Introduction
Hypokalemic periodic paralysis (HPP) is
characterized by recurrent attacks of hypokalemia and muscle
weakness and is widely recognized as a typical skeletal muscle
channelopathy. Primary HPP is categorized into either familial HPP
(FHPP) or sporadic HPP (SHPP). Thyrotoxic HPP (THPP) is
representative of all secondary HPP conditions and although
familial cases have been reported, the majority cases of THPP are
sporadic.
According to pedigree analysis, FHPP is an autosomal
dominant disease with lower penetrance in females (1). The pathogenesis of FHPP is attributed
to mutations in genes encoding ion channels in skeletal muscle,
although the underlying mechanism remains unclear. Associated
mutations have been most commonly reported in the genes encoding
the voltage-dependent L type calcium channel subunit alpha 1S
(CACNA1S) and the voltage-gated sodium channel type IV alpha
subunit (SCN4A) (2,3). Several point mutations (4,5) in
CACNA1S and SCN4A have been identified to have associations with
FHPP, including Arg1239His/Gly and Arg528His in CACNA1S, and
Arg669His, Arg672His/Gly and Arg672Ser in SCN4A. Additional rare
substitutions have also been reported (6). Some overlap in the mutations in FHPP,
SHPP (7), and THPP (8) has been reported.
To date, the majority of genetic studies of HPP have
been conducted in Western populations (2,6).
Fewer HPP cases have been reported in China compared with Western
countries, and information on HPP in Chinese populations remains
limited (2,6). The aim of the current study was to
screen for mutations in CACNA1S and SCN4A in Chinese patients with
HPP. The present study aimed to determine whether mutations
associated with HPP in Chinese cases are the same as those in
Western populations, and to provide further epidemiological
evidence for HPP.
Materials and methods
Study subjects
Informed consent was obtained from all participating
patients and the study was approved by the medical ethics committee
of the Chinese People's Liberation Army Navy General Hospital
(Beijing, China). In total, three FHPP and two THPP pedigrees were
investigated and all patients involved were free from any other
inherited systemic diseases or congenital disorders and presented
with typical periodic attacks of limb paralysis with severe
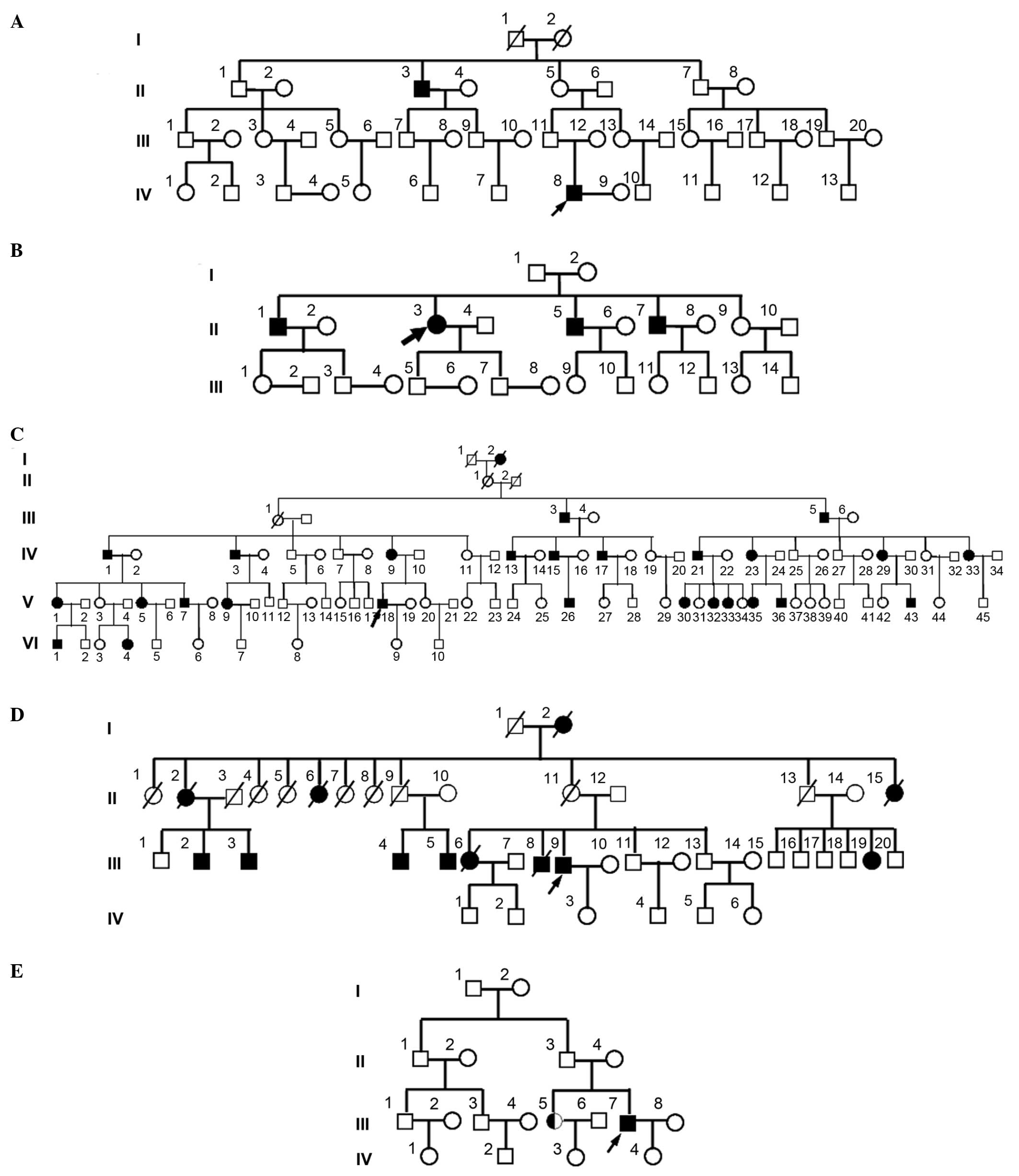
hypokalemia. The pedigree analysis is presented in Fig. 1.
