Introduction
Chronic hypoxia is one of the basic
pathophysiological processes associated with congenital heart
disease (CHD) (1). Although
patients with cyanotic CHD suffer long-term chronic hypoxia, the
majority are able to tolerate hypoxia until surgery. At present,
the underlying mechanisms of CHD remain to be fully elucidated
(2). MicroRNAs (miRNAs) are
endogenous, small (~16–29 nucleotides) non-coding, single-stranded
transcripts, which negatively regulate gene expression
post-transcriptionally by targeting the 3′-untranslated region
(UTR) of mRNAs (3). miRNAs have
been demonstrated to exert critical roles in a number of cardiac
diseases, including cardiac hypertrophy, heart failure, arrhythmia
and ischemia-reperfusion (I/R) injury (4). A previous report from our laboratory
demonstrated that miRNAs are involved in the process of
cardiomyocyte adaptation to chronic hypoxia (5). A previous study revealed that the
expression of miR-146b was higher in the heart tissue of patients
with the CHD defect, Tetralogy of Fallot (TOF), compared with that
of normal infants (6).
Furthermore, the expression of miR-146b also increased in human
pulmonary arterial smooth muscle cells following the onset of
hypoxia (7), and the levels of
miR-146b were increased in the lung tissue in an experimental
animal model of CHD compared with that of the control groups
(8). Therefore, it was
hypothesized that miR-146b may function as a regulator involved in
the cardiac chronic hypoxia process. miR-146b is the key miRNA
regulating the immune system, particularly the natural immunity
pathway (9). This miRNA
predominantly regulates the nuclear factor-κB (NF-κB) pathway by
directly targeting interleukin-1 receptor-associated kinase 1
(IRAK1), tumor necrosis factor receptor-associated factor 6 (TRAF6)
and myeloid differentiation primary response gene 88 (MyD88)
(10). However, the role of
miR-146b in cardiomyocytes under hypoxic conditions remains to be
fully elucidated. The present study aimed to investigate the
changes in the expression levels of miR-146b in cardiomyocytes
under hypoxic conditions, and to assess whether miR-146b has a
protective role in myocardial adaptation to chronic hypoxia.
Materials and methods
Sampling of the myocardial section
The sampling procedure was performed, as previously
described (5). Briefly, 21 infant
patients with CHD, with an average age of 17.1 months, were
eligible for enrollment in the present study. The inclusion
criteria were: (i) infants were aged between 3.2 and 60.0 months;
(ii) a diagnosis of TOF had been made; (iii) symptoms of a
ventricular septal defect (VSD) combined with either pulmonary
atresia or a right ventricular outflow tract obstruction were
present, which required surgical reconstruction. The diagnosis was
obtained using echocardiography, and was confirmed at the time of
surgery. The exclusion criteria implemented in the present study
were infectious diseases, and other types of congenital
malformation. A total of nine patients with TOF, and one with
pulmonary atresia and a VSD, were enrolled in the cyanotic group,
whereas 11 patients with a right ventricular outflow tract
obstruction and a VSD were enrolled in the acyanotic group. All
subjects were recruited from Xinqiao Hospital (Chongqing, China).
The local ethics committee of the Third Military Medical University
Affiliated Hospital approved the design of the present study, and
this investigation conformed to the principles of the Declaration
of Helsinki. Written informed consent was obtained from the parents
of all patients. Routine procedures for anesthesia and surgery were
performed, as previously described (1). The biopsies were obtained from the
right ventricular outflow tract and were stored at −80°C prior to
analyses.
Cell culture
The heart-derived H9c2 cell line was obtained from
American Type Culture Collection (Manassas, VA, USA) and was
maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen
Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal
bovine serum (Invitrogen Life Technologies) at 37°C. Following
incubation in serum-free medium overnight, the medium was changed
to serum-contained DMEM and the cells were placed in a modular
incubator (3131; Forma Scientific, Marietta, OH, USA), containing a
gaseous mixture of 94% N2, 5% CO2 and 1%
O2 at 37°C for 24, 48 or 72 h. The cells under
conditions of normoxia (21% O2) were classified as the
control group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was isolated using RNAiso Plus reagent
(Takara Bio, Inc., Otsu, Japan). The cDNA library was created using
a PrimeScript RT reagent kit (Takara Bio, Inc.). The RT-qPCR
analysis was performed using the SYBR® Premix Ex
Taq™ kit (Takara Bio, Inc,) on a ViiA 7 Real-Time PCR system
(Applied Biosystems, Los Angeles, CA, USA). The specific stem-loop
primers of miR-146b were obtained from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China). RT was performed at 42°C for 30 min and 85°C
for 5 sec. The PCR protocol for miR-146b consisted of 1 cycle of
95°C for 30 sec, 40 cycles of 95°C for 5 sec and 60°C for 34 sec.
