit.(Qiagen GmbH, Hilden, Germany) according to the manufacturer's
instructions. Whole-exome sequencing was performed in the proband.
The extracted genomic DNA was sonicated in order to produce
fragments and hybridized to the capture array, according to the
manufacturer's instructions. Using the NimbleGen linkers added to
the exonic DNA fragments, the enriched DNA fragments were eluted
and amplified using ligation-mediated polymerase chain reaction
(PCR). The degree of enrichment of the exonic sequences required
quantitative PCR for estimation prior to the second run of library
construction. A minimum requirement of an 80-fold enrichment was
prepared for the following procedure. The enriched exonic DNA was
ligated with DNA ligase to fragments ranging in size between 2 and
5 kb randomly. The resultant DNA fragments were cut into 200 bp, on
average, and were submitted to standard Illumina Hiseq 2000 library
preparation.
The proband DNA was sent to a commercial provider
(Shanghai Biotechnology Co, Ltd, Shanghai, China), which performed
sequencing using the Hiseq 2000 platform. Image analysis and base
calling were performed with the Illumina's Consensus Assessment of
Sequence and Variation 1.8, using the default parameters. Following
removal of reads that contained sequencing adaptors, and
low-quality reads with >5 unknown bases, high quality reads were
aligned to the NCBI human reference genome (hg19 build) using the
Burrows-Wheeler Aligner (BWA) version 0.6.2 (20). Post-alignment procedure and
duplicate removal was performed using SAM tools version 0.1.18 and
Picard version 1.49 (21). Local
realignment of the BWA-aligned and base recalibration were
performed using the Genome Analysis Tool kit.
Mutation detection and validation
All changes were filtered against exome data from
ethnic Han Chinese individuals in the 1,000 Genomes Project
(http://www.1000genomes.org/) and against
the Han Chinese small nucleotide polymorphisms in the dbSNP135
(http://ncbi.nlm.nih.gov/). In order to confirm
the candidate variants, Sanger sequencing was performed on an ABI
3730 capillary sequencing instrument (Applied Biosystems, Foster
City, CA, USA). Primers for exon 3 of PRKAG2 (NM_02449) and exon 1
of urotensin II receptor (UTS2R; NM_018949) were designed for
polymerase chain reaction amplification (Sangon Biotech Co., Ltd.,
Shanghai, China). The sequences of these primers are shown in
Table I. The PCR cycling
conditions were as follows: 98°C for 1 min, 35 cycles of 98°C for
20 sec, 56°C/68°C for 30 sec and 72°C for 30 sec, then 72°C for 10
min and 4°C until use.
 | Table IOligonucleotide primer sequences used
for gene amplification. |
Table I
Oligonucleotide primer sequences used
for gene amplification.
| Variant | Orientation | Sequence (5′-3′) | Fragment size
(bp) | Tm (°C) |
|---|
|
PRKAG2_rs121908987 | Forward |
ATTTCTAATCCCTGTATGCC | 718 | 56 |
| Reverse |
ACCCTGCCAGCAAGAATG | | |
| UTS2R/exon1 | Forward |
CCCAAGGGCTACCGCAAG | 343 | 68 |
| Reverse |
GCCAGAAGGGCAGGAAGCAG | | |
| TTN/exon15 | Forward |
AGGCATCAATAGCCGGTAG | 399 | 54 |
| Reverse |
TATGGCAAAGGAGAAAGG | | |
| TNN/exon149 | Forward |
CCGAAAATAAATATGGTG | 331 | 52 |
| Reverse |
AACTGTTCTCAGGGAAAT | | |
Results
Clinical evaluation
Affected individuals (n=6) from a four-generation
family were diagnosed with Wolff-Parkinson-White (WPW) syndrome.
Their 12-lead ECGs provided evidence of ventricular pre-excitation.
The patients also presented with dyspnea, palpitations, syncope and
fatigue symptoms (Table II).
Patient III-5 was diagnosed with WPW and died suddenly at age 28.
In addition to pre-excitation, two females (Patient II-4 and III-3)
had resting heart rates of <50 beats per minute. Patient II-4
received a permanent pacemaker implant and succumbed to lung cancer
at age 61. Patient III-3 exhibited syncope on one occasion. Cardiac
hypertrophy was identified in 2 of the 6 affected subjects (33%).