Reagents and materials
High fidelity Pyrobest DNA polymerase and dNTPs were
purchased from Takara Bio, Inc. (Otsu, Japan), and Wizard™ Genomic
DNA Purification kits, T4 DNA ligase, plasmid pGEM-T and bacteria
JM109 were all purchased from Promega Corporation (Madison, WI,
USA).
Mutation analysis
Blood samples from confirmed HPP patients were
obtained, and genomic DNA was isolated from white blood cells using
a Genomic DNA Purification kit according to the manufacturers
instructions. Sequence analysis was conducted to identify
previously reported common and infrequent mutation sites in CACNA1S
and SCN4A. CACNA1S fragments containing the sequences encoding
Arg528 and Arg1239 and a SCN4A fragment containing the sequence
encoding Arg669 and Arg672 were amplified by polymerase chain
reaction (PCR) with the primers presented in Table I. For the pedigrees free from
mutations in the above common sites, alternative PCR primers,
presented in Table II, were
designed to specifically amplify other fragments in CACNA1S and
SCN4A. All primers were synthesized by AuGCT Biotechnology Co.,
Ltd. (Beijing, China). The PCR products were separated by 2%
agarose gel electrophoresis (Beijing Huahai Henghui Technology Co.,
Ltd.) and collected using a gel extraction kit (Promega
Corporation). The PCR products were used as templates for
subsequent PCR expansion, following which the products were
collected again. PCR was performed using a reaction solution
containing 50 µl PCR buffer containing 100 µmol/l
dNTPs, 0.2 µmol/l primers, 2 units Taq DNA polymerases and
50 ng DNA template. The cycling conditions consisted of one cycle
of denaturation at a temperature of 94°C for 5 min, followed by
denaturation at a temperature of 94°C for 30 sec, and annealing at
a temperature of 55°C for 30 sec and extension at a temperature of
72°C for 30 sec, for a total of 30 cycles. A final extension step
was performed at a temperature of 72°C for 5 min. All amplified
products were sequenced using an ABI PRISM® 377 DNA
Sequencer (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Sequencing results were BLAST searched (blast.ncbi.nlm.nih.gov/)
against sequences in GenBank.
 | Table IPolymerase chain reaction primers
designed for analysis of common mutation sites. |
Table I
Polymerase chain reaction primers
designed for analysis of common mutation sites.
| Gene | Primers (5′-3′) | Amplified sites | Fragment length |
|---|
| CACNA1S |
L:CACTGAGATGCTGATGAAGATGTA | 528 | 192 bp |
|
R:GCACTCACTTGGTGATCTTGAA | | |
|
L:ATGAGAGTGCCCGCATCTCC | 1239 | 181 bp |
|
R:CTGTTGCACCTGGAAGGACTTG | | |
| SCN4A |
L:CTCTGTGACAGGGCCTCATG | 669, 672 | 250 bp |
|
R:TCCTCACCCCACCCCCATCC | | |
| Table IIPolymerase chain reaction primers for
amplification of CACNA1S and SCN4A exons. |
Table II
Polymerase chain reaction primers for
amplification of CACNA1S and SCN4A exons.
| Exon number | Left primer | Right primer |
|---|
| CACNA1S | | |
| 11 |
GGGAGTCAGGAGAATGG |
GGGAAGTCTGGGCAAGG |
| 20 |
CCAGGCTGCTGCCTCTT |
CCTTGCCGCTGCTCACT |
| 21 |
ACAGGCCTGTTCTCCAC |
TCCTTGTGCTTGAGAGT |
| 22 |
ACAGGCCTGTTCTCCAG |
TCCTTGTGCTTGAGAGT |
| 26 |
CTGTGATAACTCAATGG |
AATAAACCATAAGTGCC |
| 30 |
GCCCCTTACCCCTCTGT |
CCAGGTACGTGCAGTTT |
| SCN4A | | |
| 5 |
GACCCTGTGGTACCCCT |
GCTGCCTCTCAAACGCC |
| 12 |
CTACGCTCCTTCAGTCT |
GAAGACCCGCAGCAGAC |
| 18 |
ACGCACTGATCCCCTCG |
CCAGAGGCCCCTTCAGC |
| 19 |
GCCCTCCTAGGCGCCAT |
GTAGAGGTTCACCTCGT |
Results
Clinical features
The patients selected for the current study all
displayed clinical symptoms in accordance with the diagnostic
criteria of HPP. All patients were suffering from irregular muscle
weakness with hypokalemia and muscle weakness predominantly in the
limbs. Potassium supplements had been effective for all the
probands of these pedigrees. Patients in pedigree D and pedigree E
also exhibited symptoms of hyperthyroidism, with the
hyperthyroidism confirmed by thyroid function analysis. The
probands were IV8 of pedigree A, II3 of pedigree B, V18 of pedigree
C, III9 of pedigree D and III7 of pedigree E, and their ages of
onset were 22, 40, 15, 45 and 43 years old, respectively. As
presented in Fig. 1, the ratio of
male to female patients in these pedigrees was 2:0, 3:1, 14:13, 6:6
and 1:1, respectively, and atavism was observed in all five
pedigrees.