All reactions were repeated in triplicate and the quantitative
expression data were normalized against U6 RNA.
miRNA transfection
Upon reaching 30–50% confluence, the cells were
transfected with 100 nM miR-146b inhibitor (GenePharm, Shanghai,
China) or the negative controls (which are random sequence miRNA
inhibitor that has been widely tested in rat cell lines and shown
not to affect miRNA function), using Lipofectamine 2000™ reagent
(Invitrogen Life Technologies), according to the manufacturer's
instructions. All transfections were performed 24 h following
seeding, and the cells were incubated for a further 48 h prior to
the hypoxia experiments.
Lactate dehydrogenase (LDH) assay
The membrane integrity of the cultured H9c2
cardiomyocytes was assessed by measuring the release of LDH into
the culture medium using an LDH assay kit (Beyotime Institute of
Biotechnology, Beijing, China). The results obtained were divided
by the maximum release of LDH in each group in order to calculate
the cell mortality rate. Briefly, the assay was performed in
96-well plates, and the absorbance was measured using a microplate
reader (Varioskan® Flash; Thermo Fisher Scientific,
Waltham, MA, USA) at 490 nm.
Flow cytometric analysis
Following treatment, the cells were digested with
trypsin, washed once with cold phosphate-buffered saline (PBS),then
resuspended in 100 µl 1X Binding Buffer at a concentration
of 1×105 cells/ml. They were subsequently incubated with
5 µl annexin V-fluorescein isothiocyanate and 5 µl
propidium iodide (Annexin V:FITC Apoptosis Detection kit I; cat.
no. #556547; BD Biosciences, San Diego, CA, USA) for 20 min at room
temperature for each tube. Then 400 µl of 1X Binding Buffer
were added to each tube. The cell suspensions were analyzed by flow
cytometry (BD LSRFortessa X-20, BD Biosciences). The results were
analyzed using FlowJo 7.61 software (Treestar Inc., Ashland, OR,
USA).
Terminal deoxynucleotidyl transferase
dUTP nick-end labeling (TUNEL)
The extent of apoptosis was also analyzed by TUNEL
staining using the Cell Death Detection kit (Beyotime Institute of
Biotechnology), according to manufacturer's instructions. The
slides were visualized under a DMI3000 fluorescence microscope
(Leica, Mannheim, Germany). The total cell number in a selected
area was deter-mined on the basis of 4′,6-diamidino-2-phenylindole
(DAPI) nuclear staining. The apoptotic index was calculated as the
number of TUNEL-positive, DAPI-stained cells divided by the total
number of cells in five random fields.
Hoechst 33258 staining
The H9C2 cells were detected using Hoechst 33258
staining (Beyotime Institute of Biotechnology), according to the
manufacturer's instructions. The cells were washed with PBS and
treated with Hoechst 33258 (10 µg/ml) for 50 min at 37°C in
the dark. The Hoechst-stained nuclei were subsequently observed
under a fluorescence microscope (Leica) at a wavelength of 521
nm.
JC-1 staining
The apoptotic cells were assessed using the JC-1
assay kit (KeyGen Biotech Co., Ltd., Nanjing, China), according to
the manufacturer's instructions. The JC-1 fluorescent probe was
used to detect alterations in the mitochondrial transmembrane
potential (ΔΨm), which is one of the earliest indicators
of apoptosis occurring in cells. Following treatment, the cells
were washed with PBS twice and incubated with the JC-1 stain in the
dark at 37°C for 30 min. The cells were subsequently observed under
a fluorescence microscope (Leica).
Western blotting
The proteins were extracted from the H9C2 cells
using sodium dodecyl sulfate (SDS) lysis buffer (Beyotime Institute
of Biotechnology), containing 500 mM phenylmethylsulfonyl fluoride,
for 30 min. A total of 40 µg protein was separated by
SDS-PAGE (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
transferred onto a polyvinylidene difluoride membrane (Roche
Diagnostics, Indianapolis, IN, USA). The concentration of
separation gel was 12% for Bcl-2, Bax and cleaved-caspase 3 and 10%
for the other proteins. The membranes were blocked with 5% BSA for
1 h and incubated with primary antibodies at 4°C overnight,
followed by incubation at room temperature for 1 h with secondary
antibodies conjugated to horseradish peroxidase. The antibodies
against Bcl-2 (rabbit anti-rat; cat. no. #2870; 1:1,000), Bax
(rabbit anti-rat; cat. no. #2772; 1:1,000), caspase 3 (rabbit
anti-rat; cat. no. #9662; 1:1,000), signal transducer and activator
of transcription 3 (STAT3; (rabbit anti-rat; cat. no. #9132,
1:1,000), phosphorylated (p-)-STAT3, p65 (rabbit anti-rat, cat.
