Introduction
Laryngeal cancer is a type of malignancy that
originates in the epithelial tissue of laryngeal mucosa. Laryngeal
cancer constitutes 2.4% of all cancers. It is one of the most
common malignant tumors of the head and neck region, ranking third
after nasopharyngeal cancer and sinonasal cancer (1,2).
With increasing industrialization and air pollution, the worldwide
incidence of laryngeal cancer has shown a gradually increasing
trend (3). In the last 30 years,
novel surgical procedures, new chemotherapeutic agents, more
advanced radiotherapy and targeted drugs have been applied in the
treatment of laryngeal cancer. However, the overall survival rate
of laryngeal cancer patients has shown little improvement (4). Therefore, investigation of the
mechanisms underlying the proliferation and apoptosis of laryngeal
cancer cells is of particular importance to the development of
novel and more effective treatments for laryngeal cancer so as to
reduce the rate of mortality.
Human esophageal cancer-related gene 4 (ECRG4) is a
tumor suppressor gene that was initially identified and cloned from
human esophageal epithelial cells in 1998. ECRG4 is widely
expressed in normal human tissues (5). However, ECRG4 expression is
downregulated or lost in esophageal squamous cell carcinoma tissues
and cell lines (6). Studies have
found that in patients with breast cancer, the ECRG4 mRNA
expression level is positively correlated with the survival rate
and overall survival time (7).
Similar results have been obtained from patients with esophageal
cancer (8) and prostate cancer
(9). ECRG4 has been shown to
inhibit the growth of colon cancer cells, esophageal cancer cells
and glioma cells through the induction of cell cycle arrest
(10–12). Knockout of the ECRG4 gene in
zebrafish embryos using RNA interference technology results in an
enhanced capacity for cell proliferation (13). In addition, ECRG4 is closely
associated with apoptosis in a variety of tumor cells (14–16).
However, the effect of ECRG4 on the proliferation and apoptosis of
laryngeal cancer cells and its mechanisms are not yet clear.
In the present study, an ECRG4-overexpressing
laryngeal cancer cell line was obtained after G418 screening. The
effect of ECRG4 overexpression on the proliferation and apoptosis
of laryngeal carcinoma cells was assessed.
Materials and methods
Cell lines
Hep-2 and LSC-1 human laryngeal cancer cell lines
(Wanleibio, Shenyang, China) were cultured in RPMI-1640 medium
(Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum
(FBS, HyClone, Logan, UT, USA). The cells were passaged by
trypsinization when they reached 80–90% confluence.
Construction of the ECRG4 overexpression
vector and selection of the stably transfected cell line
The following primers were designed for
amplification of the coding region of the ECRG4 gene: ECRG4-F,
5′-ATAGAAGCTTGCCCCTCGCCCTC-3′
(HindIII restriction site underlined); and ECRG4-R,
5′-CCGGATCCTCAGAAACCAAGTAGTG-3′
(BamHI restriction site underlined). The human cDNA
preserved in the laboratory was utilized as the template. The
amplified ECRG4 gene was ligated into the pcDNA3.1 plasmid
(Invitrogen Life Technologies, Carlsbad, CA, USA). The recombinant
plasmid, pcDNA3.1-ECRG4, was digested with HindIII and
BamHI, according to the manufacturer's instructions
(Fermentas, Ontario, Canada). The recombinant plasmid was
sequence-analyzed by Sangon Biotech (Shanghai, China). After the
double restriction analysis and sequence analysis, the correct
recombinant plasmid was termed pcDNA3.1-ECRG4. Logarithmically
growing Hep-2 cells were seeded into 6-well plates. Upon reaching
~80% confluence, the cells were transfected with pcDNA3.1-ECRG4 or
negative control (pcDNA3.1 plasmid; Invitrogen Life Technologies)
using Attractene Transfection Reagent (Qiagen, Shanghai, China)
according to the manufacturer's instructions. At 24 h after
transfection, the cells were subjected to G418 selection (400
µg/ml, Invitrogen Life Technologies). After 7–14 days,
positive clones were selected and ECRG4 expression was
examined.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from all groups of cells
using the total RNA extraction kit (Tiangen Biotech, Beijing,
China) according to the manufacturer's instructions. Total RNA was
reverse transcribed into cDNA and then subjected to quantitative
fluorescence analysis on the Exicycler™ 96 quantitative
fluorescence analyzer (Bioneer, Daejeon, Korea) using the SYBR
Green MasterMix (Solarbio, Beijing, China). RT-qPCR was performed
in a volume of 20 µl, containing 0.5 µl each primer
(10 µM), 1 µl cDNA template, 10 µl SYBR GREEN
master mix and 8 µl ddH2O. Thermal cycling
conditions were as follows: Initial denaturation at 95°C for 10
min; amplification for 40 cycles of 95°C for 10 sec, 60°C for 20
sec and 72°C for 30 sec. The mixture was then cooled at 4°C for 5
min. The internal control was β-actin and each sample was tested in
triplicate. The 2−ΔΔCt method (17) was used for relative quantification
of gene expression. The sequences of the primers are listed in
Table I.