Results of DNA sequencing
Mutation analysis was first conducted in probands
and healthy controls from each pedigree. Gene fragments of CACNA1S,
containing the 528 and 1239 mutation sites, and of SCN4A,
containing the 669 and 672 mutation sites were sequenced. In
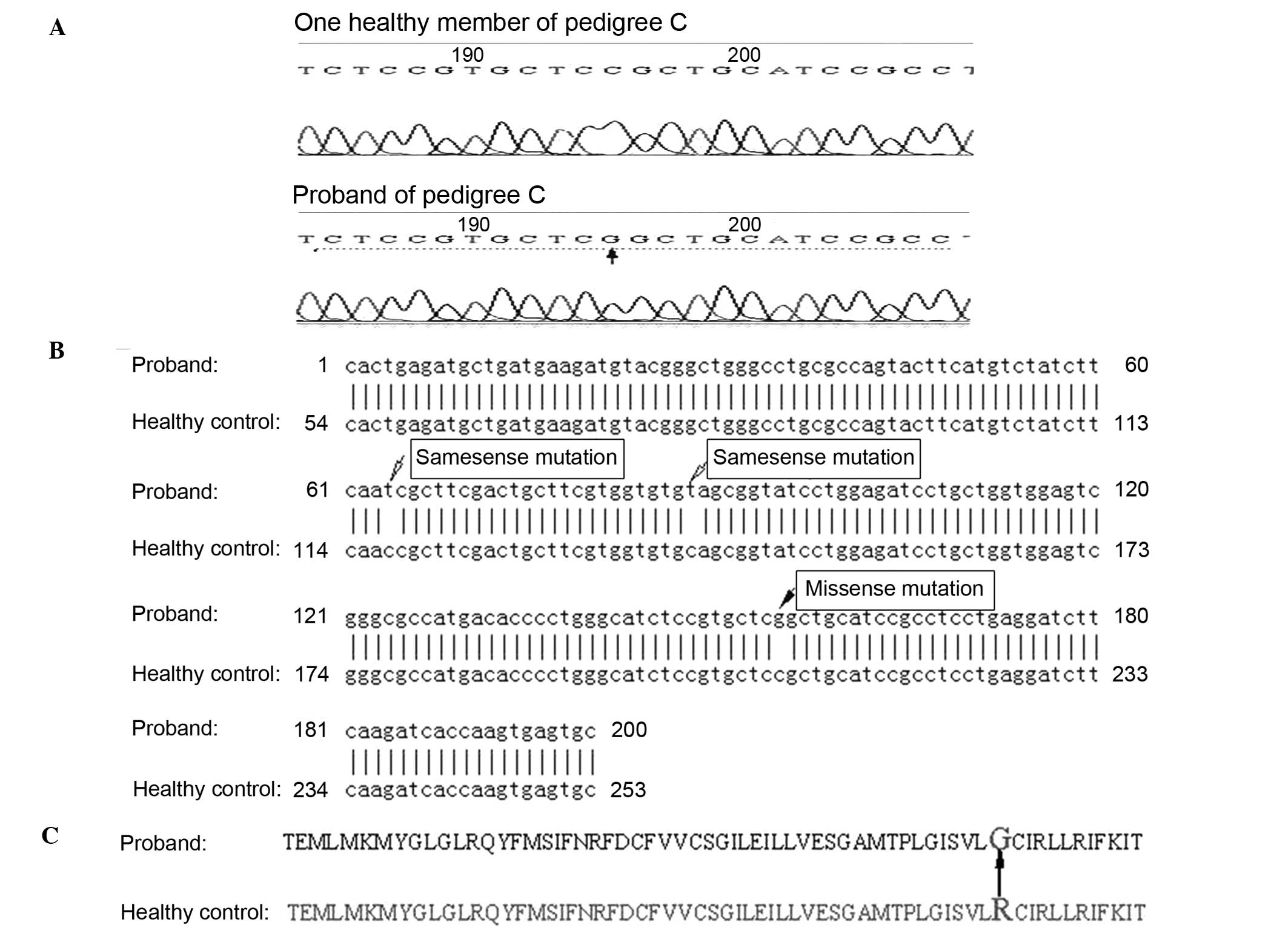
pedigree C (Fig. 2A), a cytosine
to guanine mutation was identified at site 195 in the 192 base pair
amplified fragment containing amino acid site 528 of CACNA1S.
Following sequence alignment, the mutation was identified as a
missense mutation at nucleotide 1582 in exon 11 of CACNA1S,
resulting in a substitution of glycine for arginine at site 528 in
CACNA1S (Fig. 2B and C). Two
additional mutations observed in the same fragment were confirmed
as same-sense mutations (Fig. 2B).