#8242, 1:1000) and p-p65 (rabbit anti-rat; cat. no. #3037; 1:1,000)
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA); the antibody against ribonuclease (RNase) L was obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA; rabbit
anti-rat; sc-22871; 1:700). The resultant bands were visualized
using BeyoECL Plus kit (Beyotime Institute of Biotechnology).
Luciferase reporter assay
The miR-146b promoter, or fragments generated by
mutagenesis (Guangzhou RiboBio Co., Ltd., Guangzhou, China), were
cloned into the XhoI and NotI sites downstream of the
luciferase reporter gene of pmiR-RB-Report (Guangzhou RiboBio Co.,
Ltd.). This plasmid contained Renilla luciferase as the
reporter and firefly luciferase as the internal control. The
plasmid DNA and miR-146b mimic/mimic-normal control (NC) were
co-transfected into HEK293T cells using Lipofectamine™ 2000 reagent
(Invitrogen Life Technologies). The activity of luciferase was
measured using the Dual-Glo luciferase assay system (Promega
Corporation, Madison, WI, USA) at 48 h post-transfection.
Statistical analysis
The data were analyzed using GraphPad Prism 5.0
software (GraphPad Prism Inc., La Jolla, CA, USA) and are expressed
as the mean ± standard deviation. Comparisons between groups were
made using one-way analysis of variance or two-tailed t-test. All
experiments were repeated three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of miR-146b is upregulated in
samples from patients with cyanotic CHD
A previous study reported that miR-146b is expressed
at higher levels in tissue samples of patients with TOF compared
with the normal controls (6). The
present study hypothesized that this was caused, at least in part,
by hypoxia. The clinical characteristics of the patients enrolled
in the present study were previously reported (5). A significantly lower O2
saturation in the arterial blood was observed in the cyanotic group
(74.3%) compared with the acyanotic group (98.0%; P<0.05). The
levels of hemoglobin (Hb) and hematocrit (Hct) in the arterial
blood were higher in the cyanotic group compared with the acyanotic
