Introduction
Emery-Dreifuss muscular dystrophy (EDMD) refers to a
group of inherited muscular dystrophies, characterized by slow,
progressive muscular weakness and atrophy of the humeral and
peroneal distributions, with early contractures, which affect the
Achilles tendon, spine and elbows (1). EDMD is also associated with
cardiomyopathy and abnormalities of the cardiac conduction system
(2,3). Cardiac problems are the leading cause
of sudden mortality in patients with EDMD, therefore, it is
important that cardiac symptoms are diagnosed and treated early to
improve outcomes and prolong life expectancy in patients with EDMD
(2,4).
EDMD can arise from mutations in five different
genes, EMD, LMNA, SYNE1, SYNE2 and
FHL1, and can be inherited as an X-linked, autosomal
dominant (AD) or autosomal recessive disorder (5). The autosomal forms of the disease are
predominantly caused by gene mutations in the lamin A/C gene
(LMNA) (6), and patients
with EDMD exhibiting LMNA mutations typically present with
more severe cardiac involvement, compared with patients with other
EDMD gene mutations. However, there are currently few case reports
detailing the various genetic mutations according to phenotype
(7–9). The structural/functional correlations
are further complicated by LMNA mutations, which have been
associated with several distinct clinical disorders, including
Hutchinson-Gilford progeria syndrome, Charcot-Marie-Tooth disease,
limb girdle muscular dystrophy, familial partial lipodystrophy and
dilated cardiomyopathy, in addition to EDMD (7). To further improve knowledge regarding
specific LMNA mutations and disease phenotypes, the present
case study assessed the cardiac phenotypes in two families with
EDMD, caused by the same c.1583 C→G (T528R) LMNA
mutation.
Materials and methods
Collection of clinical data
The present case study investigated two Chinese
families, which included three EDMD-affected and 12 unaffected
members, ascertained by neurological examination. Clinical data and
detailed family surveys were collected. The patients and family
members included in the present study provided written informed
consent and the study was approved by the ethics committee of the
Third Hospital of Hebei Medical University (Hebei, China).
Muscle biopsy, histochemistry and
immunohistochemical analyses
Skeletal muscle biopsies were performed for
diagnostic purposes on the three affected patients. Briefly, muscle
biopsy specimens (0.5 cm diameter, 1.0 cm length) were collected
from the biceps brachii, under local lidocaine anesthesia (Xiamen
Kang Source Biotechnology Limited, Xiamen, China). The muscle
biopsy specimens were frozen in isopentane cooled in liquid
nitrogen, and 7 µm cryostat sections were made using an EM
UC ultrathin section machine (Leica Microsystems, Mannheim,
Germany), and were used for the subsequent histochemistry and
immunofluorescence staining experiments. Morphological analysis of
the muscle was performed using the following routine histological
stains: Hematoxylin and eosin, modified Gomori trichrome,
NADH-tetrazoliumreductase, succinate dehydrogenase, adenosine
monophosphate deaminase and cytochrome c oxidase to assess
enzymatic activity, adenosine triphosphatase at pH 9.8, 4.3 and 4.6
to assess muscle fiber distribution, acid phosphatase, and oil red
O staining to assess fatty deposition (all Shanghai Bioleaf Biotech
Co., Ltd., Shanghai, China). Immunohistochemical analysis was
performed using antibodies targeting procoagulant and anticoagulant
membrane components. The following antibodies were used in 1:50
dilutions overnight at 4°C: Anti-emerin (NCL-EMERIN) and anti-lamin
A/C (NCL-LAM-A/C) (Novocastra, Newcastle upon Tyne, UK).
Biotinylated anti-mouse immunoglobulin G secondary antibodies
(Novocastra) were then used for 1 h at 37°C. The
avidin-biotin-peroxidase complex (ABC) method was used for signal
detection (ABC kit; Vector Laboratories, Inc., Burlingame, CA,
USA). After staining the muscle specimens were visualized, and the
images were captured, using a BX51 confocal scanning laser
microscope (Olympus Corporation, Tokyo, Japan).
Investigation of cardiac involvement
Electrocardiography (ECG) (FX-740; Fukuda Denshi
Co., Ltd., Tokyo, Japan), 24 h Holter (FM 800; Fukuda Denshi Co.,
Ltd.), ultrasound cardiography (UCG) (iE33; Philips, Amsterdam,
Netherlands) and 99TcM-MIBI-gated myocardiac
perfusion imaging (GPI) (Infinia Hawkeye 4; GE Healthcare
Bio-Sciences, Piscataway, NJ, USA) investigations were performed on
the three patients with EDMD (patients 1, 2 and 3).
