Introduction
Apoptosis, an active, morphologically distinct form
of programmed cell death, has a fundamental role in the normal
development and differentiation of multicellular organogenesis, in
the control of cell proliferation, in development, and in the
pathogenesis of various diseases (1,2).
Apoptosis is also characterized by cell shrinkage, blebbing of
membranes, nuclear condensation and DNA fragmentation (3–5). In
addition, induction of apoptotic cell death is an important
mechanism of numerous anti-cancer drugs (6). The intrinsic apoptotic pathway is
triggered by a range of physical and chemical stimuli causing
mitochondrial dysfunction (7,8). The
initiation of the apoptotic cascades leads to the activation of
caspase-9 and subsequent activation of effector caspases, such as
caspase-3, which in turn cleaves several specific substrates,
including poly(ADP-ribose) polymerase (PARP), as well as
architectural components of the cell, which eventually leads to
apoptosis (9). Proteins of the
B-cell lymphoma 2 (Bcl-2) family serve as critical regulators of
mitochondrial apoptosis, functioning as either inhibitors or
promoters of cell death. Bcl-2 and Bcl extra large protein
(Bcl-XL) inhibit apoptosis by blocking the release of
cytochrome C from mitochondria through prevention of channel
formation, which is mediated by Bcl-2-associated X protein (Bax)
(10,11). Mitogen-activated protein kinases
(MAPKs), including c-Jun N-terminal kinases (JNK), extracellular
signal-regulated kinases (ERK) and p38/MAPK, are activated in
response to various stimuli. Subsequent to their activation, they
participate in a variety of signaling pathways regulating diverse
cellular processes, including cell growth, differentiation and
stress responses. Activation of MAPKs is therefore closely
associated with stress stimuli-induced apoptosis (12–14).
It was previously reported that Azorella
compacta Phil. (AC), a green, compact, resinous cushion shrub
of the Apiaceae family growing in the high Andes of southern Peru
and Bolivia, northeastern Chile and northwestern Argentina,
contained mulinane and azorellane diterpenoids (15,16).
In Chilean folk medicine, AC, together with other Azorella and
Laretia species (collectively known as 'llareta') is used for the
treatment of a variety of ailments (15). Historical records indicated that AC
has been used to treat the common cold and pain, to reduce blood
sugar, and also as an ointment to treat dermatological disorders.
AC presents a valuable source of mulinane and azorellane
diterpenoids (15,17). AC has also been traditionally used
to treat colds, asthma and bronchitis, as well as conditions whose
main symptoms include inflammation and pain. Studies on AC have
demonstrated anti-bacterial (18)
and anti-plasmodial properties (19). However, to the best of our
knowledge, no scientific studies are available on their
cytotoxicity-based anti-cancer effects.
The present study was the first, to the best of our
knowledge, explore the potency of AC extract to induce apoptosis of
human leukemia HL60 cells and to elucidate its underlying
mechanisms of action. The apoptotic effects of AC extract were
assessed by examining the caspase-dependent pathway involving the
loss of mitochondrial membrane permeability, the release of
cytochrome c and the activation of the Bcl-2 family of
proteins. In addition, the involvement of MAPK-dependent signaling
was assessed using MAPK inhibitors.
Materials and methods
Chemicals
The following reagents and kits were used in the
present study: Iscove's modified Dulbecco's medium (IMDM), fetal
bovine serum (FBS) (Gibco-BRL, Carlsbad, CA, USA), Dulbecco's
modified Eagle's medium (DMEM), RPMI-1640, phosphate-buffered
saline (PBS) (Hyclone, Logan, UT, USA), MTT (Amresco, Solon, OH,
USA), trypan blue, Hoechst 33342, caspase-3 substrate
acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin
(Ac-DEVD-AFC) (Invitrogen Life Technologies, Carlsbad, CA, USA),
dimethyl sulfoxide (DMSO), adriamycin,
2′,7′-dichlorodihydrofluores-cein diacetate (DCFH-DA), formaldehyde
(Sigma-Aldrich, St. Louis, MO, USA), agarose, 5X TBE buffer
(Bioneer Corp., Daejeon, Korea), apoptotic DNA ladder kit
(BioVision, San Diego, CA, USA), Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) kit (BD Biosciences,
Franklin Lakes, NJ, USA), Omniscript RT kit (Qiagen, Hilden,
Germany), polymerase chain reaction (PCR) premix (Promega Corp.,
Madison, WI, USA), TRIzol reagent, enhanced chemiluminescence (ECL)
reagent (Thermo Fisher Scientific, Waltham, MA, USA) and
polyvinylidene difluoride (PVDF) membranes (Merck-Millipore,
Billerica, MA, USA), PD98059, SP600125 and SB203580 (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA).
Cell line and cell culture
The cell lines HepG2, MCF7, HT1080, A549, SNU-1 and
HL60 were procured from the American Type Culture Collection
(Manassas, VA, USA). HepG2, MCF7 and HT1080 cells were routinely
cultured in DMEM supplemented with 10% FBS. SNU-1 cells were
routinely cultured in RPMI-1640 supplemented with 10% FBS. HL60
cells were routinely cultured in IMDM supplemented with 20% FBS in
a humidified incubator maintaining 5% CO2 at 37°C. For
experimental purposes, all cells were harvested by centrifugation
for 5 min at 250 × g.
Preparation of AC extract
A methanolic extract of AC was obtained from the
International Biological Material Research Center (Korea Research
Institute of Bioscience and Biotechnology, Daejeon, Korea). To
produce the extracted solid (20.05 g), ground AC seeds (147 g) were
treated with methanol and sonicated several times for three days.
The dried extract was dissolved in 20 mg/ml DMSO to prepare a stock
solution, which was then diluted with PBS.
MTT assay
In the MTT assay, MTT is metabolized into a colored
formazan precipitate by mitochondrial dehydrogenases present only
in viable cells, which is utilized to quantify the number of viable
cells. Cells were seeded into 96-well plates (2.5×104
cells/well in 200 µl medium). AC extract was added to the
cells in serial concentrations (0–100 µg/ml) in quadruplets
and incubated for 20 h. Adriamycin (2 µg/ml) was used as a
positive reference drug. Subsequent to incubation with the drugs,
10 µl MTT solution (5 mg/ml in PBS) was added to each well,
followed by incubation for 4 h. The plates were then centrifuged at
250 ×g for 10 min and the medium was removed by aspiration.