 | Table ISequences of primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Sequences of primers for reverse
transcription-quantitative polymerase chain reaction.
| Primer name | Sequence (5′-3′) |
|---|
| ECRG4-F |
AACGAGAAGCACCTGTTCCAA |
| ECRG4-R |
TCGCCATAGTATTCATGTCCA |
| β-actin-F |
CCATCGTCCACCGCAAAT |
| β-actin-R |
GCTGTCACCTTCACCGTTC |
Western blot analysis
The total protein was extracted from all groups of
cells, and the protein concentration was determined using the
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Equal quantities of protein were
loaded onto each lane, separated using 10 or 13% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA).
The membranes were incubated first with the following primary
antibodies: Rabbit anti-human anti-ECRG4 polyclonal antibody
(1:200; cat. no. sc-135139; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), rabbit anti-human anti-cleaved-caspase-3 polyclonal
antibody (1:500; cat. no. bs-0081R; Bioss, Beijing, China), rabbit
anti-human anti-cleaved-poly ADP-ribose polymerase (PARP)
polyclonal antibody (1:200; cat. no. sc-23461-R; Santa Cruz
Biotechnology, Inc.), rabbit anti-human anti-Bcl-2-associated X
protein (Bax) polyclonal antibody (1:400; cat. no. BA0315; Boster,
Wuhan, China) and rabbit anti-human anti-B-cell lymphoma 2 (Bcl-2)
polyclonal antibody (1:400; cat. no. BA0412; Boster) at 4°C
overnight. The membranes were washed four times with Tween-20 in
Tris-buffered saline for 5 min. They were then incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5,000
dilution, Beyotime Institute of Biotechnology) for 45 min at 37°C.
After addition of the enhanced chemiluminescence (ECL) reagents
(Qihai Biotec, Shanghai, China), the target proteins were
visualized and scanned using Gel-Pro Analyzer software 4.0 (Media.
Cybernetics, Inc., Bethesda, MD, USA). The detected proteins were
normalized to β-actin.
Immunofluorescence staining
Cells grown on coverslips were fixed in 4%
paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100
for 30 min and incubated with rabbit anti-human anti-ECRG4
polyclonal antibody (1:100; cat. no. sc-135139; Santa Cruz
Biotechnology, Inc.) at 4°C overnight. The cells were then
incubated with Cy3-labeled goat anti-rabbit secondary antibody
(1:100 dilution; Beyotime Institute of Biotechnology) for 1 h at
room temperature. 4′,6-Diamidino-2-phenylindole (Biosharp, Hefei,
China) was added drop-wise to completely cover the cells for nuclei
staining. The coverslip with cells was inversely placed and mounted
on a slide with anti-fluorescence quenching agent (Solarbio). The
staining results were observed under a laser scanning confocal
microscope and imaged (FV1000S-SIM/IX81; Olympus, Tokyo,
Japan).
Examination of cell proliferation using
the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay
Cells from each experimental group were plated in
96-well plates at a density of 2×103 cells/well. Five
replica wells were set up for each experimental group. Blank
control wells were also included. At days 0, 1, 2, 3 and 4 after
cell inoculation, MTT solution (final concentration of 0.2 mg/ml;
Sigma-Aldrich, St. Louis, MO, USA) was added to each well
containing cells. After incubation at 37°C for 5 h, the supernatant
was removed, and 200 µl dimethylsulfoxide (Sigma-Aldrich)
was added to each well to dissolve the purple crystals. The optical
density at 490 nm (OD490) was determined using a
microplate reader (ELX-800; Bio-TEK Instruments Inc, Winooski, VT,
USA), and cell growth curves were constructed.