Gene analysis
Genomic DNA from nine family members from Family 1,
and six family members from Family 2 were extracted from peripheral
blood (500 µl) using the Genomic DNA Extraction kit 5.0
(Takara Biotechnology Co., Ltd., Dalian, China), according to the
manufacturer's instructions. Control genomic DNA samples were
obtained from 50 healthy Chinese unrelated individuals. DNA
concentration was determined using a Nanodrop ND-1000
spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE,
USA). Each reaction required 50 ng genomic DNA, 10 pmol primer sets
(sequences in Table I;
Sigma-Aldrich, St. Louis, MO, USA), dNTP and Qiagen Multiplex PCR
Master Mix (Qiagen, Hilden, Germany). The following conditions were
used for multiplex polymerase chain reaction (PCR): 15 min at 95°C;
40 cycles of amplification at 95°C for 30 sec, 60°C for 30 sec and
72°C for 1 min; 35 cycles of denaturation at 95°C for 45 sec 63.5°C
for 30 sec and 72°C for 45 sec; 10 min at 72°C. Briefly, 50 ng
genomic DNA from the patients was amplified using the hot-start PCR
method. The DNA was subjeceted to PCR using the Qiagen Multiplex
PCR Master Mix. Each PCR reac-tion consisted of 50 ng genomic DNA,
16.8 µl SYBR Green PCR Master Mix, and 250 nm specific
primer pairs, in a total 25 µl volume. Using a
pre-sequencing kit (USB Corporation, Cleveland, OH, USA), the PCR
products were purified and sequenced by dye terminator chemistry
using an ABI Prism 377 DNA Sequencer (Applied Biosystems, Foster
City, CA, USA). The resulting sequences were subsequently aligned
and mutations were determined using Sequencher version 4.9 sequence
alignment software (Gene Codes Corporation, Ann Arbor, MI,
USA).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Exon | Sense | Anti-sense |
|---|
| 1 |
5TCGGGACGCAAGAGGCAAAG3 |
5CGACTCGTTTCACGCACTCCTCA3 |
| 2 |
5CAGTGGGAGGAAGAACCATAAC3 |
5GTTATGGTTCTTCCTCCCACTG3 |
| 3 |
5GGGCGGCGTCGTAGAGTAGG3 |
5AAGTCCTACTCTACGACGCC3 |
| 4 |
5GAGTTTGAGTGCGACGAAGG3 |
5TGACCACCTCTAACTGTTACCCT3 |
| 5 |
5CCCCACCAGGTTGCTGTTCC3 |
5GGAACAGCAACCTGGTGGGG3 |
| 6 |
5AGAGGAGGAGCGGGAGGTTC3 |
5TCTGGACCTCCTGAGTGACCG3 |
| 7 |
5CCTCCTCATCCACCTCCTCCAC3 |
5GCCGCAGCAGCTTCTCACAG3 |
| 8 |
5TCAGGGTGAACTTTGGTGGG3 |
5AATGGAGATGATCCCTTGCTG3 |
| 9 |
5CAGGTGTTCTGTGCCTTCCA3 | 5
GTGGAAGGCACAGAACACCTG3 |
| 10 | 5
GTGGTGGTGATGGAGCAGGT3 | 5
TGAGGACGACGAGGATGAGG3 |
| 11 |
5CGTGACACTGGAGGCAGAAGAG3 |
5GGCAGAAGAGCCAGAGGAGATG3 |
Results
Clinical data
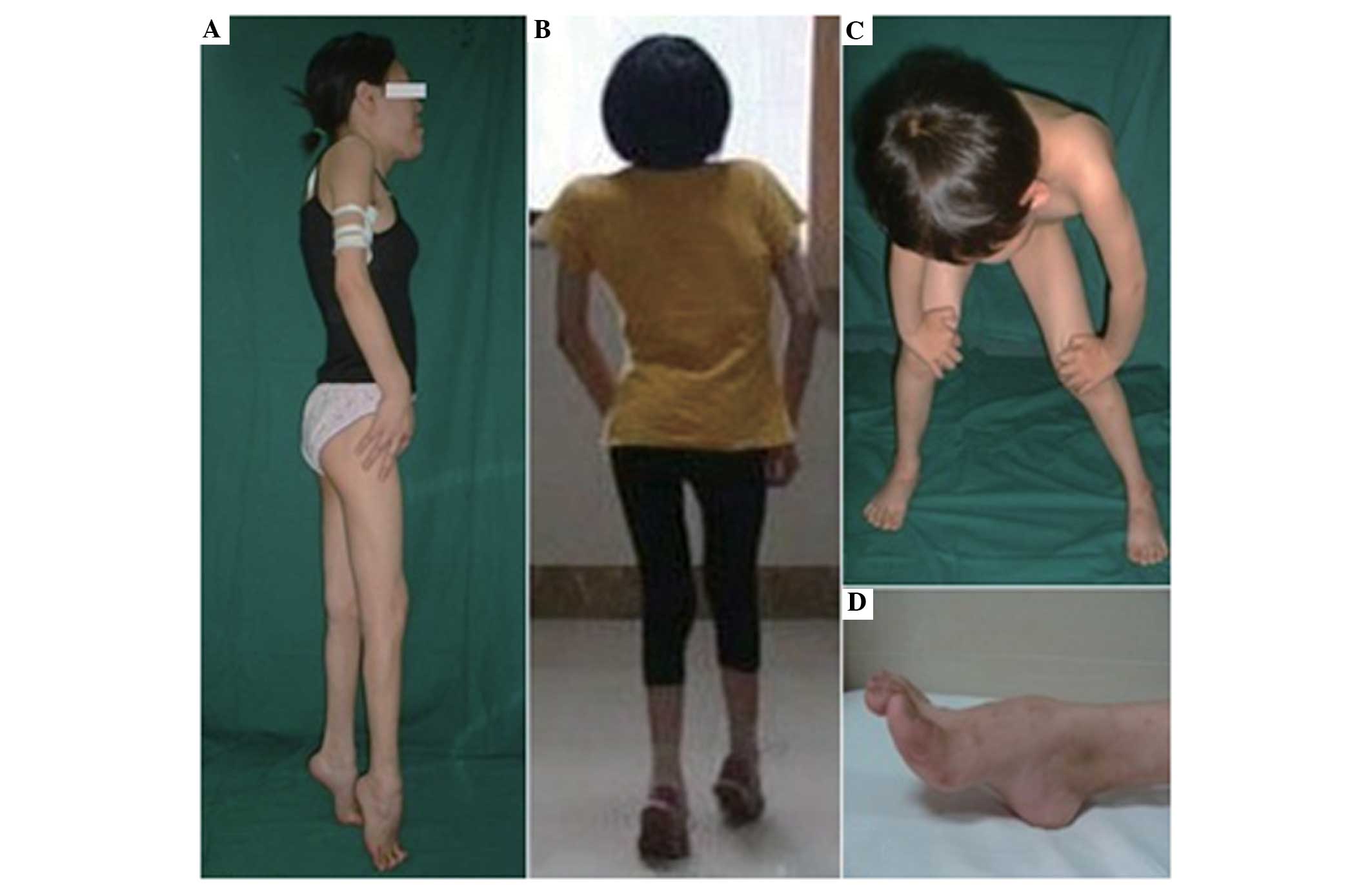
The clinical data of the three patients with EDMD
are presented and summarized in Fig.