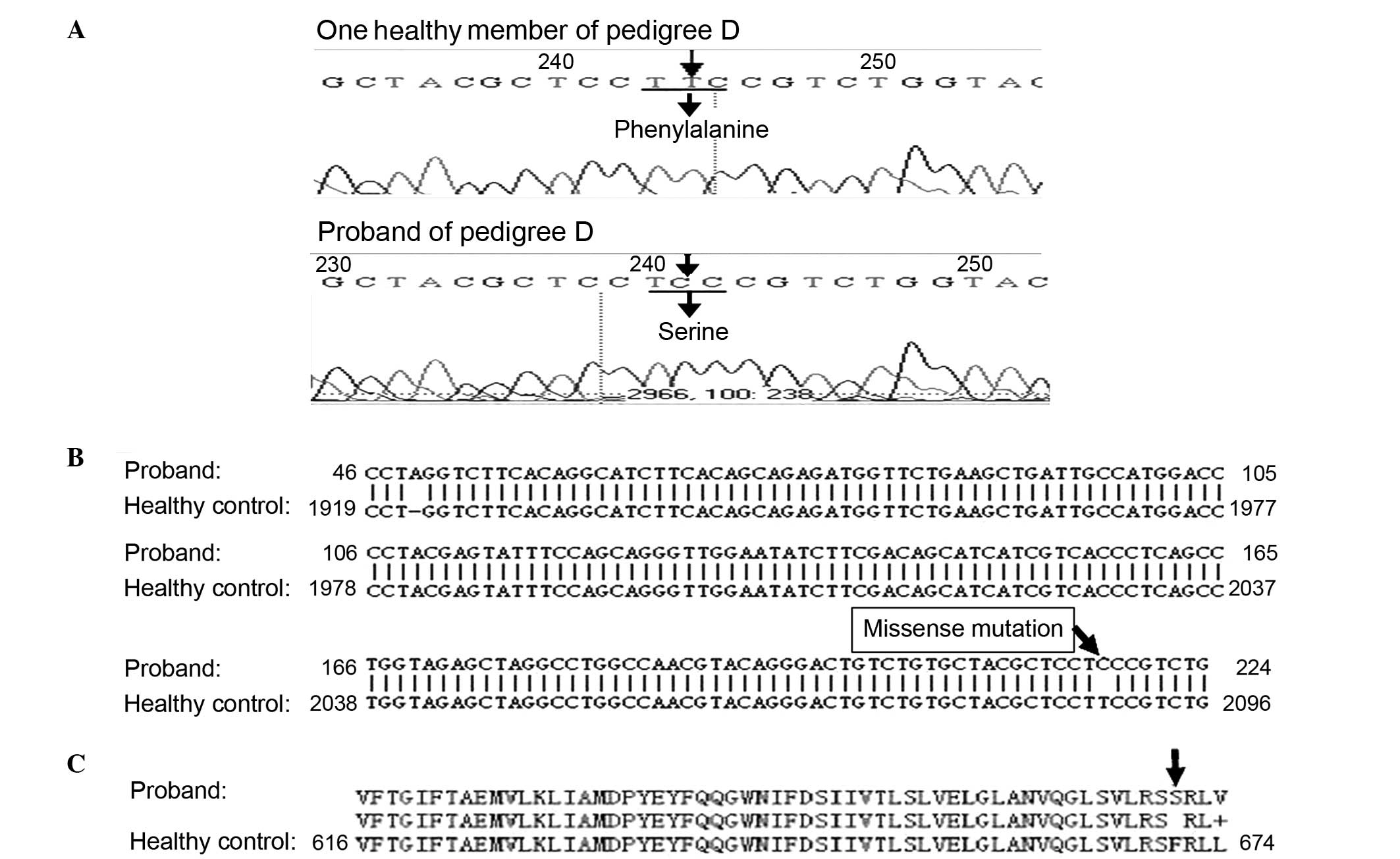
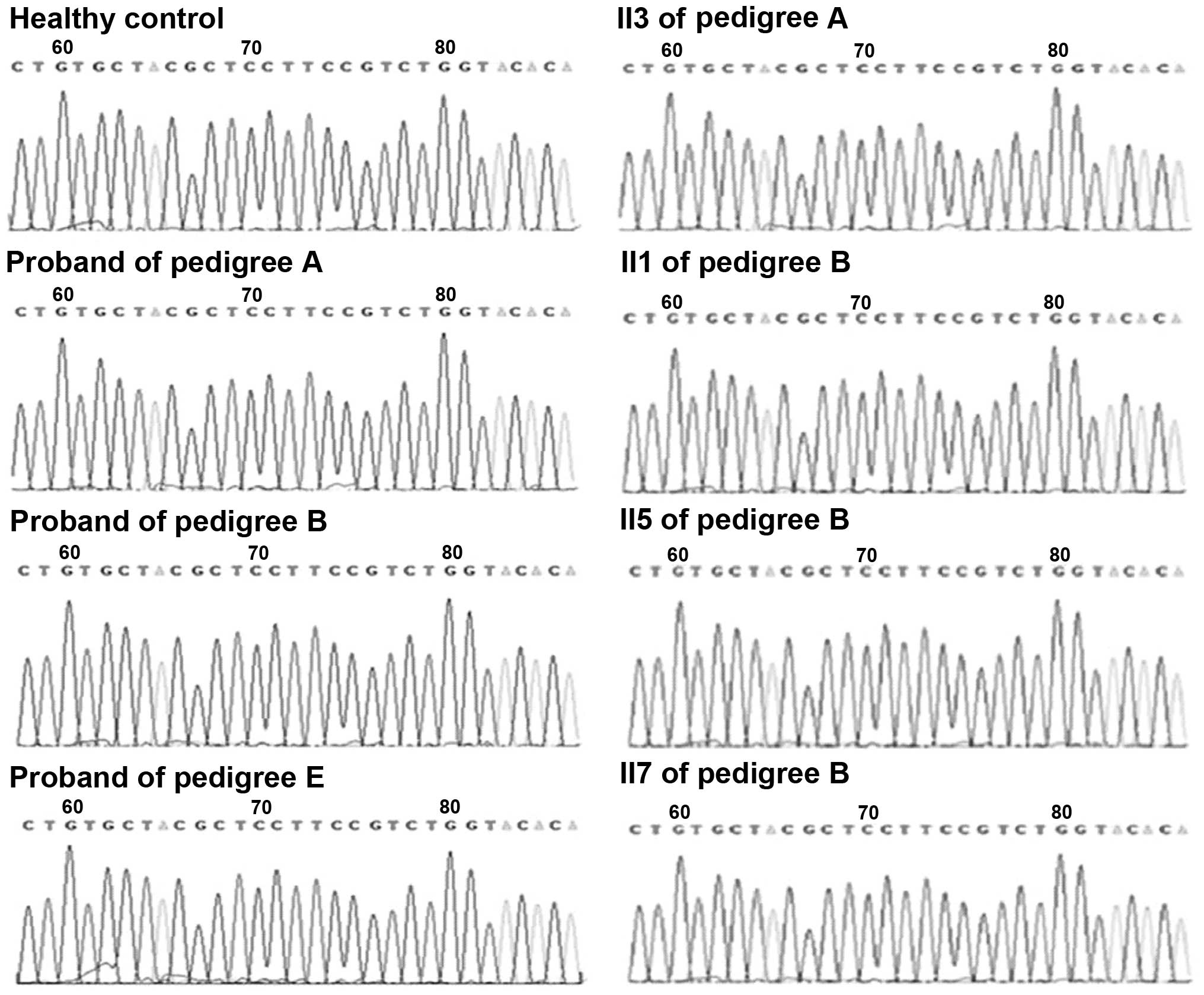
In pedigree D (Fig. 3A), a
substitution mutation of thymine to cytosine was identified in the
250 base pair SCN4A fragment containing amino acid sites 669 and
672. Following sequence alignment, the mutation was confirmed as a
missense mutation at nucleotide 2012 in exon 12 of SCN4A, resulting
in a substitution of serine for phenylalanine at site 671 (Fig. 3B and C). Two additional mutations
observed in the CACNA1S fragment containing nucleotide 1239 were
confirmed as same-sense mutations. No mutations in these fragments
containing the common mutation sites were identified in the
probands of the other three pedigrees.
Gene fragments encompassing the sites of alternative
reported mutations in CACNA1S and SCN4A were then sequenced in the