Colony formation assay
Cells from each group were seeded at ~300 cells per
60 mm Petri dish. The Petri dishes were incubated at 37°C and 5%
CO2 until visible colonies were formed. The colonies
were fixed in 4% paraformaldehyde, stained with Wright-Giemsa
staining reagent (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) and observed under a microscope (AE31; Motic
Electric, Xiamen, China). A cell cluster containing at least 50
cells was counted as a colony. The colony formation rate was
calculated based on the following formula: Colony formation rate
(%) = (number of colonies/number of seeded cells) × 100.
Analysis of the cell cycle by flow
cytometry
Cells were trypsinized, harvested and fixed in
precooled 70% ethanol at 4°C for 2 h. The fixed cells were
centrifuged at 252 × g, resuspended in 500 µl staining
buffer and mixed thoroughly with 25 µl propidium iodide (PI)
staining solution according to the instructions of the kit
(Beyotime Institute of Biotechnology). Subsequently, 10 µl
RNase A (Beyotime Institute of Biotechnology) was added to the cell
suspension. After incubation at 37°C for 30 min in the dark, the
cells were processed for flow cytometry (FACSCalibur; Becton,
Dickinson and Company, USA).
Hoechst staining
Cells from each experimental group were seeded onto
coverslips in 12-well plates at a density of 3×104
cells/well and cultured for 48 h in an incubator at 37°C and 5%
CO2. The cells were then fixed, stained with Hoechst
solution (Beyotime Institute of Biotechnology), covered with drops
of anti-quenching mounting solution and mounted onto microscope
slides. The slides were observed under a fluorescence microscope
(BX61; Olympus) and photographed.
Examination of apoptosis by flow
cytometry
Cells were trypsinized, harvested and resuspended in
500 µl Binding Buffer according to the instructions of the
apoptosis detection kit (Wanleibio). A total of 5 µl Annexin
V-fluorescein isothiocyanate was added to the cell suspension and
mixed thoroughly. Subsequently, 5 µl PI was added. After
incubation for 15 min at room temperature in the dark, the cells
were analyzed by flow cytometry (FACSCalibur; Becton, Dickinson and
Company) for determination of apoptosis rate.
Statistical analysis
Experimental data are expressed as the mean ±
standard deviation. Comparisons between the experimental groups
were conducted using one-way analysis of variance. Multiple
comparisons were conducted using the Bonferroni post hoc test. Data
analysis and image processing were performed using the Graphpad
Prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Selection of cell lines
To select a suitable human laryngeal cancer cell
line for stable transfection and subsequent ECRG4 gene-related
experiment, the total protein from Hep-2 and LSC-1 cells was
extracted. The expression levels of ECRG4 in the two cell lines
were examined using western blot analysis. The results showed that
the ECRG4 expression level in Hep-2 cells was significantly lower
than that in LSC-1 cells (Fig. 1,
P<0.05). To eliminate the effect of basal expression of ECRG4,
Hep-2 cells were selected for subsequent experiments.
Establishment and identification of the
cell line stably over-expressing ECRG4
To investigate the function of the ECRG4 gene,
pcDNA3.1-ECRG4 was transfected into Hep-2 cells, and ECRG4
expression in positive cells was analyzed by western blot analysis
and RT-qPCR. Cells transfected with empty pcDNA3.1 vector and
parental cells were used as controls. The results showed that the
expression levels of ECRG4 protein and mRNA in the pcDNA3.1-ECRG4
group were increased by 3.05 (Fig.
2A, P<0.01) and 3.07-fold (Fig.
2B, P<0.01), respectively, compared with the pcDNA3.1 group.
The immunofluorescence staining results showed that ECRG4
expression was obviously elevated in the pcDNA3.1-ECRG4 group
compared with the other two groups (Fig. 2C), which were consistent with the
western blot analysis and RT-qPCR results. Thus, a laryngeal cancer
cell line stably overexpressing ECRG4 was established.