1 and Table II. The patients
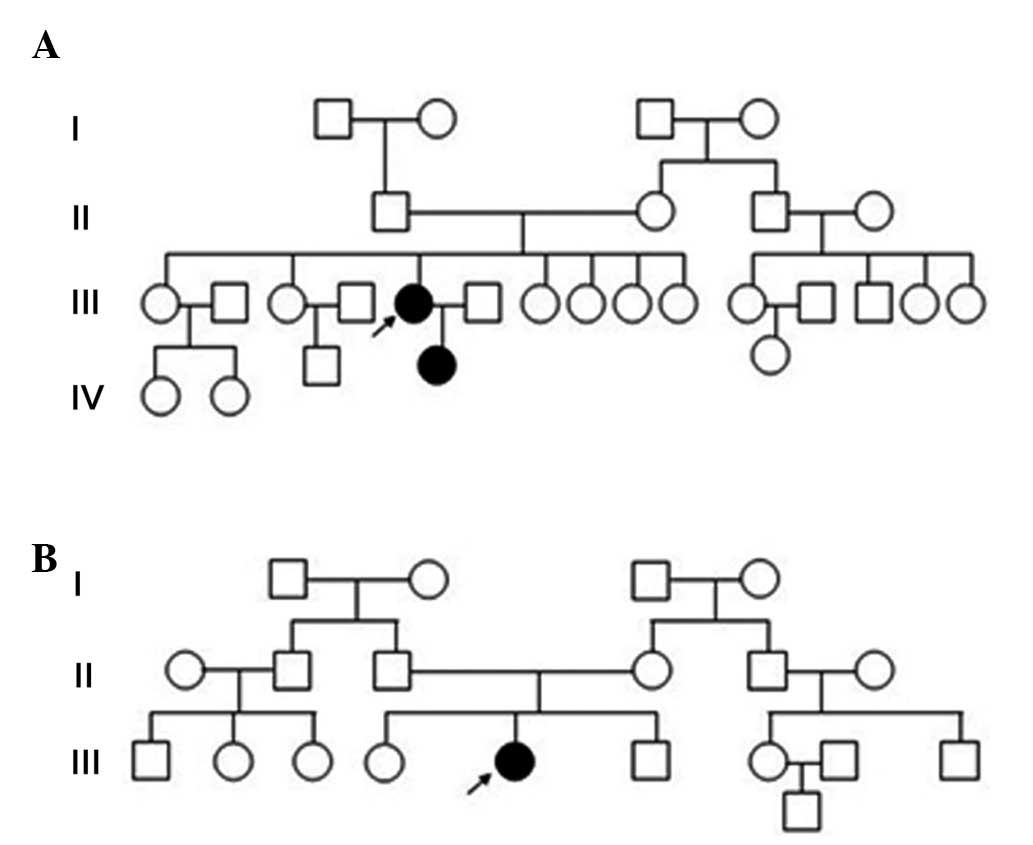
from Family 1 exhibited an AD mode of inheritance, whereas the
patient from Family 2 was a sporadic case (Fig. 2A and B). The levels of serum
creatine kinase were marginally increased in Patient 2, and were
normal in Patients 1 and 3. The electromyogram revealed myogenic
damage in each of the three patients.
| Table IIClinical data of patients 1, 2 and 3
in the two pedigrees. |
Table II
Clinical data of patients 1, 2 and 3
in the two pedigrees.