remaining three pedigrees. In pedigree A, among 43 family members
in four generations, mutations in 33 subjects, including two
patients (IV8 and II3), were detected. In pedigree B, among 26
family members in three generations, mutations in 20 subjects,
including four patients (II1, II3, II5 and II7), were detected. In
pedigree E, among 18 family members in four generations, mutations
in 12 subjects, including one confirmed patient (III7) and one
suspected patient (III5), were detected. No mutations previously
identified in exons 11, 20, 21, 22, 26 and 30 of CACNA1S, and in
exons 5, 12, 18 and 19 of SCN4A were identified in these three
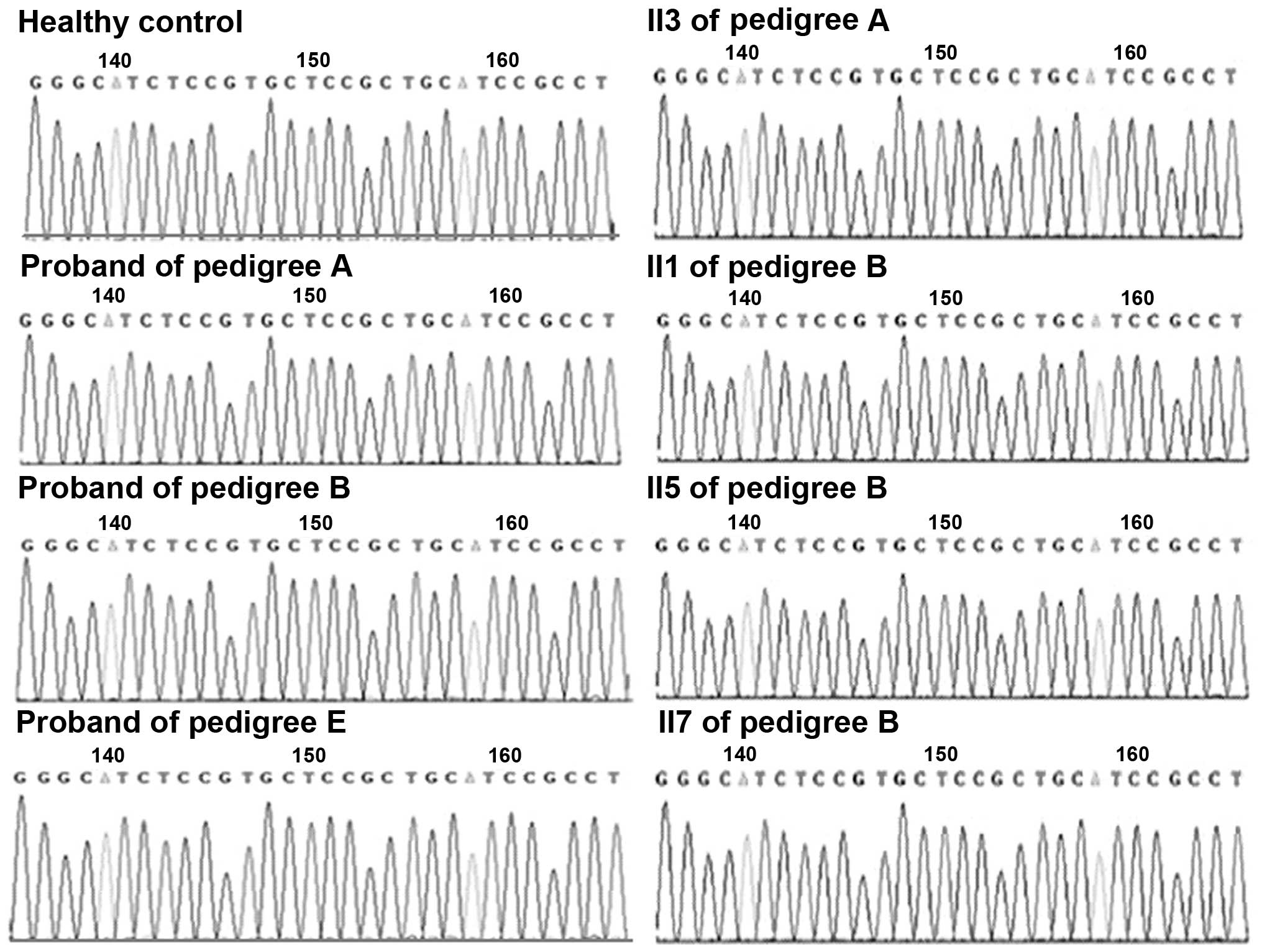
pedigrees. Sequencing maps, ranging from nucleotide 1567–1596 in
exon 11 of CACNA1S, and from nucleotide 1999–2028 in exon 12 of
SCN4A, are presented in Figs. 4
and 5, respectively.