Finally, the formazan crystals were dissolved in 100 µl DMSO
and the absorbance at 570 and 630 nm was measured using a
96-well-plate reader (VersaMax, Molecular Devices, Sunnyvale CA,
USA). The inhibitory effect of the AC extract on cell growth was
expressed as the percentage of viable cells, with the
vehicle-treated cells considered 100% viable.
Hoechst 33342 staining
The morphology of the HL60 cells exposed to AC
extract was first observed under an inverted microscope. HL60 cells
were seeded into six-well plates (2.5×105 cells/well in
1 ml medium). After treatment with AC extract for 24 h at 0, 5, 10
and 20 µg/ml, the cells were harvested, washed in ice-cold
PBS and fixed with 4% formaldehyde in PBS for 15 min at room
temperature. The fixed cells were washed with PBS and stained with
Hoechst 33342 solution (10 µg/ml) for 20 min at room
temperature in the dark. The cells were then washed two times with
PBS. Finally, they were observed under a fluorescence microscope
and images were captured (Eclipse Ti-U; Nikon, Tokyo, Japan).
DNA fragmentation assay
Apoptosis was assessed by means of electrophoresis
of genomic DNA extracted from HL60 cells treated with AC as
described previously (20), with
certain modifications. Briefly, HL60 cells (1×106
cells/well in 1 ml medium) were treated with various concentrations
of AC extract (0, 5, 10 and 20 µg/ml) for 24 h.
Subsequently, the cells were harvested and washed in ice-cold PBS.
The DNA was harvested using an Apoptotic DNA ladder kit (BioVision,
San Diego, CA, USA) following the manufacturer's instructions,
according to which the total DNA was analyzed using 1.5% agarose
gel electrophoresis.
Annexin V/PI staining and flow cytometric
analysis
Phosphatidylserine (PS) exposed on the outer
mitochondrial membrane of the apoptotic cells was determined by an
Annexin V-FITC apoptosis detection kit (BD Biosciences), as per the
manufacturer's instructions. Briefly, following treatment with AC
extract for 24 h, cells were harvested by centrifugation (250 × g,
5 min), washed twice with ice-cold PBS and re-suspended in binding
buffer at a density of 1×106 cells/ml. Next, 5 µl
Annexin V-FITC and 5 µl PI were added to 100 µl cell
suspension, which was then incubated in the dark for 15 min.
Finally, 400 µl binding buffer was added to the cell
suspension. Cells were then analyzed using a flow cytometer (BD
Biosciences, San Diego, CA, USA). The data were analyzed with
CellQuest software (BD Biosciences).
Measurement of intracellular reactive
oxygen species (ROS)
First, samples of HL60 cells were treated with AC
extract as described above. The production of reactive oxygen
species was then measured using the membrane-permeable dye DCFH-DA.
The dye was added to cells cultured in six-well plates (2.5 ×
104 cells/well in 200 µl medium) at a final
concentration of 5 µM, and the plates were incubated at 37°C
for 1 h. The fluorescence intensity was measured using a
fluorescence plate reader (Victor X3; Perkin-Elmer, Waltham, MA,
USA) with excitation at 485 nm and emission at 530 nm.
Caspase-3 activity assay
The activity of caspase-3 was determined using a
fluorometric method using the synthetic caspase-3 substrate
Ac-DEVD-AFC. Briefly, AC extract-treated or -untreated cells were
incubated for 24 h and then harvested by centrifugation. Cell
pellets were re-suspended in cold lysis buffer (0.5% Triton X-100,
10 mM EDTA, 10 mM Tris-HCl, pH 7.5) and placed on ice for 15 min.
The cell lysates were then collected and incubated with caspase-3
assay buffer (10% glycerol, 2 mM dithiothreitol, 20 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5) and
caspase-3 substrate DEVD-AFC for 1 h at 37°C. Active caspase-3 was
measured by changes in the fluorescence at 485 and 535 nm using a
microplate reader (Victor X3).
RNA isolation and reverse transcription
quantitative (RT-q) PCR
Total RNA was extracted using TRIzol after the HL60
cells had been treated with AC extract (0, 1, 5, 10 or 20
µg/ml) for 24 h. Next, 2 µg RNA from each sample was
used to generate cDNA using the Omniscript RT kit (Qiagen)
according to the manufacturer's instructions. The cycling
conditions included a denaturation step at 95°C for 5 min, followed
by 30–35 cycles of 95°C for 30 sec, 55–60°C for 30 sec and 72°C for
1 min, and a final extension step at 72°C for 10 min. The resulting
total cDNA was then used to determine the expression levels of
caspase-3, caspase-9, Bax, Bcl-XL, Bcl-2 and cytochrome
C. Equal amounts of PCR products were electrophoresed on 1.2%
agarose gels (Bioneer Corp.) and visualized by RedSafe (iNtRON
Biotechnology, Seoul, Korea) staining. Images of the gels were
captured under ultraviolet light. Expression levels of GAPDH were
used as internal control for the integrity of the mRNA. The primer
sequences were as follows: Caspase-3 forward,
5′-ACATGGCGTGTCATAAAATACC-3′ and reverse,
5′-CACAAAGCGACTGGATGAAC-3′; caspase-9 forward,
5′-ATGGACGAAGCGGATCGGCGGCTCC-3′ and reverse,
5′-GCACCACTGGGGGTAAGGTTTTCTAG-3′; Bax forward,
5′-GTGCACCAAGGTGCCGGAAC-3′ and reverse, 5′-TCAGCCCATCTTCTTCCAGA-3′;
Bcl-XL forward, 5′-CCCAGAAAGGATACAGCTGG-3′ and reverse,
5′-GCGATCCGACTCACCAATAC-3′; Bcl-2 forward,
5′-GTGAACTGGGGGAGGATTGT-3′ and reverse, 5′-GGAGAAATCAAACAGAGGCC-3′;
cytochrome C forward, 5′-CTACGGACACCTCAGGCAGT-3′ and reverse,
5′-GGTGTGGTCCAAGGAAGAGA-3′; and GAPDH forward,
5′-CAAAAGGGTCATCATCTCTG-3′ and reverse,
5′-CCTGCTTCACCACCTTCTTG-3′.