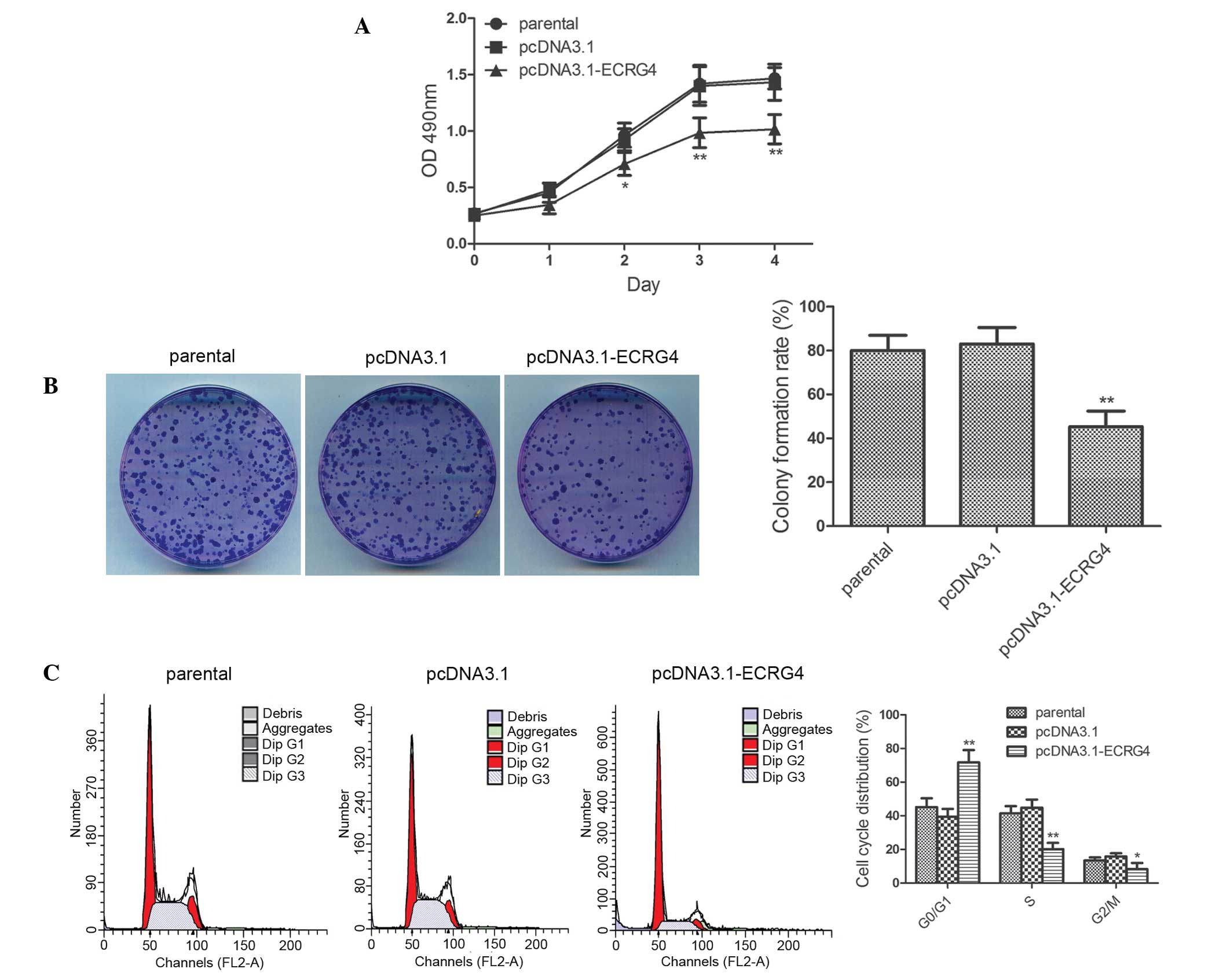
ECRG4 overexpression inhibits the
proliferation of laryngeal cancer cells
To investigate the effect of ECRG4 overexpression on
the proliferative capability of laryngeal cancer cells, cell
proliferative capability was examined using the MTT assay and
colony formation assay. The results of the MTT assay showed that at
day 2, day 3 and day 4, the proliferative capability was severely
impaired in the pcDNA3.1-ECRG4 group compared with the pcDNA3.1
group (Fig. 3A; day 2, P<0.05;
days 3 and 4, P<0.01). The effect of ECRG4 on the clonogenic
capacity of laryngeal cancer cells was further examined using the
colony formation assay. The results showed that the colony
formation rate in the pcDNA3.1-ECRG4 group was 45.27±7.19%, which
was lower than that in the pcDNA3.1 group (83.07±7.51%). The
results indicated that ECRG4 significantly reduced the colony
formation ability of laryngeal cancer cells (Fig. 3B, P<0.01). This study further
investigated the cell-cycle phase distribution in all three groups
of cells using flow cytometry. As shown in Fig. 3C, the percentage of cells in the
G0/G1 phase was significantly increased in the pcDNA3.1-ECRG4 group
compared with that in the pcDNA3.1 group (71.7 vs. 39.41%,
P<0.01). By contrast, the percentage of cells in the S phase and
G2/M phase were decreased markedly in the pcDNA3.1-ECRG4 group (S
phase, 20.14 vs. 44.83%, P<0.01; and G2/M phase, 8.17 vs.