Discussion
HPP is a disease detrimental to the quality of life
of the patients and can lead to death resulting from unexpected
respiratory paralysis or arrhythmia (9,10).
Due to variation in the age of onset, duration, frequency and
severity of attacks, the diagnosis of HPP is predominantly made
according to clinical history, attack characteristics and serum
potassium levels during attacks (16). Although the specific mechanism of
HPP remains unclear, mutations in genes encoding ion channels in
skeletal muscle have been suggested to be involved (17), and genetic analysis may aid in
elucidating the pathogenesis of the disease. The inheritance
pattern of HPP is known to be autosomal dominant, however it has
incomplete penetrance, particularly in females (16). With several family members in the
same pedigree suffering HPP, the incomplete penetrance of
pathogenic genes is considered to be involved in the atavism of the
pedigrees in the current study.
Previous studies have indicated the familial
transmission of HPP-associated mutations (2,18).
Although SCN4A mutations are also involved (19,20),
mutations in CACNA1S occur primarily in patients with HPP. In a
study involving 58 HPP pedigrees (2), the rate of mutation detection was
77.6%, including 40 pedigrees with CACNA1S mutations (69.0%) and
five pedigrees with SCN4A mutations (8.6%) (no mutations were
observed in the remaining 13 pedigrees). The two most common
mutations in CACNA1S were R528H and R1239H, which account for 45.0
and 24.0% of mutations detected, respectively. In an additional
study involving 83 cases of HPP (6), the rate of mutation was 87.9%,
including 65 cases with CACNA1S mutations, accounting for 78.3%,
and eight cases with SCN4A mutations, accounting for 9.6%.
Compared with Western populations, mutations
observed in Eastern populations are very similar, however the rate
of mutation detection is markedly lower. In a study of Chinese
patients (21), one case with the
R1239H mutation and two cases with the R672H mutation were
identified in the probands of 14 pedigrees, the mutation rate of
which was 21.4%, and only one case with the R672C mutation was
observed in 71 sporadic patients. Mutation screening in Taiwanese
patients with HPP detected one case with R528H in 12 sporadic
patients, accounting for 8.3%, with no cases detected in 36
patients with THPP (7). An
additional study in Taiwanese patients reported four cases with
mutations among 60 sporadic patients with HPP, including one case
with R1239H, two cases with R669H and one case with R1135H, with
the mutation rate in the current study at 6.6% (22). Mutations have been rarely reported
in populations from China (23,24).
Accordingly, the present study additionally demonstrated a lower
rate of HPP-associated gene mutations.
The known mutations in CACNA1S and SCN4A are
presented in Table III. The
majority of mutation sites in the table have been identified in
Western populations, excluding R528G, R900G, R916Q and R1239G. In
studies of Asian populations, mutations in CACNA1S were
predominantly reported in case studies (9,13,25–31)
and pedigree studies (7,14,32–34).
In the current study, a rare R528G mutation in CACNA1S in a Chinese
HPP pedigree was observed, and this mutation has been previously
reported in another Chinese pedigree with HPP (27). THPP shares certain common
characteristics with HPP, however, the identification of pathogenic
genes between them are not similar (35–37).
In contrast with HPP, mutations in CACNA1S are not associated with
THPP (35). To date, gene
mutations have been confirmed in three THPP pedigrees, including a
pedigree with an R83H mutation in KCNE3 (38), a pedigree with an M58V mutation in
KCNE4 (36), and a Chinese
pedigree with an F671S mutation in SCN4A (the current study). R83H
and M58V have not been identified in Chinese patients with THPP,
however it remains controversial whether R83H is a causative
mutation in THPP, due to the fact that it is additionally present
in ~2% of healthy individuals (39).
| Table IIIVerified mutation sites in CACNA1S
and SCN4A in patients with hypokalemic periodic paralysis. |
Table III
Verified mutation sites in CACNA1S
and SCN4A in patients with hypokalemic periodic paralysis.