However, subject III-6 manifested no clinical symptoms. As a result
of the mortality of the first generation, it is unknown whether
these patients exhibited WPW or any other phenotype.
| Table IIClinical characteristics of affected
individuals. |
Table II
Clinical characteristics of affected
individuals.
| ID number | Age/sex | ECG | LVWT mm | Clinical |
|---|
| II-4 | 61/F | PR | N/A | C, P, V |
| III-2 | 36/M | PR, LVH | 11.7 | C, P, D |
| III-3 | 37/F | PR, LVH, RBBB | 18.2 | P, S |
| III-5 | 28/M | PR | N/A | SCD |
| III-6 | 42/F | PR | 10 | A |
| IV-1 | 10/M | PR | 14.7 | C, P |
Identification of PRKAG2 mutations
through exome sequencing
Whole-exome sequencing of genomic DNA obtained from
the peripheral blood of the proband was performed. There were 126,
101 and 848 total reads produced by exome sequencing. Following
elimination of PCR repetition, the number of unique mapped reads
was 110, 840 and 477, which held 89.02% of the mapped reads. The
coverage of 1x, 10x and 50x was measured in the capturing target
area, accounting for 99.96, 99.4 and 88.92%, respectively. Finally,
20 and 449 exonic variations were identified in the protein-coding
region. According to the screening criteria, 85 variants, including
84 non-synonymous single nucleotide variants (SNVs) and 1 stop-gain
SNV (UTS2R), were identified. However, none of these SNVs revealed
any association with cardiac hypertrophy or WPW, while UTS2R
(p.S241X) appeared to be closely associated with heart failure.
According to OMIM (http://www.ncbi.nlm.nih.gov/omim), a list of genes
associated with cardiac hypertrophy and WPW syndrome was
identified, which contained MYH7, MYL3, MYL2, ACTC, TPM1, TNNT2,
TNNI3, TNNC1, MYBPC3, ACTN2, TTN, PRKAG2, GAA, LAMP-2, PTPN11,
SOS1, KRAS, RAF1 and NKX2.5. A comparison between these candidate
genes and the whole-exome sequencing of the propositus, identified
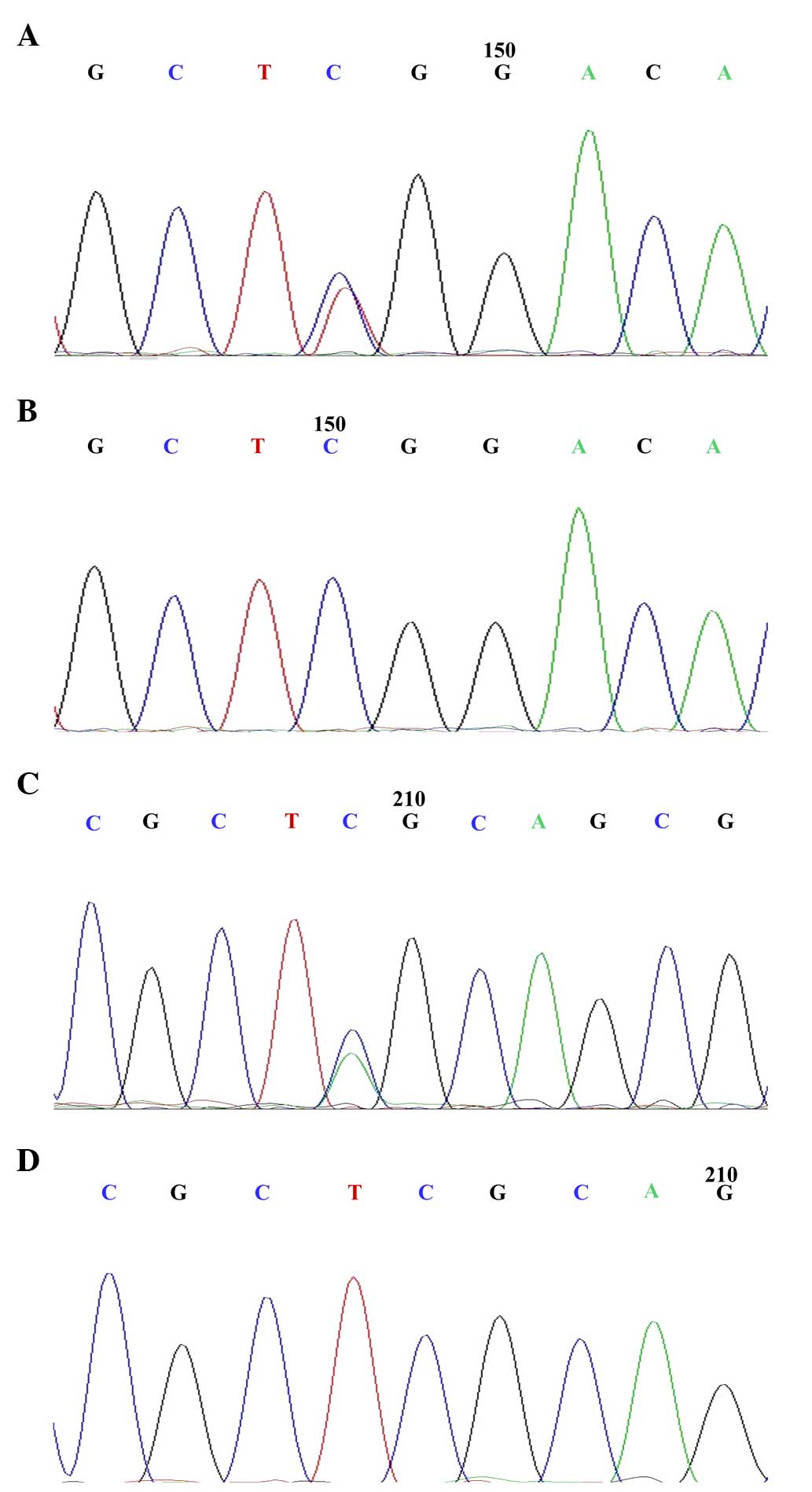
mutations in PRKAG2 (p.R302Q), TTN (p.D20198E), TTN (p.K811T) and
UTS2R (p.S241X). Since PRKAG2 (p.R302Q) has been previously
reported, the present study aimed to confirm whether TTN and UTS2R