| Patient | Gender/age
(years) | Weakness
| Atrophy
| Contracture
| EMG myogenic | UCG
| CK (U/l) |
|---|
| Prox | Dist | Limbs | Body | Neck | Shoulder | Elbow | Ankle | Spine | EDV/ESV (ml) | LVEF (%) |
|---|
| 1 | F/33 | + | + | + | + | + | + | + | + | + | + | 163/70 | 40 | 140 |
| 2 | F/12 | + | + | + | − | − | − | − | + | − | + | 80/41 | 69 | 255 |
| 3 | F/24 | + | + | + | + | + | + | + | + | + | + | 135/66 | 55 | 127 |
Pathological analysis of muscle
biopsies
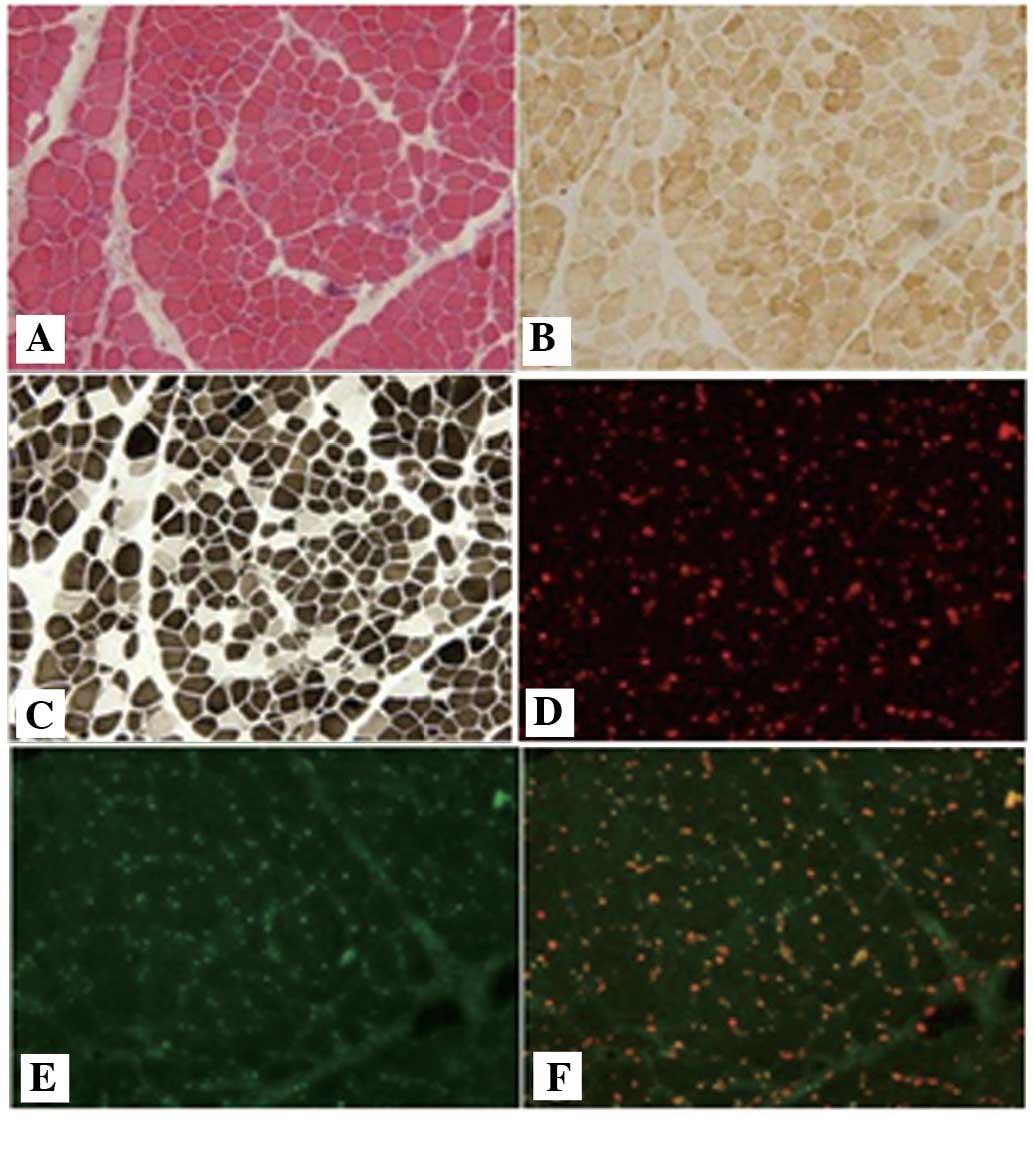
Routine histological analysis revealed dystrophic
features in the muscle tissues, with degeneration, polyfocal
necrosis, fiber splitting and moderate to marked fibrosis. Adipose
tissue infiltration was also observed. In addition, atrophic and
scattered regenerating fibers were noted. The ATP enzyme staining
revealed a predominance of type I fibers. Immunofluorescence
staining for emerin and lamin A/C revealed normal labeling along
the membrane in all three cases (Fig.
3).
Cardiac evaluation
Patient 1 (33 years old) presented with panicky,
shortened breathing and dyspnea. The heart border expended to the
left and a heart rate of 42 beats per min (bpm) was recorded.
Patient 2 (12 years old), the daughter of Patient 1, presented with
no clear cardiac abnormalities upon initial clinical examination.
Patient 3 (24 years old) presented with panicky and transient
amaurosis with a heart rate of 52 bpm.