Protein extraction and western blot
analysis
Following treatment with AC extract for 24 h, HL60
cells were harvested, washed with ice-cold PBS and lysed in lysis
buffer containing a protease-inhibitor-cocktail tablet (Roche
Diagnostics, Basel, Switzerland). The supernatant was obtained by
centrifuging at 2,000 x g for 15 min. Total protein was extracted
and protein concentration was determined using a bicinchoninic acid
assay kit (Thermo Fisher Scientific). For immunoblotting, 30
µg protein from each sample was subjected to 10% SDS-PAGE
and separated proteins were transferred onto a PVDF membrane. The
membrane was blocked with 5% skimmed milk at room temperature for 1
h and then incubated with the primary antibodies against caspase-3
(cat. no. 9661), caspase-9 (cat. no. 9505), Bcl-XL (cat.
no. 2762) (1:1,000; Cell Signaling Technology, Danvers, MA, USA),
PARP (cat. no. ab194217) (1:1,000; Abcam, Cambridge, MA, USA),
phosphorylated (p)-JNK (cat. no. sc-6254), p-ERK (cat. no.
sc-7383), p-p38 (cat. no. sc-7973) (1:1,000; Santa Cruz
Biotechnology, Inc.) and GAPDH (cat. no. sc-32233) (1:2,000; Santa
Cruz Biotechnology, Inc.), respectively, at 4°C overnight. After
washing, the membrane was incubated with anti-rabbit (cat. no.
sc-2030) or anti-mouse (cat. no. sc-2005) secondary antibody
(1:2,000; Santa Cruz Biotechnology, Inc.). GAPDH was used as an
internal control to monitor equal protein loading and transfer of
proteins from the gel to the membranes; for this, blots were
stripped with GAPDH antibody. Signals were detected using an
enhanced ECL reagent, and an LAS 4000 imaging system (Fujifilm,
Tokyo, Japan). The results shown are representative of three
independent experiments.
Statistical analysis
All in vitro experiments were performed in
triplicate, and each data point represents the average of at least
three independent experiments. Values are expressed as the mean ±
standard deviation. The comparisons were made between controls and