| Gene | Exon number | Mutation sites |
|---|
| CACNA1S | 11 | R528H (6), R528G (6) |
| 20 | V876E (11), R897S (12) |
| 21 | R900G (13), R900S (6) |
| 22 | H916Q (14) |
| 26 | R1086C (6) |
| 30 | R1239H (6), R1239G (6) |
| SCN4A | 5 | R222W (6), R669H (6) |
| 12 | F671S (6), R672H (6), R672G (6), |
| | R672C (6), R672S (6) |
| 18 | R1132Q (6), R1135H (6) |
| 19 | P1158S (15) |
In conclusion, mutations in CACNA1S and SCN4A are
relatively rare in Chinese HPP cases compared with cases in Western
individuals. This indicates a racial difference and the possibility
of other pathogenic genes or factors in Chinese patients. Due to
the fact that the sample sizes in Asian pedigree studies have been
relatively small, and the newly identified mutations in Chinese
cases are often identified in case studies (13,14),
further research is required to investigate whether R528G in HPP
and F671S in THPP are common in Chinese populations. Future studies
should aim to identify novel mutations in Chinese patients in
CACNA1S and SCN4A in addition in other genes, and to establish
animal models with known mutations to further clarify the
pathophysiological mechanisms of HPP.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant nos. 30671006 and
81170800). The authors would like to thank Edanz China for the
English Language Service.
References
|
1
|
Kawamura S, Ikeda Y, Tomita K, Watanabe N
and Seki K: A family of hypokalemic periodic paralysis with CACNA1S
gene mutation showing incomplete penetrance in women. Intern Med.
43:218–222. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sternberg D, Maisonobe T, Jurkat-Rott K,
Nicole S, Launay E, Chauveau D, Tabti N, Lehmann-Horn F, Hainque B
and Fontaine B: Hypokalaemic periodic paralysis type 2 caused by
mutations at codon 672 in the muscle sodium channel gene SCN4A.
Brain. 124:1091–1099. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Links TP, Ginjaar HB and van der Hoeven
JH: From gene to diseases; hypokalemic periodic paralysis. Ned
Tijdschr Geneeskd. 148:1035–1038. 2004.In Dutch. PubMed/NCBI
|
|
4
|
Kuzmenkin A, Muncan V, Jurkat-Rott K, Hang
C, Lerche H, Lehmann-Horn F and Mitrovic N: Enhanced inactivation
and pH sensitivity of Na(+) channel mutations causing hypokalaemic
periodic paralysis type II. Brain. 125:835–843. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng WY, Lui KF, Thai AC and Cheah JS:
Absence of ion channels CACN1AS and SCN4A mutations in thyrotoxic
hypokalemic periodic paralysis. Thyroid. 14:187–190. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matthews E, Labrum R, Sweeney MG, Sud R,
Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB,
et al: Voltage sensor charge loss accounts for most cases of
hypokalemic periodic paralysis. Neurology. 72:1544–1547. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin SH, Hsu YD, Cheng NL and Kao MC:
Skeletal muscle dihydropyridine-sensitive calcium channel (CACNA1S)
gene mutations in chinese patients with hypokalemic periodic
paralysis. Am J Med Sci. 329:66–70. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lane AH, Markarian K and Braziunene I:
Thyrotoxic periodic paralysis associated with a mutation in the
sodium channel gene SCN4A. J Pediatr Endocrinol Metab.
17:1679–1682. 2004. View Article : Google Scholar
|
|
9
|
Kil TH and Kim JB: Severe respiratory
phenotype caused by a de novo Arg528Gly mutation in the CACNA1S
gene in a patient with hypokalemic periodic paralysis. Eur J
Paediatr Neurol. 14:278–281. 2010. View Article : Google Scholar
|
|
10
|
Stunnenberg BC, Deinum J, Links TP, Wilde
AA and Franssen H: Hypokalemia as only cause? Muscle Nerve.
50:327–332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ke T, Gomez CR, Mateus HE, et al: Novel
CACNA1S mutation causes autosomal dominant hypokalemic periodic
paralysis in a South American family. J Hum Genet. 54:660–664.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chabrier S, Monnier N and Lunardi J: Early
onset of hypokalaemic periodic paralysis caused by a novel mutation
of the CACNA1S gene. J Med Genet. 45:686–688. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hirano M, Kokunai Y, Nagai A, Nakamura Y,
Saigoh K, Kusunoki S and Takahashi MP: A novel mutation in the
calcium channel gene in a family with hypokalemic periodic
paralysis. J Neurol Sci. 309:9–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li FF, Li QQ, Tan ZX, Zhang SY, Liu J,
Zhao EY, Yu GC, Zhou J, Zhang LM and Liu SL: A novel mutation in
CACNA1S gene associated with hypokalemic periodic paralysis which
has a gender difference in the penetrance. J Mol Neurosci.
46:378–383. 2012. View Article : Google Scholar
|
|
15
|
Webb J and Cannon SC: Cold-induced defects
of sodium channel gating in atypical periodic paralysis plus
myotonia. Neurology. 70:755–761. 2008. View Article : Google Scholar
|
|
16
|
Vicart S, Sternberg D, Arzel-Hézode M,
Franques J, Bendahhou S, Lory P, Hainque B, Fournier E, Nicole S
and Fontaine B: Hypokalemic Periodic Paralysis. GeneReviews®
[Internet]. Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A,
Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH and Stephens K:
University of Washington; Seattle, WI: 2002, updated 2014.
|
|
17
|
Matthews E and Hanna MG: Muscle
channelopathies: Does the predicted channel gating pore offer new
treatment insights for hypokalaemic periodic paralysis? J Physiol.
588:1879–1886. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Surtees R: Inherited ion channel
disorders. Eur J Pediatr. 159:S199–S203. 2000. View Article : Google Scholar
|
|
19
|
Incecik F, Hergüner MO, Altunbaşak S and
Lehman-Horn F: Hypokalemic periodic paralysis due to the SCN4A
R672H mutation in a Turkish family. Turk J Pediatr. 52:409–410.