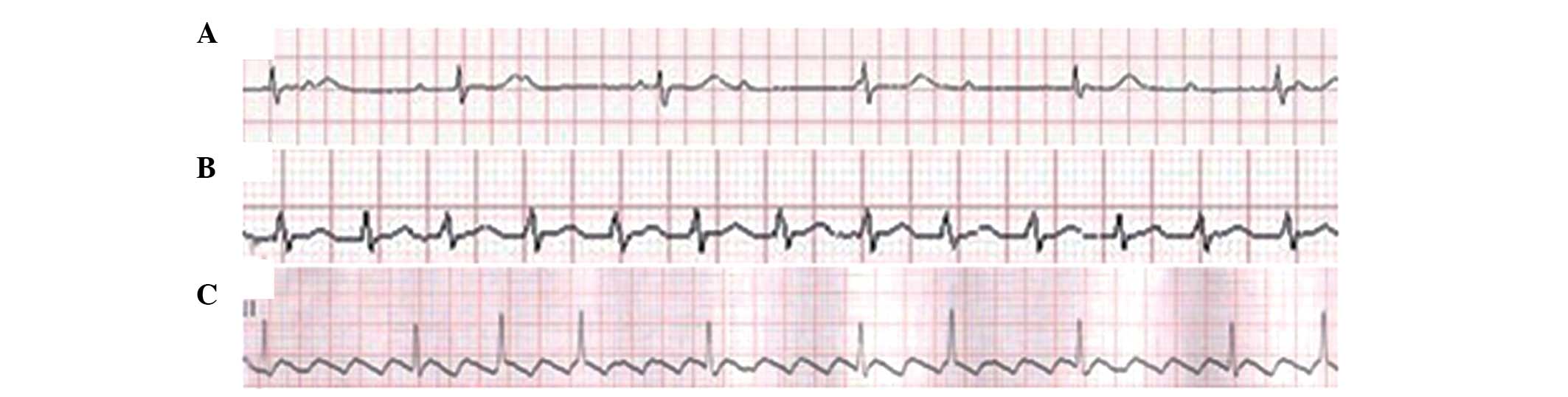
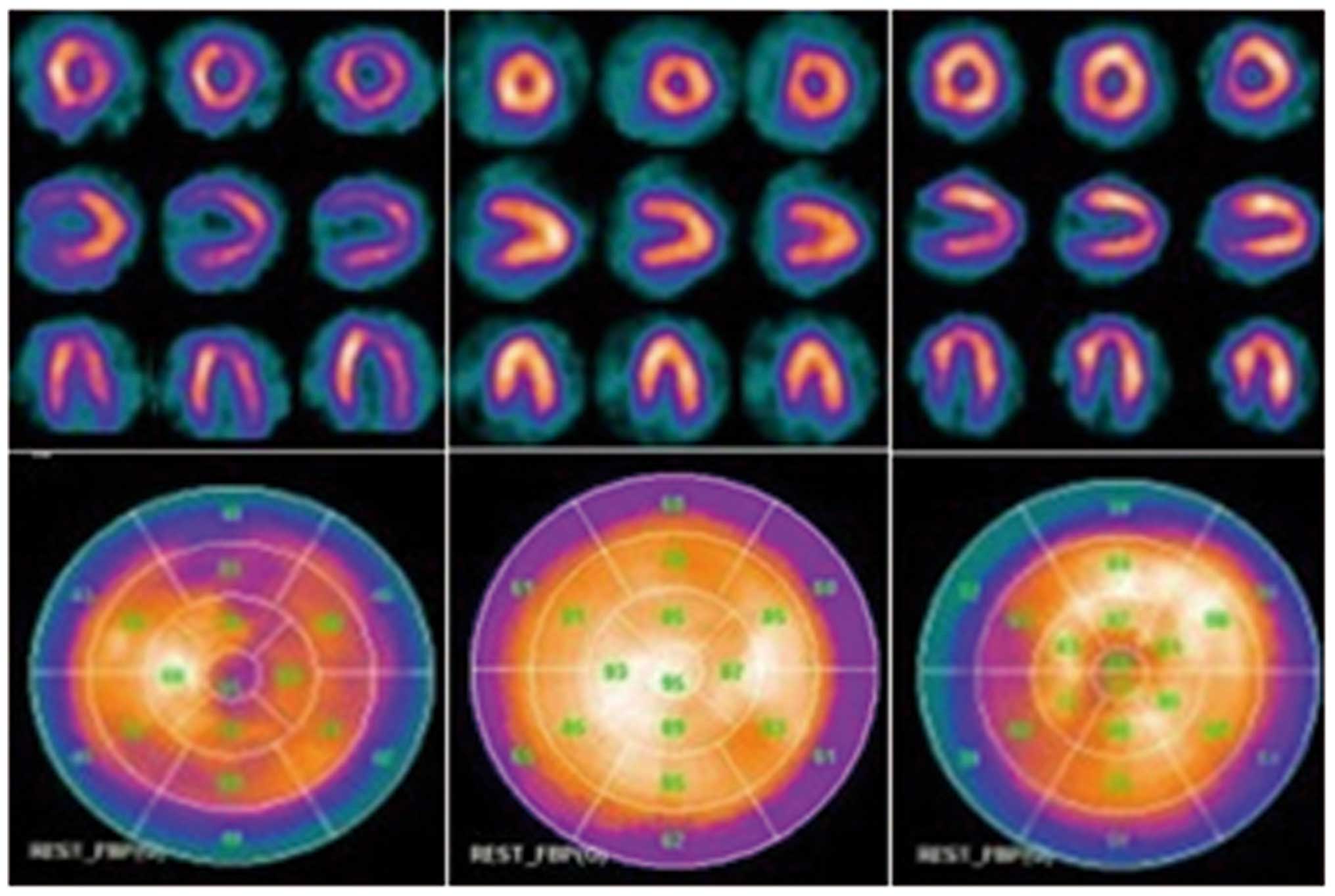
Further cardiac assessments revealed a variety of
abnormalities in each patient (Figs.