treated cultures using an unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference between values.
Results
AC extract inhibits cell growth and
induces apoptosis in human leukemic HL60 cells
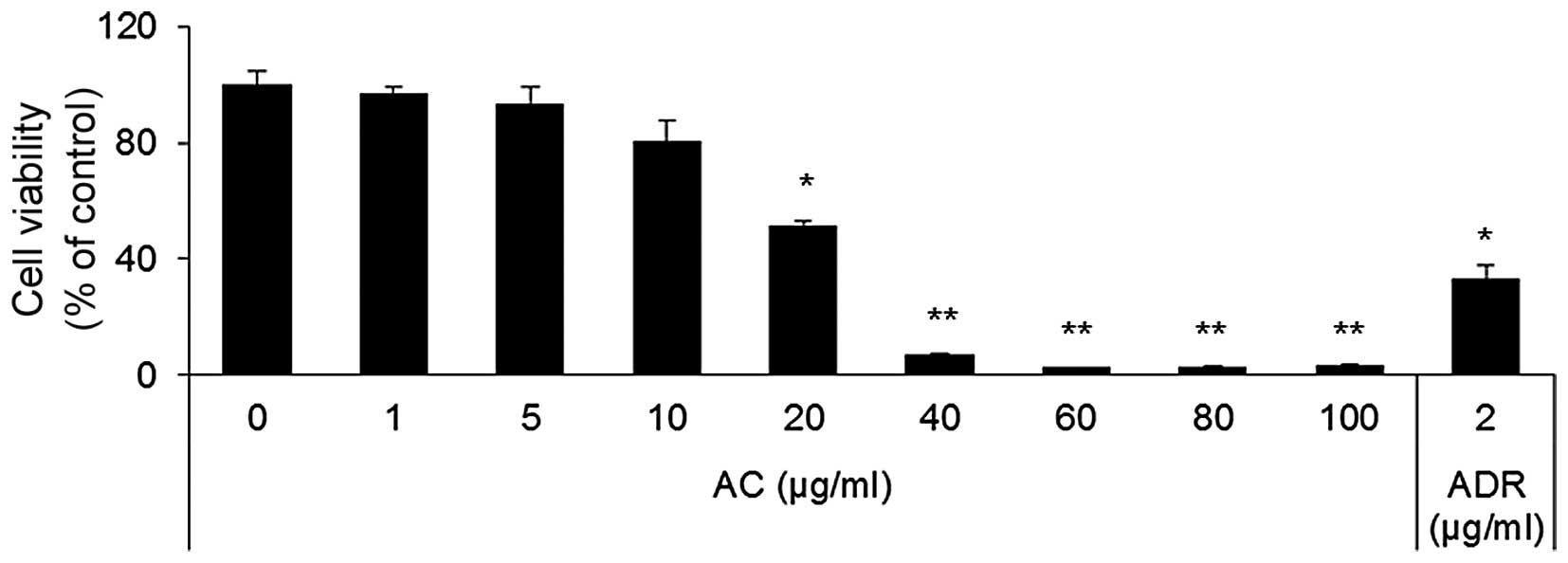
To determine the effects of AC extract on the growth
of cancer cells, HepG2, SNU-1, MCF7, HT1080, A549 and HL60 cells
were treated with AC extract at concentrations of 1–100
µg/ml for 24 h, after which cell proliferation was assessed
by MTT assay. AC extract inhibited the growth of HL60 cells in a
dose-dependent manner (Fig. 1). At
concentrations of 40 µg/ml and higher, AC extract almost
completely inhibited the growth of HL60 cells. The IC50
of AC extract was determined to be 34±4.06 µg/ml on HL60
cells. Next, the present study investigated whether the observed
inhibitory effects of AC extract on cell viability resulted from
apoptotic cell death. Apoptosis is a process of programmed cell
death, which is characterized by various biochemical and
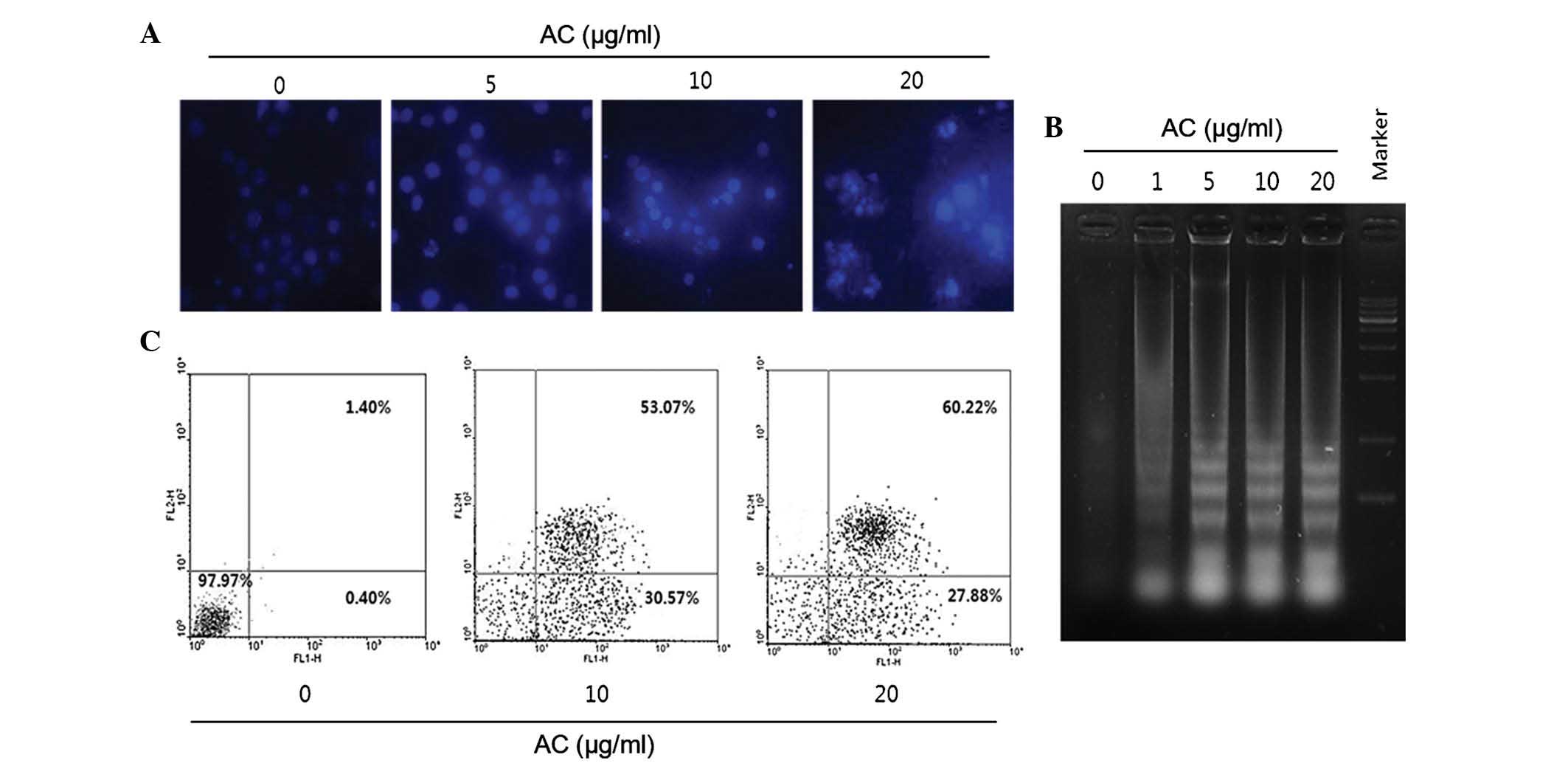
morphological changes. To evaluate the effects of AC extract on
nuclear morphology, Hoechst 33342 staining was performed. The
nuclei of cells treated with AC extract at 5, 10 and 20
µg/ml were deeply stained and exhibited bright fluorescence,
which indicated the condensation of chromatin (Fig. 2A). In addition, as shown in
Fig. 2B, DNA ladder formation was
confirmed by agarose gel electrophoresis. Efficient DNA laddering
was observed in HL60 cells treated with >10 µg/ml AC
extract for 24 h. With increasing concentration, a more intense
pattern of DNA laddering was detected (Fig. 2B). The apoptosis-inducing effects
of the AC extract were also evaluated using Annexin V/PI staining.
As show in in Fig. 2C, the
apoptotic rate increased in an AC concentration-dependent manner.
The early and late apoptotic fractions in control cells were 0.40
and 1.40%, respectively, but increased to 23.80 and 58.82% after
treatment with 20 µg/ml AC extract (Fig. 2C). PI-positive cells also increased
marginally at higher AC concentrations.