15.76%, P<0.05, respectively). These results indicated that
overexpression of ECRG4 inhibited laryngeal cancer cell
proliferation and arrested cells in the G0/G1 phase of the cell
cycle.
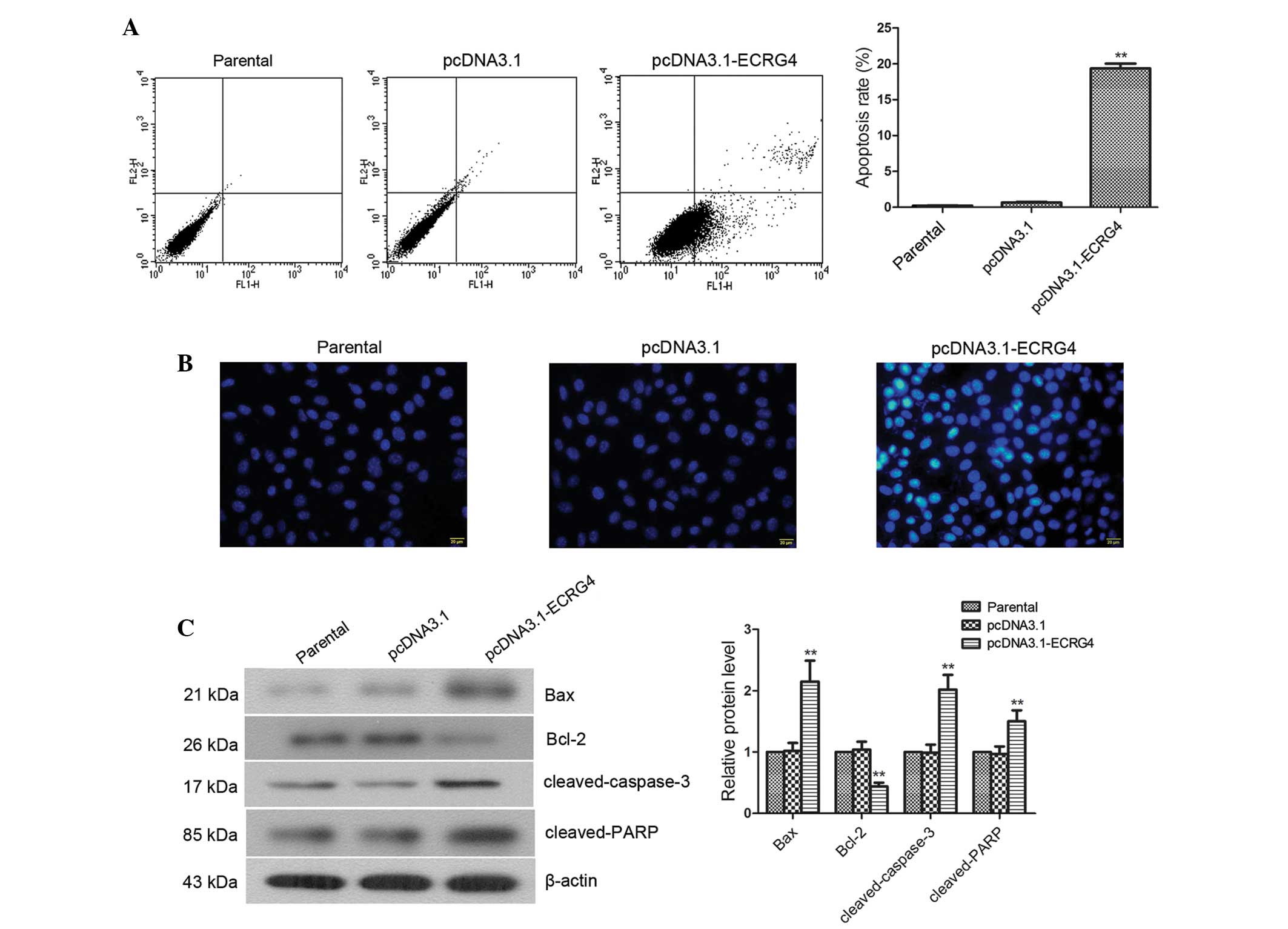
Overexpression of ECRG4 effectively
induces apoptosis in laryngeal cancer cells
Cell apoptosis was measured by flow cytometry and
fluorescence microscopy using Annexin V/PI and Hoechst staining, as
well as the analysis of the expression of apoptosis-related
factors. The results of flow cytometric analysis showed that the
apoptotic rate was significantly elevated in the pcDNA3.1-ECRG4
group compared with the pcDNA3.1 group (19.37±0.67 vs. 0.66±0.09%;
Fig. 4A, P<0.01). Hoechst
staining showed that compared with cells in the parental group and
the pcDNA3.1 group, cells in the pcDNA3.1-ECRG4 group exhibited
significantly increased chromatin condensation and more densely
stained nuclei (Fig. 4B). To
determine whether ECRG4-induced apoptosis of laryngeal cancer cells
affected the expression of apoptosis-related factors, western blot
analysis was performed to examine the expression of Bax, Bcl-2,
cleaved-caspase-3 and cleaved-PARP. The results showed that the
expression levels of cleaved-PARP, cleaved-caspase-3 and Bax were
significantly elevated in cells from the pcDNA3.1-ECRG4 group
compared with that from the pcDNA3.1 group (Fig. 4C, P<0.01). By contrast, the
expression level of Bcl-2 was markedly decreased (P<0.01). These
results demonstrated that overexpression of ECRG4 significantly
induced apoptosis in laryngeal cancer cells.