4 and 5). The ECG and Holter
of Patient 1 demonstrated complete atrioventricular inhibition and
premature ventricular contractions of 26 beats/24 h. The UCG
revealed that this individual had generalized cardiac enlargement,
mitral and tricuspid valve regurgitation and decreased function of
the left ventricle (Table II).
The 99TcM-MIBI GPI assessment indicated left
ventricular hypertrophy and variegated changes in the left
ventricular myocardium. Patient 2 suffered from sinus tachycardia,
as determined by ECG and Holter. The UCG and
99TcM-MIBI GPI scan was normal. The ECG and
Holter of Patient 3 revealed atrial flutter and the UCG
demonstrated mild left ventricular enlargement and decreased
diastolic function of the left ventricle (Table II). The
99TcM-MIBI GPI assessment revealed that
myocardial perfusion was moderately decreased in the apex of the
left ventricle.
Genetic analysis
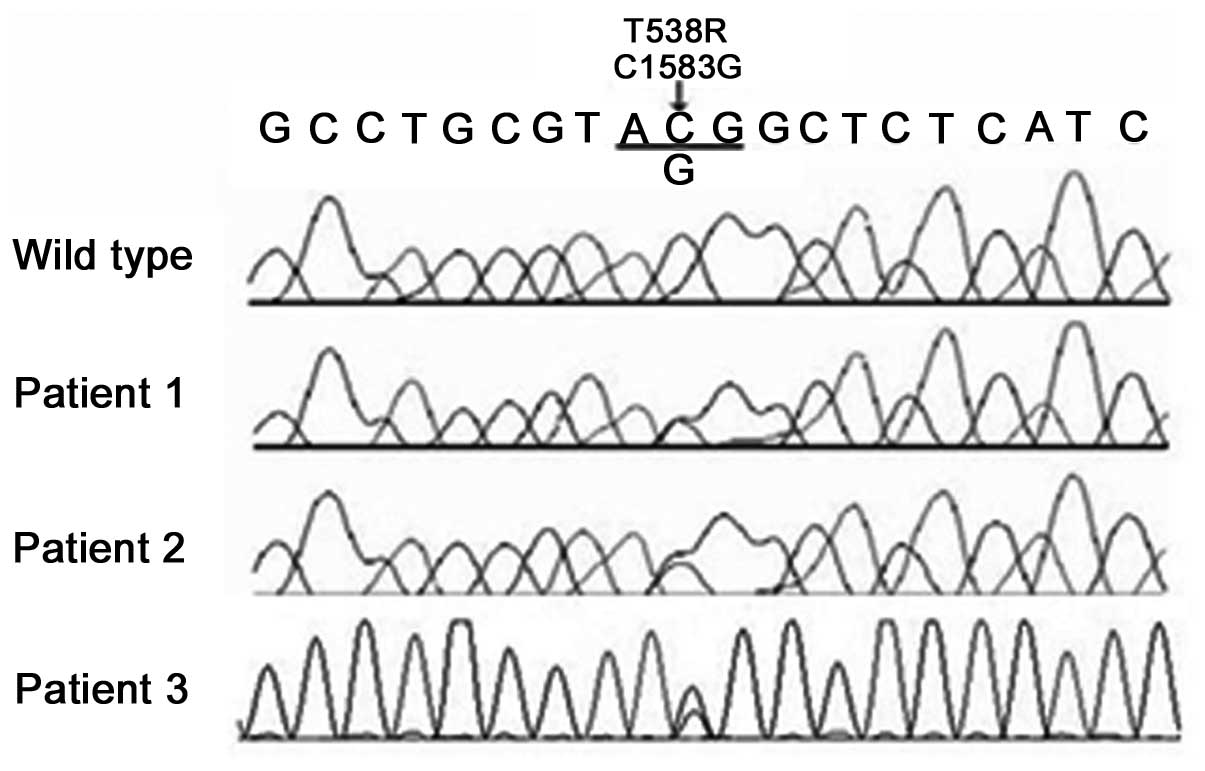
The genetic analysis in the present study identified
one missense mutation, c.1583C→G, in exon 9 of LMNA. This
change caused a T528R mutation in the LMNA protein. The mutation
was heterozygous in the three affected individuals examined in the
present study, and none of the 12 unaffected family members (seven
from Family 1 and five from Family 2) exhibited this base change on
either allele (Fig. 6). In
addition, the mutation was not identified in 50 Chinese control
chromosomes.
Discussion
The LMNA gene is located on chromosome 1q21
and encodes the A-type lamins, lamin A and C, and two minor
isoforms, C2 and AΔ10, which arise through alternative splicing.
The first 566 amino acids are identical in lamins A and C (10). Structurally, the protein is
composed of a central α-helical dimerization domain flanked by a
short amino-terminal head and a larger carboxy-terminal tail
(11). Lamins A and C are located
in the inner nuclear lamina and are widely expressed in skeletal
and cardiac muscle, fat, blood vessels, skin and nerve tissue
(12). Lamins A and C are
considered to be important in maintaining the size and shape of the
nucleus, and contribute to DNA replication, chromatin formation,
RNA splicing, cell differentiation and signal transduction
(10,11,13).