AC extract induces ROS production and
activation of caspases in human leukemic HL60 cells
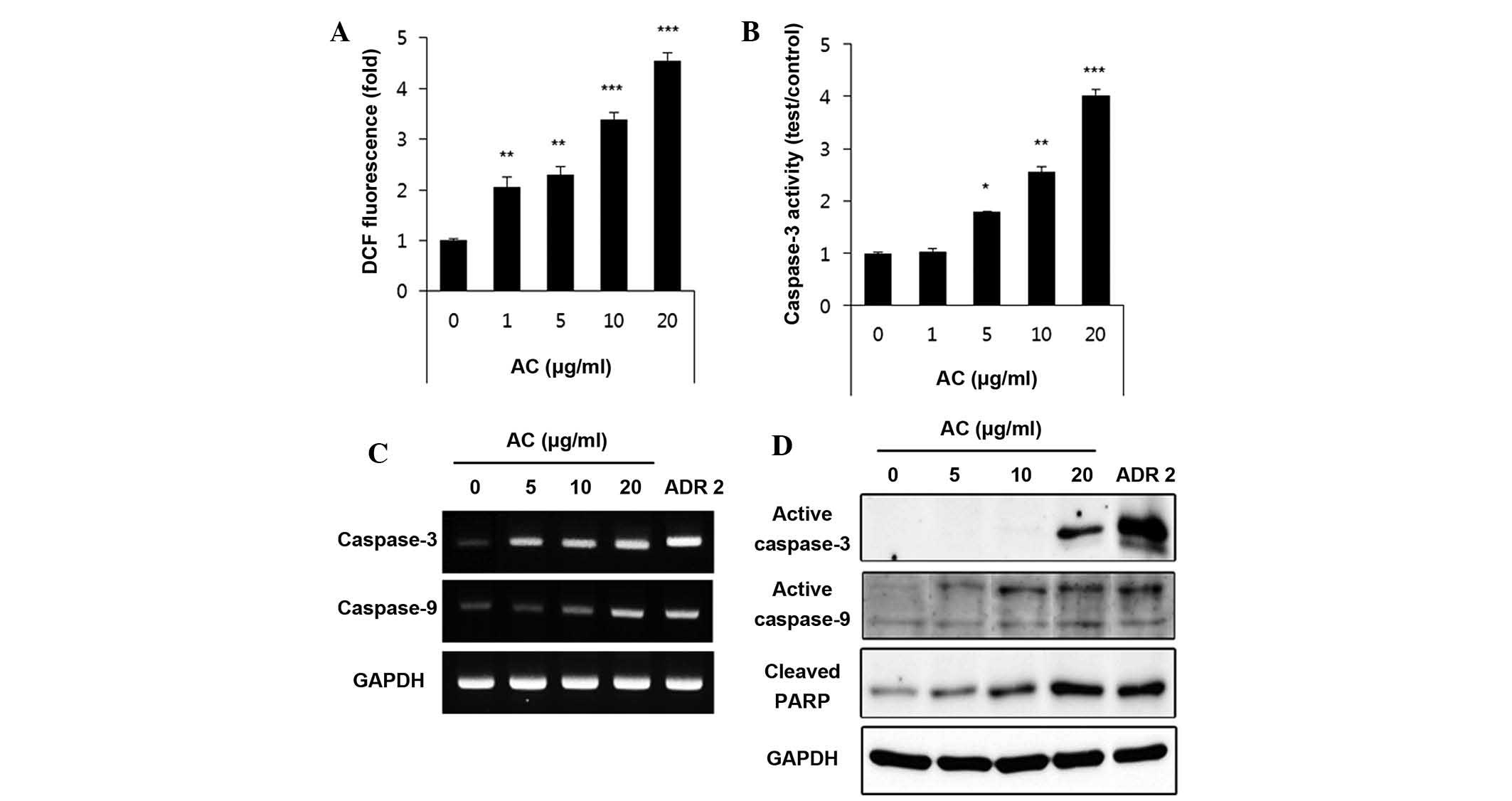
ROS levels in AC-extract-treated HL60 cells were
quantified 1 h after the addition of DCFH-DA. As displayed in
Fig. 3A, intracellular ROS levels
significantly increased in an AC concentration-dependent manner.
These results suggested that ROS production may be the cause of AC
extract-induced apoptosis in HL60 cells. As AC-extract treatment
led to enhanced ROS generation, it is possible that alterations in
the cell may have a role in AC extract-induced apoptosis. Caspase-3
and -9, known to serve as important mediators of intrinsic
apoptotic pathways, also contribute to general apoptotic morphology
through the cleavage of various cellular substrates, including PARP
(21). As indicated in Fig. 3B, western blot analysis showed that
AC extract induced the activation of caspase-3 in a
concentration-dependent manner. RT-PCR was used to detect the mRNA
expression of caspase-3 and caspase-9 24 h after AC-extract
treatment. The change in mRNA expression was normalized against
GAPDH expression. Fig. 3C shows
that the mRNA expression of caspase-3 and caspase-9 increased in a
manner that was AC-extract dose-dependent. Furthermore, the western
blots in Fig. 3 D showed that
AC-extract treatment induced the activation of caspase-3 and -9 in
a concentration-dependent manner. In addition, western blot
analysis revealed that progressive proteolytic cleavage products of
PARP protein, a downstream target of activated caspase-3, occurred
in HL60 cells treated with AC extract.
AC extract activates apoptosis-associated
signaling in human leukemia HL60 cells
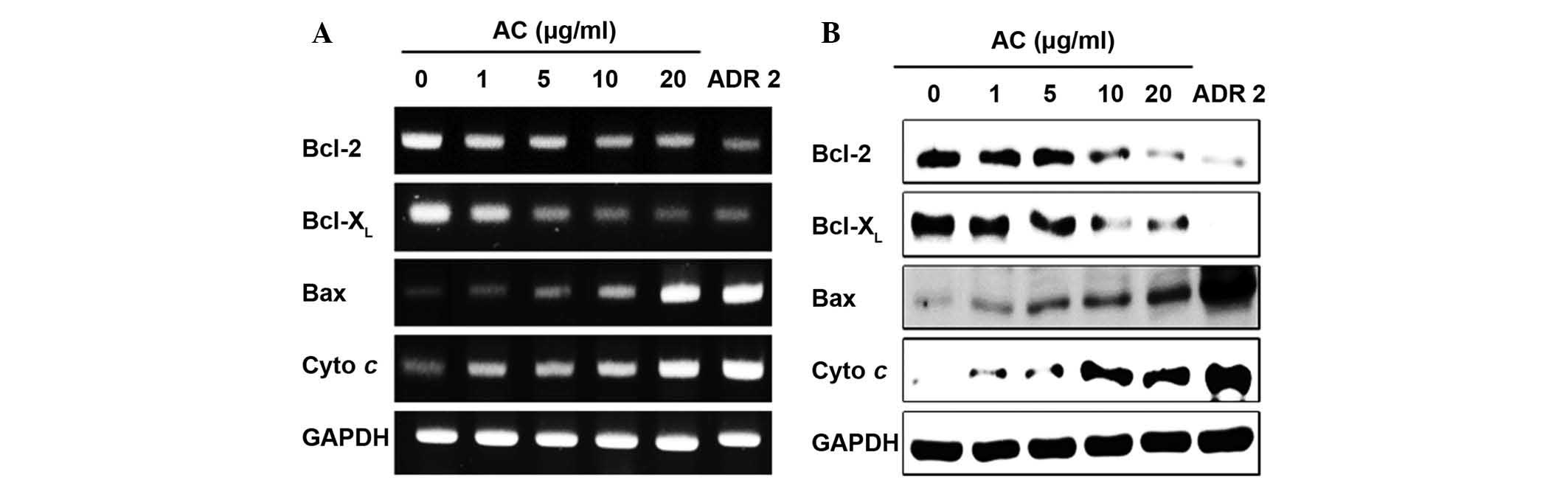
The underlying mechanism by which AC extract induced
apoptosis of HL60 cells was delineated by RT-PCR and western blot
analyses. As shown Fig. 4A and B,
examination of Bax, Bcl-2, Bcl-XL and cytochrome C
expression during apoptosis indicated that AC treatment at 1, 5,
10, or 20 µg/ml dose-dependently increased the expression of
cytochrome C and pro-apoptotic Bax, whereas the expression of
anti-apoptotic Bcl-2 and Bcl-XL was downregulated with
increasing concentrations of AC extract.