were polymorphic in the affected individuals in this family. Normal
DNA samples (n=100) were sequenced to confirm whether UTS2R
(p.S241X), TTN (p.D20198E) and TTN (p.K811T) were polymorphisms.
Family members of the patients III-2, III-3, III-6, IV-1, II-4,
III-5 and IV-3 were shown to exhibit the PRKAG2 mutation, while
others revealed no mutation, with the exception of the members who
had died, I-1 and I-2 (Fig.
2).
Discussion
In the present study of one family, in which the
proband exhibited cardiac hypertrophy and WPW syndrome, 6/25 family
members, excluding the first generation, were found to be affected.
All affected members exhibited electrocardiographic evidence of
pre-excitation. The patients also exhibited clinical symptoms,
including dyspnea, palpitations, syncope and fatigue. A previous
study demonstrated that WPW is likely to be responsible for 10.5%
of cases of SCD in affected individuals <35 years (22). The risk of SCD for patients with
WPW syndrome is 0.02% per year (23). Only one SCD had occurred in the
family involved in the present study, in which the incidence of SCD
was therefore 16.6%, which is marginally higher than 0.02%
(22). Although the prevalence of
SCD appears to have improved, no clear difference was shown, due to
the limited number of cases examined.
The results were in accordance with the results of a
study by Gollb et al (24),
which first demonstrated mutations in the PRKAG2 gene in a family
with an inherited form of WPW, who presented with cardiac
hypertrophy, ventricular pre-excitation and AV nodal disease.
PRKAG2 encodes the regulatory subunit of AMP-activated protein
kinase, and >11 PRKAG2 mutations have been identified, including
a frame-shift mutation (L351Ins) and 10 missense mutations (R302Q,
H383R, T400 N, Y487H, N488I, E506 K, R531G, R531Q, S548P and G100S)
(25). The cardiac phenotype of
the PRKAG2 mutation is similar to that of glycogen storage
cardiomyopathy and HCM. Therefore, other genes associated with
these conditions may also be candidates for causing myocardial
hypertrophy, including NKX2.5, BMP2, MYH7, MYL3, MYL2, ACTC1, TPM1,
TNNT2, TNNI3, TNNC1, MYBPC3, ACTN2, TTN, PRKAG2, LAMP-2 and GAA.