The three patients with EDMD identified in the
present study were members of two families. The first family
presented with an AD inheritance pattern, while the single patient
with EDMD in Family 2 was the only known affected member,
suggesting a sporadic mutation. Patients 1, 2 and 3 all presented
with the typical clinical features of EDMD, including joint
contractures, muscle weakness and cardiac involvement. The
contractures initially affected the shoulders, elbows or ankles and
impacted the posterior cervical muscles, thereby limiting neck
flexion and forward spine flexion. Muscle weakness was slowly
progressive and typically targeted to the peroneal and humeral
distribution. With time, a limb-girdle distribution of muscle
weakness developed. Immunostaining for emerin and lamin A/C
revealed normal labeling along the nuclear membrane in all three
cases. Although routine histological analyses revealed dystrophic
features, these phenotypes lacked specificity for EDMD and,
therefore, gene analysis was required.
It was revealed that each patient possessed the same
heterozygous missense mutation, c.1583C→G, in exon 9 of
LMNA, which caused a T528R amino acid change in the LMNA
protein. A previous study suggested that the majority (76%) of
AD-EDMD cases arise from sporadic mutations (14). The mutation identified in the
present study was previously described in reports of Italian and
German individuals, suggesting that this change exhibits an
increased incidence despite ethnic differences (15,16).
In the present study, Patient 3 was the only known individual from
Family 2 to exhibit the T528R LMNA mutation, which supported
the suggestion that it was a sporadic mutation.
Cardiac involvement is a frequent and serious
complication in EDMD, with a high risk of sudden mortality
(4). The degree of cardiac
involvement negatively correlates with the severity of skeletal
muscle weakness, however, it demonstrates a positive correlation
with age and disease course (17).
The predominant cardiac manifestations are progressive conduction
system defects and/or sinus node dysfunction. The incidence of
atrial fibrillation and atrial flutter is the highest, followed by
atrioventricular inhibition (18).
A number of patients present with ventricular tachycardia,
ventricular fibrillation and other malignant arrhythmias, which are
the leading causes of sudden mortality in patients with EDMD
(15,17). Consistent with previous reports,
the present study revealed various ECG findings, which were
positively correlated with age and disease course in all three
patients. The ECG demonstrated only sinus tachycardia in the
youngest patient (Patient 2; 12-years old). By contrast, atrial
flutter was observed in Patient 3 (24-years old), supporting sinus
node dysfunction and complete atrioventricular inhibition in the
oldest patient (Patient 1; 33-years old). This latter finding
suggested severe conduction system defects.
It has been previously reported that cardiac
involvement in AD-EDMD caused by LMNA mutations may be more
severe, compared with other forms of EDMD (17,19,20)
and are often accompanied by dilated cardiomyopathy (DCM), left
ventricular dysfunction and heart failure (15,20).
Pathologically, the normal myocardium is progressively replaced by
fibro-adipose tissue, resulting in atrial and ventricular
enlargement with progression towards clinical heart failure
(15,21). The cardiac imaging of the three
patients reported in the present study revealed that myocardial
damage was progressive with increasing age and course of disease.
The oldest patient (Patient 1) presented with left ventricular
dysfunction, enlarged heart chambers and severe myocardial
perfusion defects, which suggested dilated cardiomyopathy and heart
failure. Patient 3 also developed decreased myocardial perfusion in
the left ventricular apex. By contrast, the youngest patient
(Patient 2) revealed no abnormal findings on cardiac imaging
examination, therefore, follow-up observations are required as this
patient ages.
Amino acid 528 is located on the highly conserved
carboxy terminal tail of lamin A/C, suggesting that the T528R
mutation may negatively impact the structure of the carboxy
terminal tail and decrease protein stability (22,23).
It has been reported that patients with AD-EDMD caused by the R541H
LMNA mutation develop severe DCM, requiring heart transplant
during childhood (9,24). Analysis of protein function has
revealed that the lamin A/C p.R541H mutation causes the highly
conserved sequence of the carboxy terminal tail to mis-fold,
leading to severe cardiac damage, which is consistent with the
observations of the present study (25,26).
In conclusion, the present study identified three
patients with EDMD exhibiting the same dominant LMNA
mutation. These patients presented with a spectrum of severe
cardiac abnormalities, including cardiac conduction system defects,
cardiomyopathy and heart failure. Since LMNA mutations have
been associated with at least six clinical disorders, including
EDMD, the present case study provides additional mutational and
functional data, which may assist in further establishing LMNA
mutational variation and disease pathogenesis.
References
|
1
|
Dreifuss FE and Hogan GR: Survival in x
chromosomal muscular dystrophy. Neurology. 11:734–737. 1961.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Emery AE: Emery Dreifuss muscular
dystrophy-a 40 year retrospective. Neuromuscul Disord. 10:228–232.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Emery AE and Dreifuss FE: Unusual type of
benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry.
29:338–342. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Becane HM, Bonne G, Varnouss, Muchir A,
Ortega V, Hammouda EH, Urtizberea JA, Lavergne T, Fardeau M, Eymard
B, et al: High incidence of sudden death with conduction system and
myocardial disease due to lamins A and C gene mutation. Pacing Clin
Electrophysiol. 23:1661–1666. 2000. View Article : Google Scholar
|
|
5
|
Emery AE: The muscular dystrophies.