Activation of MAPK is involved in
AC-extract-induced apoptosis in human leukemia HL60 cells
Next, the effect of AC-extract treatment on the
expression and activities of MAPKs was investigated in order to
determine whether these signaling pathways have a role in mediating
the observed apoptotic response. To confirm an association between
the activation of MAPKs and the induction of apoptosis by the AC
extract, the cells were pre-treated with MAPK inhibitors and their
viability was assessed using an MTT assay. As shown in Fig. 5A, the levels of p-p38, p-ERK and
p-JNK proteins increased in an AC concentration-dependent manner.
As Fig. 5B demonstrates,
pre-treatment with SB203580 (inhibitor of p38), PD98059 (inhibitor
of ERK) and SP600125 (inhibitor of JNK) increased the viability of
cells treated with AC extract. To further determine the mechanism
of MAPKs activation in HL60 cells by AC extract, the effects of
these MAPK inhibitors on the release of Bax and cleaved PARP
proteins were also investigated (Fig.
5C). The results showed that MAPK inhibitor blocked
pro-apoptotic protein and cleaved PARP protein. These results
indicated that AC extract induced apoptosis via these two proteins
of the intrinsic apoptotic pathway.
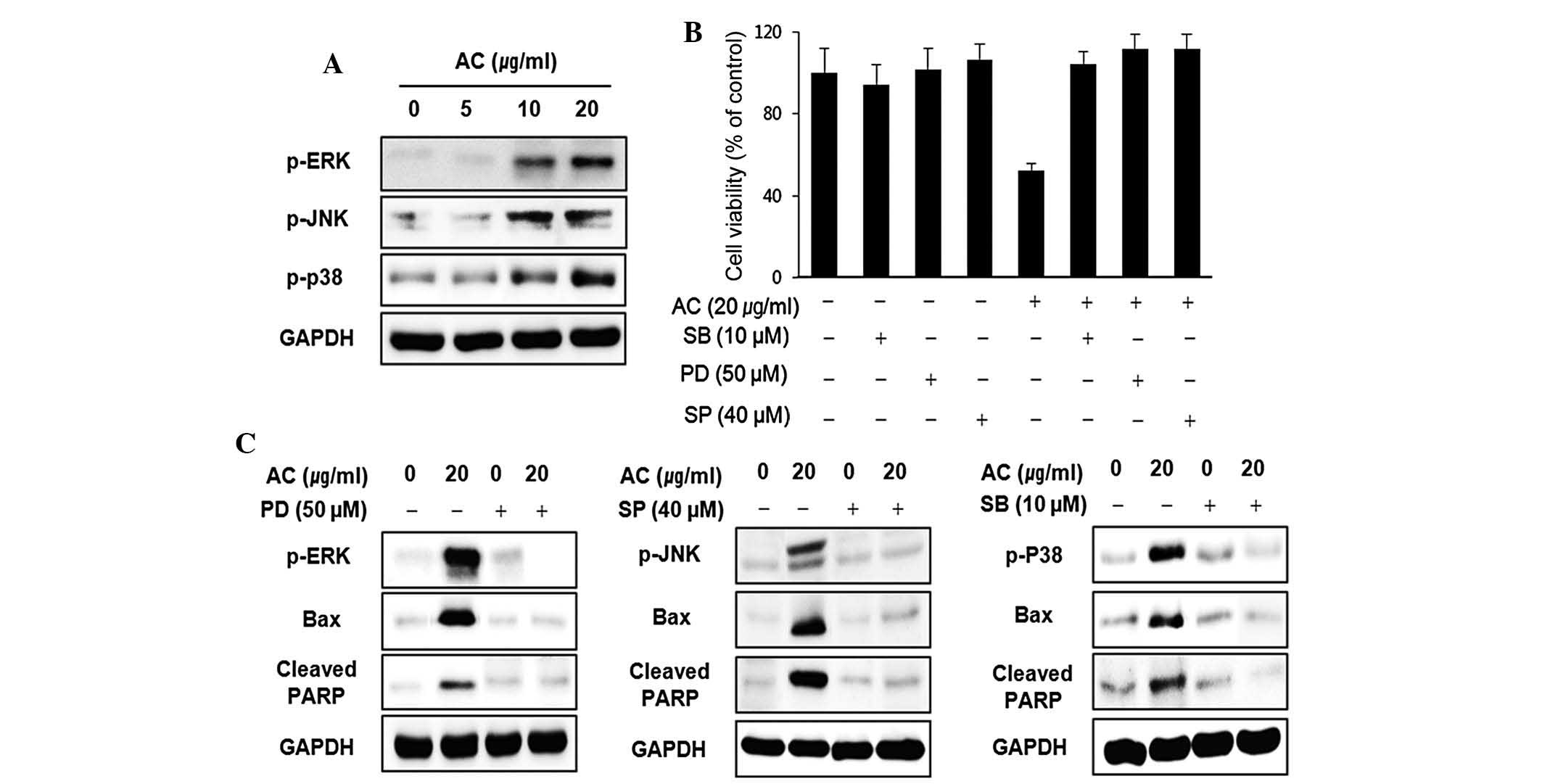 | Figure 5MAPKs were involved in AC
extract-induced apoptosis of HL60 cells. (A) HL60 cells were
treated with AC extract at indicated concentrations for 24 h, and
the activation of MAPKs family proteins was determined by western
blotting. (B) Cells pre-treated with SP600125 (40 µM),
SB203580 (10 µM) or PD98059 (50 µM) for 1 h were
incubated with AC extract for 24 h and cell viability was assessed
using an MTT assay. (C) The effects of AC and/or SP600125, SB203580
and PD98059 on the activation of MAPK family proteins PARP and Bax
were determined by western blot analysis. MAPK, mitogen-activated
protein kinase; p-ERK, phosphorylated extracellular
signal-regulated protein kinase; JNK, c-Jun N-terminal kinase; Bax,
B-cell lymphoma 2-associated X protein; ARP, poly(adenosine
diphosphate ribose) polymerase; PD, ERK inhibitor PD98059; SP, JNK
inhibitor SP600125; SB, p38 inhibitor SB203580; AC, Azorella
compacta. |
Discussion
The ability to induce tumor-cell apoptosis is an
important property of candidate anti-cancer drugs and serves to
discriminate between anti-cancer drugs and compounds with toxicity.