Discussion
ECRG4 is expressed at low or undetectable levels in
a variety of malignant tumor tissues and cell lines. The expression
level of ECRG4 is closely associated with tumor proliferation and
apoptosis. However, the role of ECRG4 in laryngeal cancer has not
been reported. In the present study, a laryngeal cancer cell line
stably overexpressing ECRG4 was established. This study found that
upregulation of ECRG4 induced cell cycle arrest and inhibited
laryngeal cancer cell proliferation. In addition, upregulation of
ECRG4 accelerated apoptosis in laryngeal cancer cells by regulating
apoptosis-related factor expression. This study preliminarily
clarified the role of ECRG4 in the proliferation and apoptosis of
laryngeal cancer cells and its mechanisms during apoptosis.
The Hep-2 and LSC-1 cells were selected from the
available human laryngeal cancer cell lines and the expression
levels of ECRG4 were compared in the two cell lines. The results
showed that the ECRG4 expression level in Hep-2 cells was
significantly lower than that in LSC-1 cells. As high basal ECRG4
expression would interfere with subsequent experiments, Hep-2 cells
were selected for further experiments.
Attenuated ECRG4 expression levels have been
confirmed in esophageal squamous cell carcinoma (8), prostate cancer (9), colon cancer and glioma (10), while enhanced expression levels of
ECRG4 were detected in normal tissues. ECRG4 efficiently inhibits
the growth of colon cancer cells, glioma cells and esophageal
cancer cells. ECRG4 induces cell cycle arrest at the G0/G1 phase
(10–12), which is hypothesized to be the key
determinant that leads to proliferation inhibition in tumor cells
(18–20). Based on the above findings, this
study investigated the impact of ECRG4 on the proliferation and
cell cycle of the human laryngeal cancer cells. The results showed
that overexpression of ECRG4 significantly inhibited laryngeal
cancer cells proliferation. Further cell cycle analyses by flow
cytometry revealed that ECRG4 overexpression induced G0/G1 cell
cycle arrest. The results demonstrated that ECRG4 inhibited the
growth of laryngeal cancer cells through arresting cells in the
G0/G1 phase and delaying cell cycle progression from the G0/G1
phase to the S phase and G2/M phase. The present results are
consistent with the findings of previous studies (11,12).
Apoptosis can be initiated through the death
receptor- or the mitochondria-dependent pathways (21). Stimulated by apoptotic signals, the
proapoptotic member of the Bcl-2 family, Bax, undergoes a
conformational change in the mitochondrial pathway of apoptosis.
Bax translocates from the cytoplasm to the mitochondria and inserts
into the mitochondrial membrane, which induces an increase in
mitochondrial membrane permeability and results in the release of
cytochrome c. Cytochrome c then activates caspase-3,
thereby inducing apoptosis (22).
The level of Bcl-2, an important antiapoptotic protein, is
correlated with tumor cell apoptosis. Bcl-2 is overexpressed in a
variety of tumor cells, which conveys a certain degree of
resistance to apoptosis-inducing drugs (23–25).
Additionally, downregulation of Bcl-2 abolishes the resistance and
promotes apoptosis in tumor cells (26,27).