Lancet. 359:687–695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonne G, Di Barletta MR, Varnouss, Bécane
HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F,
Urtizberea JA, et al: Mutations in the gene encoding lamin A/C
cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat
Genet. 21:285–288. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rankin J and Ellard S: The laminopathies:
A clinical review. Clin Genet. 70:261–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Waters DD, Nutter DO, Hopkins LC and
Dorney ER: Cardiac features of an unusual X-linked humeroperoneal
neuromuscular disease. N Engl J Med. 293:1017–1022. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fatkin D, MacRae C, Sasaki T, Wolff MR,
Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De
Girolami U, et al: Missense mutations in the rod domain of the
lamin A/C gene as causes of dilated cardiomyopathy and conduction
system disease. N Engl J Med. 341:1715–1724. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fisher DZ, Chaudhary N and Blobel G: cDNA
sequencing of nuclear lamins A and C reveals primary and secondary
structural homology to intermediate filament proteins. Proc Natl
Acad Sci USA. 83:6450–6454. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goldman RD, Gruenbaum Y, Moir RD, Shumaker
DK and Spann TP: Nuclear lamins: Building blocks of nuclear
architecture. Genes Dev. 16:533–547. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moir RD, Spann TP and Goldman RD: The
dynamic properties and possible functions of nuclear lamins. Int
Rev Cytol. 162B:141–182. 1995.PubMed/NCBI
|
|
13
|
Lin F and Worman HJ: Structural
organization of the human gene encoding nuclear lamin A and nuclear
lamin C. J Biol Chem. 268:16321–16326. 1993.PubMed/NCBI
|
|
14
|
Bonne G, Mercuri E, Muchir A, Urtizberea
A, Bécane HM, Recan D, Merlini L, Wehnert M, Boor R, Reuner U, et
al: Clinical and molecular genetic spectrum of autosomal dominant
Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C
gene. Ann Neurol. 48:170–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sanna T, Dello Russo A, Toniolo D, Vytopil
M, Pelargonio G, De Martino G, Ricci E, Silvestri G, Giglio V,
Messano L, et al: Cardiac features of Emery-Dreifuss muscular
dystrophy caused by lamin A/C gene mutations. Eur Heart J.
24:2227–2236. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vytopil M, Benedettis, Ricci E, Galluzzi
G, Dello Russo A, Merlini L, Boriani G, Gallina M, Morandi L,
Politano L, et al: Mutation analysis of the lamin A/C gene (LMNA)
among patients with different cardiomuscular phenotypes. J Med
Genet. 40:e1322003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brodsky GL, Muntoni F, Miocic S, Sinagra
G, Sewry C and Mestroni L: Lamin A/C gene mutation associated with
dilated cardiomyopathy with variable skeletal muscle involvement.
Circulation. 101:473–476. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Voit T, Krogmann O, Lenard HG, Neuen-Jacob
E, Wechsler W, Goebel HH, Rahlf G, Lindinger A and Nienaber C:
Emery-Dreifuss muscular dystrophy: Disease spectrum and
differential diagnosis. Neuropediatrics. 19:62–71. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Funakoshi M, Tsuchiya Y and Arahata K:
Emerin and cardiomyopathy in Emery Dreifuss muscular dystrophy.
Neuromuscul Disord. 9:108–114. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Berlo JH, Duboc D and Pinto YM: Often
seen but rarely recognised: Cardiac complications of lamin A/C
mutations. Eur Heart J. 25:812–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wehnert MS and Bonne G: The nuclear
muscular dystrophies. Semin Pediatr Neurol. 9:100–107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Garg A, Cogulu O, Ozkinay F, Onay H and
Agarwal AK: A novel homozygous Ala529Val LMNA mutation in Turkish
patients with mandibuloacral dysplasia. J Clin Endocrinol Metab.
90:5259–5264. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen JJ, Brown CA, Lupski JR and Potocki
L: Mandibuloacral dysplasia caused by homozygosity for the R527H
mutation in lamin A/C. J Med Genet. 40:854–857. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sébillon P, Bouchier C, Bidot LD, Bonne G,
Ahamed K, Charron P, Drouin-Garraud V, Millaire A, Desrumeaux G,
Benaïche A, et al: Expanding the phenotype of LMNA mutations in
dilated cardiomyopathy and functional consequences of these
mutations. J Med Genet. 40:560–567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cenni V, Sabatelli P, Mattioli E,
Marmirolis, Capanni C, Ognibene A, Squarzoni S, Maraldi NM, Bonne G
and Columbaro M: et al Lamin A N-terminal phosphorylation is
associated with myoblast activation: Impairment in Emery-Dreifuss
muscular dystrophy. J Med Genet. 42:214–220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krimm I, Ostlund C, Gilquin B, Couprie J,
Hossenlopp P, Mornon JP, Bonne G, Courvalin JC, Worman HJ and
Zinn-Justins: The Ig-like structure of the C-terminal domain of
lamin A/C, mutated in muscular dystrophies, cardiomyopathy and
partial lipodystrophy. Structure. 10:811–823. 2002. View Article : Google Scholar : PubMed/NCBI
|