group (Hb, 171, vs. 123 g/l; Hct, 54.1, vs. 40.3%; P<0.05).
Other factors, including gender, age and body weight, were similar
between the groups. The present study revealed that the expression
levels of miR-146b were 40% higher in cyanotic hearts compared with
acyanotic hearts (P<0.05; Fig.
1A).
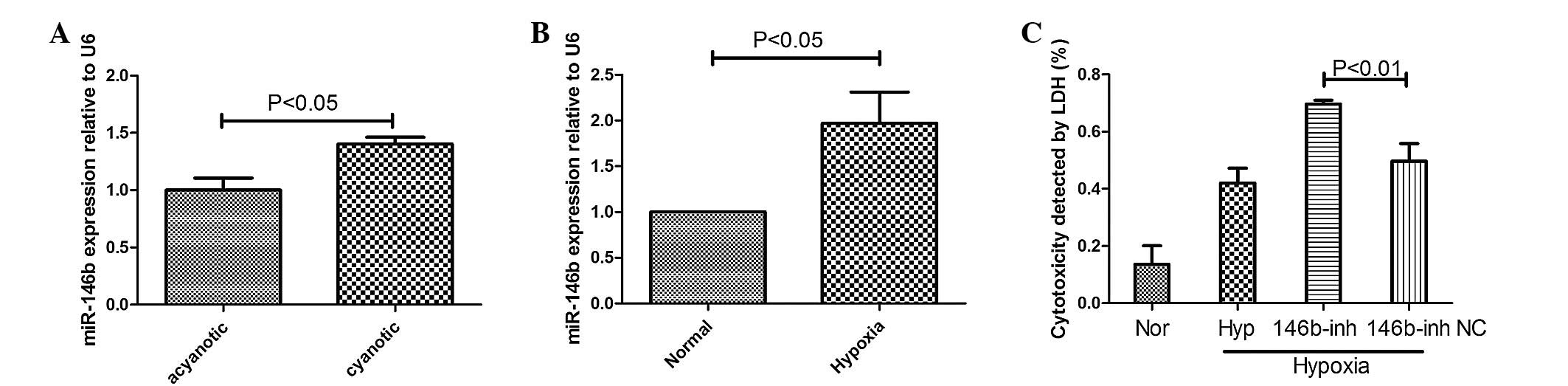 | Figure 1Expression of miR-146b in heart
samples from patients with CHD and hypoxic cardiomyocytes. (A)
Reverse transcription-polymerase chain reaction revealed that the
expression of miR-146b was significantly upregulated in the heart
tissue of patients with cyanotic CHD compared with acyanotic CHD
(P<0.05). (B) The H9c2 cells were cultured in 1% O2
for 72 h and the expression of miR-146b increased when the cells
were exposed to hypoxic conditions (P<0.05). (C) Cell mortality
was determined by measuring the release of LDH under normoxic or
hypoxic conditions after 72 h, and with or without addition of a
146b-inh. The data are expressed as the mean ± standard deviation
(P<0.01, 146b-inhibitor-treated cells compared with the NC). NC,
normal control; Nor, normal; Hyp, hypoxia; inh, inhibitor; LDH,
lactate dehydrogenase; miR, microRNA; 146b-inh, inhibitor of
miR-146b; CHD, congenital heart disease. |
Expression of miR-146b increases in
cardiomyocytes under hypoxic conditions
The H9c2 cells were cultured in 1% O2 for
72 h. The expression of miR-146b was assessed by RT-qPCR. The
expression of miR-146b was significantly upregulated 0.97-fold
compared with that of the normal group (P<0.05) at 72 h
(Fig. 1B).
Effect of miR-146b on cell apoptosis
under hypoxic conditions
The induction of hypoxia increased the levels of LDH
in the culture medium, and the downregulation of miR-146b resulted
in a further increase of LDH (Fig.
1C). In order to confirm whether miR-146b was involved in
hypoxia-induced apoptosis, several methods were used. Flow
cytometric analysis revealed a higher level of cell apoptosis at 72
h following the induction of hypoxia, and transfection with the
miR-146b inhibitor increased the apoptosis rate (17.3, vs. 7.3%
under hypoxic conditions; P<0.05; Fig. 2A). To further assess the effect of