The caspase family of proteases has been shown to exhibit a
critical in apoptosis (28).
Caspase-3 is a member of the caspase family and a key protease in
apoptosis. Once activated, caspase-3 triggers the activation of the
downstream proteins and inevitably leads to apoptosis. Therefore,
caspase-3 is known as the death protease (29). Caspase-3 is normally present in the
cytoplasm in the form of an inactive zymogen. Apoptotic signals
induce caspase-3 cleavage and activation through a variety of
proteolytic enzymes, resulting in the generation of
cleaved-caspase-3. PARP, the substrate of caspase-3, is activated
and subsequently induces apoptosis (30–32).
Studies have shown that ECRG4 effectively induces apoptosis in
esophageal squamous cell carcinoma cells (14), head and neck squamous cell
carcinoma cells (15) and gastric
cancer cells (16); accompanied by
upregulation of Bax and downregulation of Bcl-2 (15). This study further investigated
whether ECRG4 overexpression induced apoptosis in human laryngeal
cancer cells and examined the expression levels of a number of key
apoptosis-related factors. The results of this study also
demonstrated that overexpression of ECRG4 activated caspase-3 and
PARP, and ultimately induced apoptosis through upregulating the
expression of proapoptotic protein Bax and downregulating the
expression of antiapoptotic protein Bcl-2.
In conclusion, ECRG4 suppresses the proliferation of
laryngeal cancer cells through the induction of G0/G1 cell cycle
arrest. In addition, ECRG4 induces apoptosis via regulation of the
expression of Bax, Bcl-2, cleaved-caspase-3 and cleaved-PARP.
Therefore, overexpression of ECRG4 may become an effective gene
therapy strategy for the treatment of laryngeal cancer.
Acknowledgments
This study was supported by a grant from the
Medicine Summit Project of Liaoning Province (grant no.
4010218).
References
|
1
|
Hoffman HT, Porter K, Karnell LH, Cooper
JS, Weber RS, Langer CJ, Ang KK, Gay G, Stewart A and Robinson RA:
Laryngeal cancer in the United States: changes in demographics,
patterns of care and survival. Laryngoscope. 116(Suppl 111): 1–13.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jaseviciene L, Gurevicius R, Obelenis V,
Cicenas S and Juozulynas A: Trends in laryngeal cancer incidence in
Lithuania: A future perspective. Int J Occup Med Environ Health.
17:473–477. 2004.
|
|
4
|
Almadori G, Bussu F, Cadoni G, Galli J,
Paludetti G and Maurizi M: Molecular markers in laryngeal squamous
cell carcinoma: Towards an integrated clinicobiological approach.
Eur J Cancer. 41:683–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuzaki J, Torigoe T, Hirohashi Y,
Tamura Y, Asanuma H, Nakazawa E, Saka E, Yasuda K, Takahashi S and
Sato N: Expression of ECRG4 is associated with lower proliferative
potential of esophageal cancer cells. Pathol Int. 63:391–397. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue CM, Deng DJ, Bi MX, Guo LP and Lu SH:
Expression of ECRG4, a novel esophageal cancer-related gene,
downregulated by CpG island hypermethylation in human esophageal
squamous cell carcinoma. World J Gastroenterol. 9:1174–1178.
2003.PubMed/NCBI
|
|
7
|
Sabatier R, Finetti P, Adelaide J, Guille
A, Borg JP, Chaffanet M, Lane L, Birnbaum D and Bertucci F:
Down-regulation of ECRG4, a candidate tumor suppressor gene, in
human breast cancer. PLoS One. 6:e276562011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mori Y, Ishiguro H, Kuwabara Y, Kimura M,
Mitsui A, Kurehara H, Mori R, Tomoda K, Ogawa R, Katada T, et al:
Expression of ECRG4 is an independent prognostic factor for poor
survival in patients with esophageal squamous cell carcinoma. Oncol
Rep. 18:981–985. 2007.PubMed/NCBI
|
|
9
|
Vanaja DK, Ehrich M, Van den Boom D,
Cheville JC, Karnes RJ, Tindall DJ, Cantor CR and Young CY:
Hypermethylation of genes for diagnosis and risk stratification of
prostate cancer. Cancer Invest. 27:549–560. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Götze S, Feldhaus V, Traska T, Wolter M,
Reifenberger G, Tannapfel A, Kuhnen C, Martin D, Müller O and
Sievers S: ECRG4 is a candidate tumor suppressor gene frequently
hypermethylated in colorectal carcinoma and glioma. BMC Cancer.