Taking these factors into account, the present study performed
whole-exome sequencing of the proband and analyzed genes, which
were known to be associated with cardiac hypertrophy and WPW.
PRKAG2 (p.R302Q) was found to be the causative mutation in the
affected individuals and a search for mutations within the other
candidate genes failed to reveal sequence alternations. An
identical mutation was identified in all affected members and was
absent in all unaffected members of the family, with the exception
of patient IV-3. Sidhu et al (26) created PRKAG2 transgenic mice model
in order to identify the conduction accessory pathway, and Tan
et al (27) reported that
ventricular pre-excitation, caused by PRKAG2 (p.R302Q), was
associated with Mahaim fibers. In the present study, it was
observed that patient IV-3 (12-years old) expressed the identical
mutation, whilst remaining asymptomatic. Penetrance may be affected
by mutation-specific phenotypes resulting from the patient's own
genetic and environmental background. As the gene carrier is at
risk of developing WPW, they should be closely monitored in the
future. The association between genotype and phenotype remains to
be elucidated.
Notably, the current study identified a stop-gain
mutation, termed UTS2R (p.S241X), in patients III-2 and IV-1, by
Sanger sequencing. Human urotensin II has several cardiovascular
effects. A previous study demonstrated marked expression of
urotensin II in cardiomyocytes, with reduced expression in patients
with early-stage congestive cardiac failure (28). The symptoms of the two members with
this mutation, were more severe than those of other individuals in
this family, and the two carriers may have a tendency to develop
heart failure. Massive exome sequencing may provide a reliable
basis for the identification of genes associated with an increased
risk of cardiac events, although the financial cost of such an
undertaking would be significant.
A number of patients manifest hypertrophic
cardiomyopathy as an initial finding. The present study of a single
family suggested that it may be beneficial to assess patient gene
mutations, particularly in children whose echocardiography reveals
cardiac hypertrophy, and who exhibit a short PR interval on ECG.
Gene carriers who have no clinical manifestations are often
encountered clinically. Exome sequencing methods may provide
genetic evidence to identify pathogenic genes. In addition, the
identification of relevant risk factors to avoid the occurrence of
SCD is required. With the development of next generation sequencing
technology, this approach may make medical genomics a reality.
Acknowledgments
This study was supported by a Project supported by
the National Natural Science Foundation of China (grant no.
81070134/30772349), the National Basic Research Program of China
(grant no. 2010CB529501), the Joint health research program for
major diseases in Shanghai (grant no. 2013ZYJB0016) and the
Shanghai Committee of Science and Technology China (grant no.
124119a3900). The authors would like to thank all the staff at
Xinhua Hospital (Shanghai, China) for their assistance, and all the
members of the family involved in this study.
References
|
1
|
Marin-Garcia J, Ananthakrishnan R,
Goldenthal MJ, Filiano JJ and Perez-Atayde A: Cardiac mitochondrial
dysfunction and DNA depletion in children with hypertrophic
cardiomyopathy. J Inherit Metab Dis. 20:674–680. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scaglia F, Towbin JA, Craigen WJ, et al:
Clinical spectrum, morbidity, and mortality in 113 pediatric
patients with mitochondrial disease. Pediatrics. 114:925–931. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erdmann J, Daehmlow S, Wischke S, et al:
Mutation spectrum in a large cohort of unrelated consecutive
patients with hypertrophic cardiomyopathy. Clin Genet. 64:339–349.
2001. View Article : Google Scholar
|
|
4
|
Richard P, Charron P, Carrier L, et al:
Distribution of disease genes in 102 genotyped families with
hypertrophic cardiomyopathy. Circulation. 104:521. 2001.
|
|
5
|
Sachdev B, Takenaka T, Teraguchi H, Tei C,
Lee P, McKenna WJ and Elliott PM: Prevalence of Anderson-Fabry
disease in male patients with late onset hypertrophic
cardiomyopathy. Circulation. 105:1407–1411. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patra S, Subramaniun A, Mahimaiha J,
Sastry UM and Nanjappa MC: Apical hypertrophic cardiomyopathy in an
infant: first presentation of pompe's disease. World J Pediatr
Congenit Heart Surg. 5:491–493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moak JP and Kaski JP: Hypertrophic
cardiomyopathy in children. Heart. 98:1044–1054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marian AF and Roberts R: The molecular
genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol.