miR-146b, TUNEL was performed. As shown in Fig. 2B, the apoptotic index was
significantly increased to 9.75±1.3% following the induction of
hypoxia (P<0.05, compared with the NC). Transfection with the
miR-146b inhibitor further increased the apoptotic rate
(16.00±1.2%; P<0.05). Nuclear morphological changes were
observed by Hoechst 33258 staining. A larger number of abnormal
nuclei were detected in the inhibitor group compared with the NC
group at 72 h following hypoxia induction (Fig. 2C). JC-1 staining was performed to
determine the proportion of cells with a normal ΔΨm. A
decreased ΔΨm was observed following transfection with
the miR-146b inhibitor, as revealed by a larger number of cells
yielding only a green emission upon excitation (Fig. 2D).
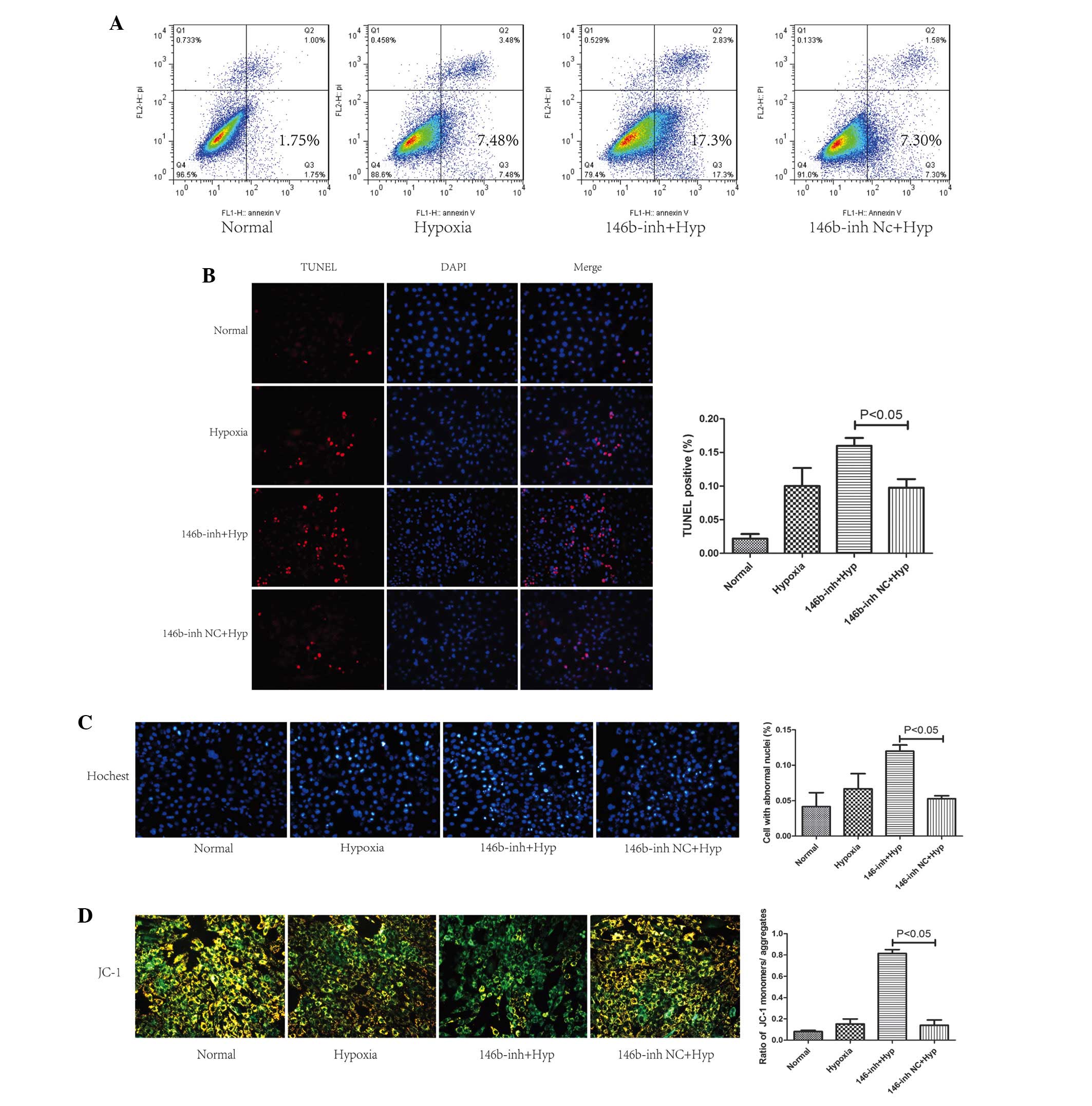 | Figure 2Addition of the miR-146b inhibitor
augments the hypoxia-induced apoptosis observed in the H9c2 cells.
Following exposure to hypoxia for 72 h, cell apoptosis was detected
by (A) annexin V/PI flow cytometric analysis, (B) TUNEL, (C)
Hoechst 33258 and (D) JC-1 staining. Each value was counted in five
random fields. The data are expressed as the mean ± standard
deviation (P<0.05, 146b-inh-treated cells compared with the NC
under hypoxic conditions). NC, normal control; Hyp, hypoxia; PI,
propidium iodide; TUNEL, terminal deoxynucleotidyl transferase dUTP
nick-end labeling; 146b-inh, miR-146b inhibitor; miR, microRNA. |
The expression levels of apoptosis-associated
proteins were measured by western blotting. The Bcl-2/Bax ratio was
decreased in the hypoxia model and was further decreased upon
transfection with the miR-146b inhibitor compared with that of the
control group (Fig. 3A–D).
Transfecting the cells with the miR-146b inhibitor resulted in a
significant increase in the levels of cleaved-caspase 3 (Fig. 3A and E) and Bax (Fig. 3A and C), and a decrease in the
level of Bcl-2 (Fig. 3A and B),
compared with that in the inhibitor-NC group (P<0.05). These
findings indicated that the addition of miR-146b increases the
extent of apoptosis in the H9c2 cells under hypoxic conditions.