2010.PubMed/NCBI
|
|
20
|
Park YH and Kim JB: An atypical phenotype
of hypokalemic periodic paralysis caused by a mutation in the
sodium channel gene SCN4A. Korean J Pediatr. 53:909–912. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ke QWW, Xu QG, Huang DH, Yu SY and Huang
XS: Correlating phenotype and genotype in the familial hypokalemic
periodic paralysis. Chin J Neurol. 39:323–327. 2006.
|
|
22
|
Sung CC, Cheng CJ, Lo YF, Lin MS, Yang SS,
Hsu YC and Lin SH: Genotype and phenotype analysis of patients with
sporadic periodic paralysis. Am J Med Sci. 343:281–285. 2012.
View Article : Google Scholar
|
|
23
|
Kung AW, Lau KS, Fong GC and Chan V:
Association of novel single nucleotide polymorphisms in the calcium
channel alpha 1 subunit gene (Ca(v)1.1) and thyrotoxic periodic
paralysis. J Clin Endocrinol Metab. 89:1340–1345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kung AW, Lau KS, Cheung WM and Chan V:
Thyrotoxic periodic paralysis and polymorphisms of sodium-potassium
ATPase genes. Clin Endocrinol (Oxf). 64:158–161. 2006. View Article : Google Scholar
|
|
25
|
Kusumi M, Kumada H, Adachi Y and Nakashima
K: Muscle weakness in a Japanese family of Arg1239His mutation
hypokalemic periodic paralysis. Psychiatry Clin Neurosci.
55:539–541. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim JB, Lee KY and Hur JK: A Korean family
of hypokalemic periodic paralysis with mutation in a voltage-gated
calcium channel (R1239G). J Korean Med Sci. 20:162–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Q, Liu M, Xu C, Tang Z, Liao Y, Du R,
Li W, Wu X, Wang X, Liu P, et al: Novel CACNA1S mutation causes
autosomal dominant hypokalemic periodic paralysis in a Chinese
family. J Mol Med Berl. 83:203–208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ke Q, Wu WP, Guo XH, Xu QG, Huang DH, Mao
YL and Huo CN: R1239H mutation of CACNA1S gene in a Chinese family
with hypokalaemic periodic paralysis. Zhonghua Yi Xue Yi Chuan Xue
Za Zhi. 23:272–274. 2006.In Chinese. PubMed/NCBI
|
|
29
|
Kageyama K, Terui K, Tsutaya S, Matsuda E,
Shoji M, Sakihara S, Nigawara T, Takayasu S, Moriyama T, Yasujima
M, et al: Gene analysis of the calcium channel 1 subunit and
clinical studies for two patients with hypokalemic periodic
paralysis. J Endocrinol Invest. 29:928–933. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ke Q, Xu QG, Huang DH, Yuan HJ, Zhao YL
and Wu WP: The mutation R672H in SCN4A gene exists in Chinese
patients with hypokalaemic periodic paralysis. Zhonghua Yi Xue Za
Zhi. 86:724–727. 2006.In Chinese. PubMed/NCBI
|
|
31
|
Kim H, Hwang H, Cheong HI and Park HW:
Hypokalemic periodic paralysis; two different genes responsible for
similar clinical manifestations. Korean J Pediatr. 54:473–476.
2011. View Article : Google Scholar
|
|
32
|
Kim JB, Kim MH, Lee SJ, Kim DJ and Lee BC:
The genotype and clinical phenotype of Korean patients with
familial hypokalemic periodic paralysis. J Korean Med Sci.
22:946–951. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang W, Jiang L, Ye L, Zhu N, Su T, Guan
L, Li X and Ning G: Mutation screening in Chinese hypokalemic
periodic paralysis patients. Mol Genet Metab. 87:359–363. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim SH, Kim UK, Chae JJ, Kim DJ, Oh HY,
Kim BJ and Lee CC: Identification of mutations including de novo
mutations in Korean patients with hypokalaemic periodic paralysis.
Nephrol Dial Transplant. 16:939–944. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dias da Silva MR, Cerutti JM, Tengan CH,
Furuzawa GK, Vieira TC, Gabbai AA and Maciel RM: Mutations linked
to familial hypokalaemic periodic paralysis in the calcium channel
alpha1 subunit gene (Cav1.1) are not associated with thyrotoxic
hypokalaemic periodic paralysis. Clin Endocrinol (Oxf). 56:367–375.
2002. View Article : Google Scholar
|
|
36
|
Silva MR, Chiamolera MI, Kasamatsu TS,
Cerutti JM and Maciel RM: Thyrotoxic hypokalemic periodic
paralysis, an endocrine emergency: Clinical and genetic features in
25 patients. Arq Bras Endocrinol Metabol. 48:196–215. 2004.In
Portuguese. PubMed/NCBI
|
|
37
|
Lin SH and Huang CL: Mechanism of
thyrotoxic periodic paralysis. J Am Soc Nephrol. 23:985–988. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dias Da Silva MR, Cerutti JM, Arnaldi LA
and Maciel RM: A mutation in the KCNE3 potassium channel gene is
associated with susceptibility to thyrotoxic hypokalemic periodic
paralysis. J Clin Endocrinol Metab. 87:4881–4884. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sternberg D, Tabti N, Fournier E, Hainque
B and Fontaine B: Lack of association of the potassium
channel-associated peptide MiRP2-R83H variant with periodic
paralysis. Neurology. 61:857–859. 2003. View Article : Google Scholar : PubMed/NCBI
|