33:655–670. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seidman CE, Seidman JG, InScriver CR, et
al: The Metabolic and Molecular Basis of Inherited Disease. pp.
433–5418. 2001
|
|
10
|
Xu Q, Dewey S, Nguyen S and Gomes AV:
Malignant and benign mutations in familial cardiomyopathies:
Insights into mutations linked to complex cardiovascular
phenotypes. J Mol Cell Cardiol. 48:899–909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Howell RR, Byrne B, Darras BT, Kishnani P,
Nicolino M and van der Ploeg A: Diagnostic challenges for Pompe
disease: an under-recognized cause of floppy baby syndrome. Genet
Med. 8:289–296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lalani SR, Thakuria JV, Cox GF, et al:
20p12.3 microdeletion predisposes to Wolff-Parkinson-White syndrome
with variable neurocognitive deficits. J Med Genet. 46:168–175.
2009. View Article : Google Scholar :
|
|
13
|
Jay PY, Harris BS, Maguire CT, et al:
Nkx2–5 mutation causes anatomic hypoplasia of the cardiac
conduction system. J Clin Invest. 113:1130–1137. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nishino I, Fu J, Tanji K, et al: Primary
LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and
myopathy (Danon disease). Nature. 406:906–910. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tartaglia M, Mehler EL, Goldberg R, et al:
Mutations in PTPN11, encoding the protein tyrosine phosphatase
SHP-2, cause Noonan syndrome. Nat Genet. 29:465–468. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tartaglia M, Pennacchio LA, Zhao C, et al:
Gain-of-function SOS1 mutations cause a distinctive form of Noonan
syndrome. Nat Genet. 39:75–79. 2007. View
Article : Google Scholar
|
|
17
|
Schubbert S, Zenker M, Rowe SL, et al:
Germline KRAS mutations cause Noonan syndrome. Nat Genet.
38:331–336. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Metzker ML: Sequencing technologies-the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar
|
|
19
|
Mamanova L, Coffey AJ, Scott CE, et al:
Target-enrichment strategies for next-generation sequencing. Nat
Methods. 7:111–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Y, Schmidt B and Maskell DL: CUSHAW: a
CUDA compatible short read aligner to large genomes based on the
Burrows-Wheeler transform. Bioinformatics. 28:1830–1837. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li H, Handsaker B, Wysoker A, et al: The
sequence alignment/map format and SAMtools. Bioinformatics.
25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gollob MH, Green MS, Tang AS and Roberts
R: PRKAG2 cardiac syndrome: Familial ventricular preexcitation,
conduction system disease and cardiac hypertrophy. Curr Opin
Cardiol. 17:229–234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fitzsimmons PJ, McWhirter PD, Peterson DW
and Kruyer WB: The natural history of Wolff-Parkinson-White
syndrome in 228 military aviators: A long-term follow-up of 22
years. Am Heart J. 142:530–536. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gollb MH, Green MS, Tang AS, et al:
Identification of a gene responsible for familial
Wolff-Parkinson-White syndrome. N Engl J Med. 344:1823–1831. 2001.
View Article : Google Scholar
|
|
25
|
Zhang BL, Ye Z, Xu RL, et al:
Overexpression of G100S mutation in PRKAG2 causes
Wolff-Parkinson-White syndrome in zebrafish. Clin Genet.
86:287–291. 2014. View Article : Google Scholar
|
|
26
|
Sidhu JS, Rajawat YS, Rami TG, et al:
Transgenic mouse model of ventricular preexeitation and
atrioventricular reentrant achycardia induced by an AMP-activated
protein kinase loss-of-function mutation responsible for
Wolff-Parkinson-White syndrome. Circulation. 111:21–29. 2005.
View Article : Google Scholar
|
|
27
|
Tan HL, van der Wal AC, Campian ME, et al:
Nodoventricular accessory pathways in PRKAG2-dependent familial
preexcitation syndrome reveal a disorder in cardiac development.
Circ Arrhythm Electrophysiot. 1:276–281. 2008. View Article : Google Scholar
|
|
28
|
Douglas SA, Tayara L, Ohlstein EH, Halawaa
N and Giaid A: Congestive heart failure and expression of
myocardial urotensin II. Lancet. 359:1990–1997. 2002. View Article : Google Scholar : PubMed/NCBI
|