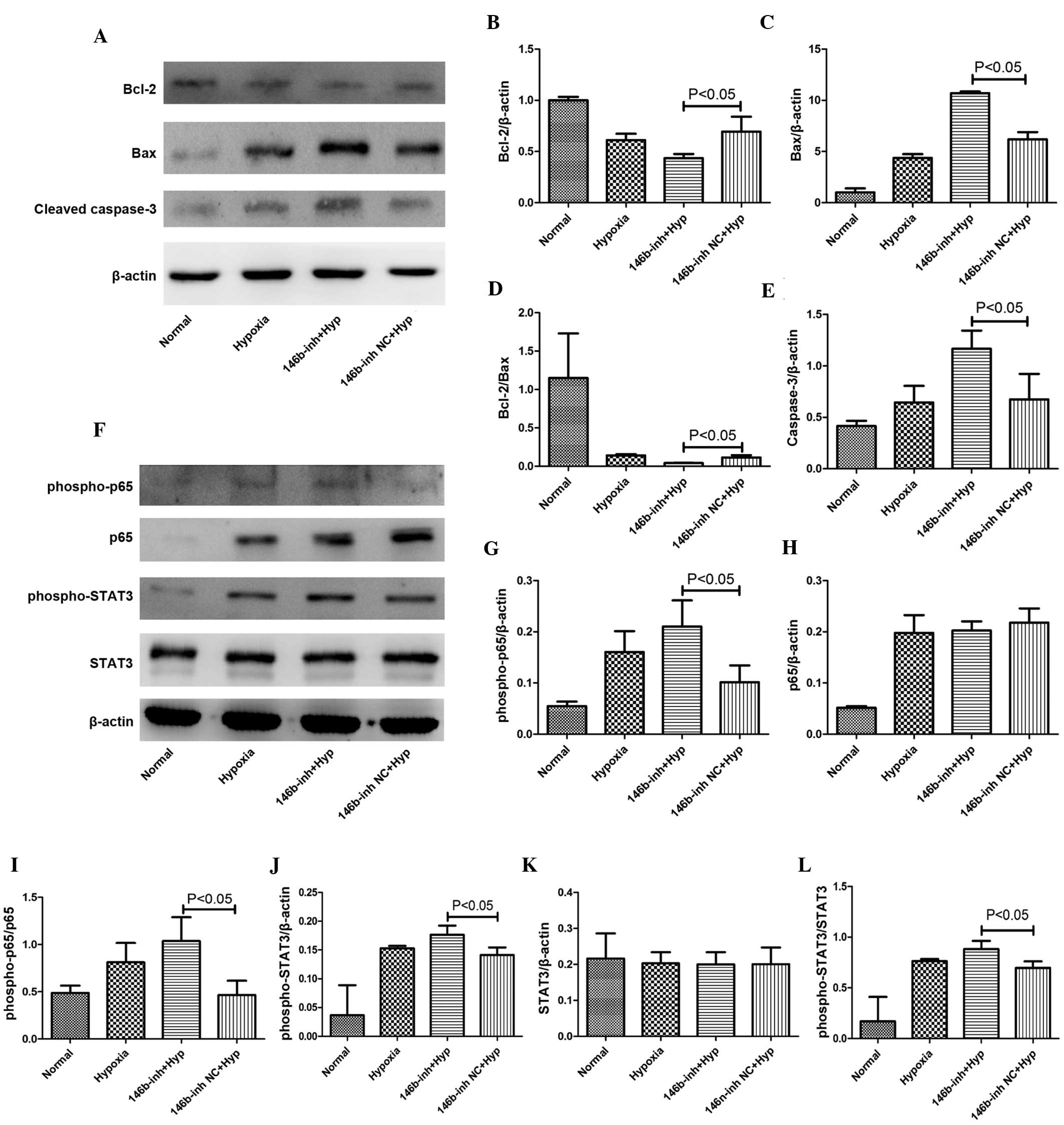 | Figure 3Addition of the miR-146b inhibitor
causes changes in the levels of apoptosis-associated proteins and
the nuclear factor-κB/interleukin-6 pathway. (A) A representative
western blot is shown for the Bcl-2, Bax and cleaved-caspase 3
proteins under conditions of normoxia and hypoxia at 72 h following
transfection with the miR-146b inhibitor. The miR-146b inhibitor
(B) markedly reduced the expression of Bcl-2, whereas the
expression of (C) Bax, (D) the Bcl-2/Bax ratio and (E)
cleaved-caspase 3 were increased. (F) Western blot assays were
performed for the p65, phospho-p65, STAT3 and phospho-STAT3
proteins under conditions of normoxia and hypoxia at 72 h following
transfection with the miR-146b inhibitor or with NC. The addition
of the 146b-inh (G) significantly increased the expression of
phospho-p65, although (H) no significant change in the level of p65
was observed. The levels of (I) phospho-p65/p65 and (J)
phospho-STAT3 were significantly increased by the 146b-inh, whereas
(K) the level of STAT3 was not significantly altered, although (L)
the level of phopsho-STAT3/STAT3 was significantly increased. The
values are expressed as the mean ±standard deviation (P<0.05,
146b-inhib -treated cells compared with the NC under hypoxic
conditions). NC, normal control; inh, inhibitor; Hyp,
hypoxia;phospho, phosphorylated; STAT3, signal transducer and
activator of transcription 3; 146b-inhib, miR-146b inhibitor; miR,
microRNA. |
Inhibition mediated by miR-146b increases
the activation of NF-κB and STAT3
miR-146b has been widely recognized to attenuate the
activation of the NF-κB signaling pathway. Newly emerging data have
revealed a connection between STAT3 and miR-146b (11). The present study revealed that the
miR-146b inhibitor failed to cause any significant change in the
protein levels of p65 and STAT3 at 72 h following the induction of
hypoxia, although the levels of p-p65 and p-STAT3 were increased
compared with the NC group (P<0.05; Fig. 3F–L).