9:4472009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li LW, Yu XY, Yang Y, Zhang CP, Guo LP and
Lu SH: Expression of esophageal cancer related gene 4 (ECRG4), a
novel tumor suppressor gene, in esophageal cancer and its
inhibitory effect on the tumor growth in vitro and in vivo. Int J
Cancer. 125:1505–1513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li W, Liu X, Zhang B, Qi D, Zhang L, Jin Y
and Yang H: Overexpression of candidate tumor suppressor ECRG4
inhibits glioma proliferation and invasion. J Exp Clin Cancer Res.
29:892010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gonzalez AM, Podvin S, Lin SY, Miller MC,
Botfield H, Leadbeater WE, Roberton A, Dang X, Knowling SE,
Cardenas-Galindo E, et al: Ecrg4 expression and its product augurin
in the choroid plexus: impact on fetal brain development,
cerebrospinal fluid homeostasis and neuroprogenitor cell response
to CNS injury. Fluids Barriers CNS. 8:62011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Zhang C, Li X, Lu S and Zhou Y: The
candidate tumor suppressor gene ECRG4 inhibits cancer cells
migration and invasion in esophageal carcinoma. J Exp Clin Cancer
Res. 29:1332010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu T, Xiao D and Zhang X: ECRG4 inhibits
growth and invasiveness of squamous cell carcinoma of the head and
neck in vitro and in vivo. Oncol Lett. 5:1921–1926. 2013.PubMed/NCBI
|
|
16
|
Jiang CP, Wu BH, Wang BQ, Fu MY, Yang M,
Zhou Y and Liu F: Overexpression of ECRG4 enhances chemosensitivity
to 5-fluorouracil in the human gastric cancer SGC-7901 cell line.
Tumour Biol. 34:2269–2273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Hsiao YC, Hsieh YS, Kuo WH, Chiou HL, Yang
SF, Chiang WL and Chu SC: The tumor-growth inhibitory activity of
flavanone and 2′-OH flavanone in vitro and in vivo through
induction of cell cycle arrest and suppression of cyclins and CDKs.
J Biomed Sci. 14:107–119. 2007. View Article : Google Scholar
|
|
19
|
Ayyagari VN and Brard L: Bithionol
inhibits ovarian cancer cell growth in vitro-studies on
mechanism(s) of action. BMC Cancer. 14:612014. View Article : Google Scholar
|
|
20
|
He L, Lu N, Dai Q, Zhao Y, Zhao L, Wang H,
Li Z, You Q and Guo Q: Wogonin induced G1 cell cycle arrest by
regulating Wnt/β-catenin signaling pathway and inactivating CDK8 in
human colorectal cancer carcinoma cells. Toxicology. 312:36–47.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gross A, Jockel J, Wei MC and Korsmeyer
SJ: Enforced dimerization of BAX results in its translocation,
mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang D, Chen MB, Wang LQ, Yang L, Liu CY
and Lu PH: Bcl-2 expression predicts sensitivity to chemotherapy in
breast cancer: A systematic review and meta-analysis. J Exp Clin
Cancer Res. 32:1052013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moldoveanu T, Follis AV, Kriwacki RW and
Green DR: Many players in BCL-2 family affairs. Trends Biochem Sci.
39:101–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
26
|
Hall C, Troutman SM, Price DK, Figg WD and
Kang MH: Bcl-2 family of proteins as therapeutic targets in
genitourinary neoplasms. Clin Genitourin Cancer. 11:10–19. 2013.
View Article : Google Scholar
|
|
27
|
Barillé-Nion S, Bah N, Véquaud E and Juin
P: Regulation of cancer cell survival by BCL2 family members upon
prolonged mitotic arrest: opportunities for anticancer therapy.
Anticancer Res. 32:4225–4233. 2012.PubMed/NCBI
|
|
28
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cryns V and Yuan J: Proteases to die for.
Genes Dev. 12:1551–1570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Visconti R and D'Adamio L: Functional
cloning of genes regulating apoptosis in neuronal cells. Methods
Mol Biol. 399:125–131. 2007. View Article : Google Scholar
|
|
31
|
Kuribayashi K, Mayes PA and El-Deiry WS:
What are caspases 3 and 7 doing upstream of the mitochondria?
Cancer Biol Ther. 5:763–765. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lockshin RA: Programmed cell death:
History and future of a concept. J Soc Biol. 199:169–173. 2005.In
French. View Article : Google Scholar
|