Much effort has been directed toward identifying compounds that
influence apoptosis and toward understanding their mechanisms of
action. Extracts prepared from a large variety of plants have been
demonstrated to possess the ability to trigger the activation of
apoptotic pathways (22,23). The mechanisms of apoptosis
induction are complex and not fully known, but certain key events
have been identified, which appear essential for the cell to enter
apoptosis (24). The notion that
apoptosis represents a critical element in control of cell-number
in physiological and pathological situations has been well reviewed
and its role in oncogenesis is now well established (25). Apoptosis has an important function
in the normal development and differentiation of multicellular
organisms and is characterized by morphological and biological
changes, including chromatin condensation, cytoplasmic shrinkage
and DNA degradation (26).
Apoptosis also serves as a critical protective mechanism against
carcinogenesis caused by mutations of the genetic material in
normal cells and against various other forms of carcinogenesis. A
variety of stimuli can trigger apoptosis, including death
receptor-mediated signaling (extrinsic pathway) or intracellular
stresses (intrinsic pathway) (27). The present study aimed to determine
the capacity of AC extract to induce apoptosis and to identify the
associated biochemical mechanisms in human leukemic cells. The
results demonstrated that AC extract inhibits leukemic cell growth
by induction of apoptotic cell death, which appeared to account for
its anti-proliferative action. Depending on the cell line, AC
extract exerted growth-inhibitory effects on cancer cells with
IC50-values of 20–100 µg/ml after 24 h treatment.
In HL60 cells, a promising level of cytotoxicity was observed;
therefore, the present study pursued the effect of AC extract on
human leukemia HL60 cells. It was demonstrated that AC extract
exerted a significant anti-proliferative effect against HL60 cells
in a dose-dependent manner. Further cellular and biochemical
analysis indicated that the proliferation of inhibitory activity of
AC extract was associated with the induction of apoptosis. Several
sensitive methods for detecting apoptosis have been developed.
Staining of apoptotic cells with the fluorescent dye Hoechst 33342
is considered to be a suitable method for evaluating changed
nuclear morphology (28). One of
the earliest events indicating apoptosis is the loss of plasma
membrane polarity, accompanied by translocation of PS from the
inner to outer membrane leaflets, thereby exposing PS to the
external environment. The phospholipid-binding protein Annexin V
has a high affinity for PS and can bind to apoptotic cells with an
inverted mitochondrial membrane; the quantity of
fluorescently-labeled Annexin-V correlates with the loss of
membrane polarity during apoptosis. The perforation and inversion
of the mitochondrial membrane precedes the complete loss of
membrane integrity that accompanies later stages of cell death,
resulting from either apoptosis or necrosis. By contrast, PI only
enters cells after loss of membrane integrity. Thus, dual staining
with Annexin V and PI allows for a clear discrimination between
affected cells, early apoptotic cells and late apoptotic or
necrotic cells (29). HL60 cells
treated with AC extract at lower concentrations contained a
population of Annexin V-positive cells indicating early apoptosis,
while Annexin V- and PI-positive cells were present at higher AC
concentrations, which revealed the occurrence of post-apoptotic
necrosis. The impairment of mitochondrial function has been
considered to be a key event in the ROS-mediated apoptotic pathway
(30). In the present study ROS
generation was indicated to be involved in AC extract-induced cell
death. ROS levels were determined in HL60 cells after AC-extract
treatment using the peroxide-sensitive fluorescent probe, DCFH-DA,
and a four-fold increase was evidenced after 24 h. In general, the
mitochondria-mediated intrinsic pathway and the
death-receptor-triggered extrinsic pathway can lead to caspase-3
activation (31). In the system of
the present study, caspase-9 was significantly activated, which
implicated mitochondrial involvement, since caspase-9 is the
initiator caspase for the intrinsic apoptotic pathway (32). In the intrinsic pathway, the ratio
of the expression of pro-apoptotic proteins such as Bax and
anti-apoptotic proteins, including Bcl-2 and Bcl-XL,
ultimately determines cell death or survival through regulation of
mitochondrial-permeability transition. This leads to activation of
caspase-3 for induction of apoptosis via release of cytochrome C to
the cytosol (11). The results of
the present study indicated that AC extract induced Bax
translocation from the cytosol to the mitochondria, leading to the
release of cytochrome C, apoptosome formation, and finally,
induction of apoptosis in HL60 cells.
MAPKs, including ERK, JNK and p38, have critical
roles in cell survival and apoptosis in various types of cancer
cell. It is known that activation of ERK, JNK and p38 leads to
induction of apoptosis (33,34).
Activation of ERK, JNK and p38 can induce mitochondrial dysfunction
with subsequent release of apoptotic proteins, such as cytochrome
C, from the mitochondria into the cytosol, and finally activate
caspase-9 and caspase-3. In the present study, treatment with AC
extract resulted in upregulation of ERK, JNK and p38
phosphorylation. Therefore, the involvement of ERK, JNK, and p38
activation in the MAPK-signaling pathway during AC-extract-induced
apoptosis in HL60 cells was further investigated. As the results
indicated, PD98059 (inhibitor of ERK), SP600125 (inhibitor of JNK)
and SB203580 (inhibitor of p38) blocked AC-extract-induced
apoptosis of HL60 cells by inhibiting the interaction between Bax
and activated PARP. The results therefore suggested an association
of AC-extract-induced apoptosis with activation of ERK, JNK and
p38/MAPK.
In conclusion, the present study demonstrated that
AC extract significantly induced apoptosis in leukemia cells by
increasing the generation of ROS, by causing translocation of Bax
to the mitochondria from the cytosol, and by initiating the release
of cytochrome C followed by activation of caspase-9 and caspase-3.