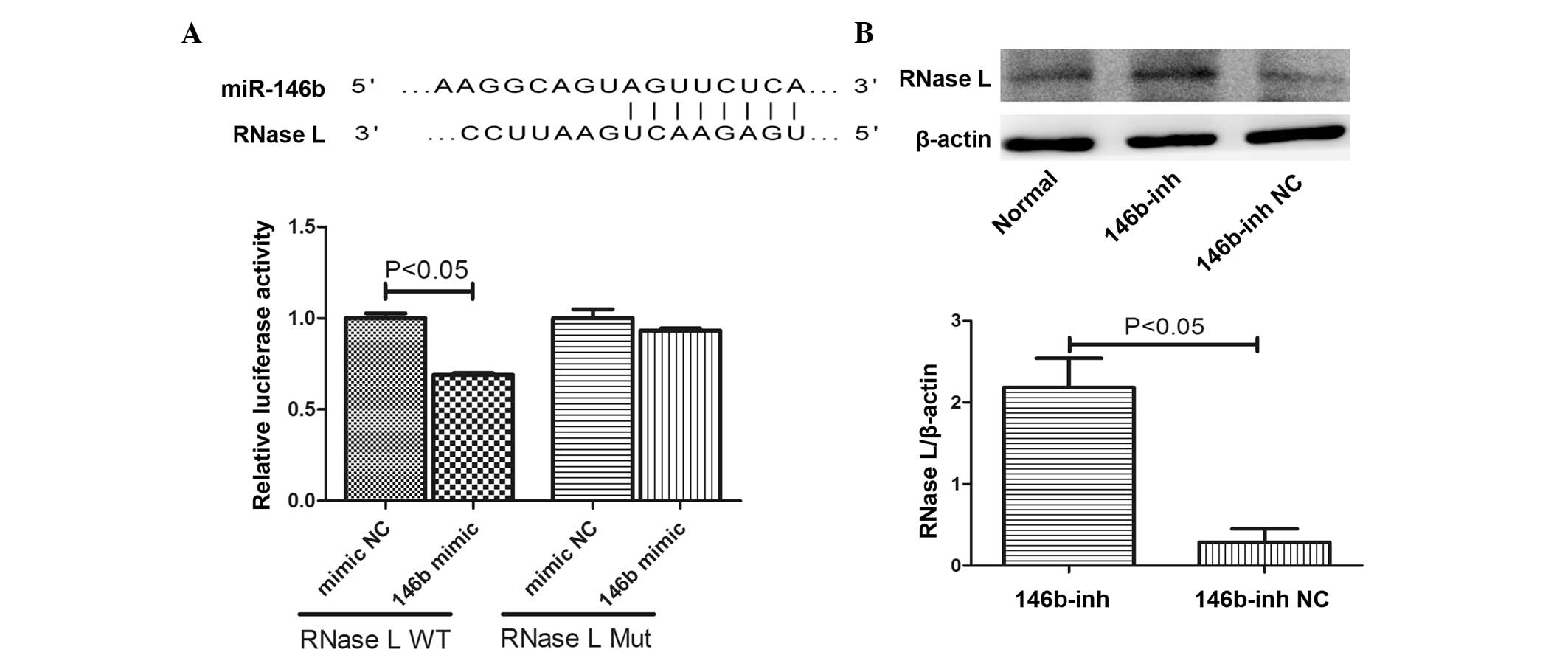
RNase L is a target of miR-146b
A bioinformatics analysis using TargetScan predicted
that RNase L is a target of miR-146b, as the 3′-UTR of the RNase L
mRNA contains a theoretical miR-146b binding site (Fig. 4A). To determine whether miR-146b
regulates the expression of RNase L, the 3′-UTR of wild-type or
mutated RNase L was cloned into the pmiR-RB-Report vector to
generate a luciferase reporter. The miR-146b mimic was subsequently
co-transfected with the vector into HEK293T cells and a luciferase
activity assay was performed following a further 48 h incubation.
Compared with the control, the miR-146b mimic significantly reduced
the normalized luciferase activity (P<0.05; Fig. 4B). Furthermore, the protein
expression level of RNase L was increased by the miR-146b
inhibitor, suggesting that RNase L is a target of miR-146b by
directly binding to its 3′-UTR (Fig.