AC extract was found to exert its anti-cancer effects via the ERK,
JNK and p38/MAPK-mediated intrinsic apoptotic pathway in human
leukemia HL60 cells. Future studies will examine the effects of AC
on upstream signaling pathways of MAPKs and evaluate its
anti-cancer efficacy in vivo using nude mouse models. As the
present study did not observe any side effects, AC is expected to
be a promising anti-cancer drug. The potential use of AC in
combination with other drugs may also be investigated.
Acknowledgments
This study was supported by the Ministry of Science,
ICT and Future Planning (no. FGC1011433) and the KRIBB Initiative
Program (no. KGM1221521) of the Republic of Korea. The authors
would like to thank the International Biological Material Research
Center (Korea Research Institute of Bioscience and Biotechnology,
Daejeon, Korea) for providing the AC extract.
References
|
1
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: the significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Earnshaw WC: Nuclear changes in apoptosis.
Curr Opin Cell Biol. 7:337–343. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nagata S: Apoptosis by death factor. Cell.
88:355–365. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly (ADP-ribose)
polymerase by a proteinase with properties like ICE. Nature.
371:346–347. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kyosseva SV: Mitogen-activated protein
kinase signaling. Int Rev Neurobiol. 59:201–220. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Keshet Y and Seger R: The MAP kinase
signaling cascades: a system of hundreds of components regulates a
diverse array of physiological functions. Methods Mol Biol.
661:3–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sumbayev VV and Yasinska IM: Regulation of
MAP kinase-dependent apoptotic pathway: implication of reactive
oxygen and nitrogen species. Arch Biochem Biophys. 436:406–412.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Loyola LA, Bórquez J, Morales G,
San-Martín A, Darias J, Flores N and Giménez A: Mulinane-type
diterpenoids from Azorella compacta display antiplasmodial
activity. Phytochemistry. 65:1931–1935. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wachter GA, Franzblau SG, Montenegro G,
Suarez E, Fortunato RH, Saavedra E and Timmermann BN: A new
antitu-bercular mulinane diterpenoid from Azorella madreporica
Clos. J Nat Prod. 61:965–968. 1998. View Article : Google Scholar
|
|
17
|
Areche C, Rojas-Alvarez F, Campos-Briones
C, Lima C, Pérez EG and Sepúlveda B: Further mulinane diterpenoids
from Azorella compacta. J Pharm Pharmacol. 65:1231–1238. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wachter GA, Matooq G, Hoffmann JJ, Maiese
WM, Singh MP, Montenegro G and Timmermann BN: Antibacterial
diterpenoid acids from Azorella compacta. J Nat Prod. 62:1319–1321.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fuentes NL, Sagua H, Morales G, Borquez J,
San Martin A, Soto J and Loyola LA: Experimental antihyperglycemic
effect of diterpenoids of llareta Azorella compacta (Umbelliferae)
Phil in rats. Phytother Res. 19:713–716. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee K, Kwon OK, Xia Y and Ahn KS: Effect
of AC-264, a novel indole derivative, on apoptosis in HL-60 cells.
Bull Korean Chem Soc. 31:3777–3781. 2010. View Article : Google Scholar
|
|
21
|
Pink JJ, Wuerzberger-Davies S, Tagliarino
C, Planchon SM, Yang X, Froelich CJ and Boothman DA: Activation of
a cysteine protease in MCF-7 and T47D breast cancer cells during
beta-lapachone-mediated apoptosis. Exp Cell Res. 255:144–155. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ren G, Zhao YP, Yang L and Fu CX:
Anti-proliferative effect of clitocine from the mushroom
Leucopaxillus giganteus on human cervical cancer HeLa cells by
inducing apoptosis. Cancer Lett. 262:190–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu SJ, Ng LT, Chen CH, Lin DL, Wang SS and
Lin CC: Antihepatoma activity of Physalis angulata and P. peruviana
extracts and their effects on apoptosis in human Hep G2 cells. Life
Sci. 74:2061–2073. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Andersson B, Janson V, Behnam-Motlagh P,
Henriksson R and Grankvist K: Induction of apoptosis by
intracellular potassium ion depletion: Using the fluorescent dye
PBFI in a 96-well plate method in cultured lung cancer cells.
Toxicol In vitro. 20:986–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hall PA: Assessing apoptosis: A critical
survey. Endocr Relat Cancer. 6:3–8. 1999. View Article : Google Scholar
|
|
26
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Galluzzi L, Larochette N, Zamzami N and
Kroemer G: Mitochondria as therapeutic targets for cancer
chemotherapy. Oncogene. 25:4812–4830. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Belloc F, Dumain P, Boisseau MR,
Jalloustre C, et al: A flow cytometric method using Hoechst 33342
and propidium iodide for simultaneous cell cycle analysis and
apoptosis determination in unfixed cells. Cytometry. 17:59–65.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Wu S, Zhang Q, Liu F and Wu P:
23,24-Dihydrocucurbitacin B induces G2/M cell-cycle arrest and
mitochondria-dependent apoptosis in human breast cancer cells
(Bcap37). Cancer Lett. 256:267–278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang R, Li C, Song D, Zhao G, Zhao L and
Jing Y: Ethacrynic acid butyl-ester induces apoptosis in leukemia
cells through a hydrogen peroxide mediated pathway independent of
glutathione S-transferase P1-1 inhibition. Cancer Res.
67:7856–7864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cullen SP and Martin SJ: Caspase
activation pathways: Some recent progress. Cell Death Differ.
16:935–938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng TS, Hunot S, Kuida K, Momoi T,
Srinivasan A, Nicholson DW, Lazebnik Y and Flavell RA: Deficiency
in caspase-9 or caspase-3 induces compensatory caspase activation.
Nat Med. 6:1241–1247. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moon DO, Kim MO, Choi YH, Kim ND, Chang JH
and Kim GY: Bcl-2 overexpression attenuates SP600125-induced
apoptosis in human leukemia U937 cells. Cancer Lett. 264:316–325.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cross TG, Scheel-Toellner D, Henriquez NV,
Deacon E, Salmon M and Lord JM: Serine/threonine protein kinases
and apoptosis. Exp Cell Res. 256:34–41. 2000. View Article : Google Scholar : PubMed/NCBI
|