4B).
Discussion
Hypoxia causes apoptosis, and a linear association
exists between the severity of hypoxic-ischemic cardiomyocyte
injury and the number of apoptotic cells (12). In the present study, an increase in
the expression of miR-146b was observed in tissue samples of
cyanotic CHD and hypoxic H9C2 cells, and the inhibition mediated by
miR-146b was revealed to diminish the ability of the H9c2 cells to
adapt to chronic hypoxia.
miR-146b differs from miR-146a by having two
different nucleotides at its 3′-end. Although the two miRNAs have
similar functions, previous studies revealed that the miR-146
isoforms do exhibit certain specific differences in terms of their
expression and biological action (10,13).
miR-146a/b repressed the activation of the NF-κB pathway by
directly targeting IRAK-1, TRAF6 and MyD88 (10). A sustained activation of NF-κB was
revealed to be proapoptotic in H9c2 cardiomyocytes (14). Therefore, it was hypothesized that
inhibition of the NF-κB pathway caused by upregulated levels of
miR-146b may reduce hypoxia-induced cardiomyocyte apoptosis.
Furthermore, miR-146a/b targets multiple genes, which are harmful
to cardiomyocytes under hypoxic conditions. miR-146a/b targets
human antigen R, which suppresses the expression of endothelial
nitric oxide synthase (eNOS) (15). Increasing the levels of eNOS
inhibited hypoxia-induced apoptosis, therefore, leading to a lower
rate of apoptosis (16).
miR-146a/b targets the endothelial growth factor receptor (17), which may increase hypoxia-induced
cardiomyocyte injury. miR-146a was previously identified as having
a protective role in cardiac I/R injury. It was demonstrated that,
although the expression of miR-146a does not notably increase
following I/R injury, the overexpression of miR-146a may
significantly reduce the I/R injury, which is partly caused by the
attenuation of both the activation of NF-κB and inflammatory
cytokine production by suppressing IRAK1 and TRAF6 (18). miR-146 is a therapeutic target and
biomarker for peripartum cardio-myopathy (19). In the present study, miR-146b was
revealed to protect cardiomyocytes from hypoxia-induced
apoptosis.
A previous report revealed that miR-146b exerts a
critical role in the feedback communication between NF-κB and the
interleukin-6 (IL-6) signaling axis (20). miR-146b acts as a feedback
regulator of this pathway, leading to the inhibition of NF-κB
(11). A previous study in our
laboratory reported that the expression levels of p65, p-p65 and
p-STAT3 were increased following the onset of chronic hypoxia in
cardiomyocytes (21), findings
which are corroborated by the results in the present study. STAT3
exerts a cardioprotective role in the ischemic heart (22), and miR-146b is a target gene of
STAT3 (11). The upregulation of
miR-146b may finely adjust the activation of NF-κB in the IL-6
signaling axis, in addition to other target genes, thereby acting
as a protective regulator in chronic hypoxia.
RNase L is a member of the 2-5A system, and it
belongs to the Mx family. RNase L may induce apoptosis in a
caspase-dependent manner. Furthermore, RNase L may change the
ΔΨm and elicit the generation of reactive oxygen species
(23). RNase L-deficient cells are
highly resistant to apoptosis, which is partly mediated through the
c-Jun N-terminal kinase/c-jun pathway (24). RNase L may also affect the NF-κB
signaling pathways by indirectly affecting the IκB kinases-α and -ε
(25). As revealed in the
Tissue-specific Gene Expression and Regulation database, the mRNA
expression levels of RNase L are relatively high in heart tissue,
suggesting that it may fulfil an important role in
pathophysiological processes of the heart.
The primary role of the Bcl-2 protein family is to
regulate cell apoptosis. In this family, Bax is proapoptotic,
whereas Bcl-2 is antiapoptotic (26). The ratio of Bcl-2/Bax acts as an
indicator of the mortality or survival of the cells, following an
apoptotic stimulus (27). The
caspase protein family are essential mediators of apoptosis. Among
the family members, caspase 3 is the key protease, which is
involved in the final execution phase of apoptosis (28). It was previously reported that the
Bcl-2 family of proteins and caspase-3 are involved in the RNase
L-mediated apoptotic pathway. The activation of RNase L led to the
release of cytochrome c from the mitochondria, and adding a
caspase 3 inhibitor or overexpressing Bcl-2 suppressed the RNase
L-mediated apoptosis (29). The
present study revealed that the inhibition mediated by miR-146b
regulated these apoptosis-associated proteins, causing an increased
expression of cleaved-caspase 3 and Bax and a decreased expression
of Bcl-2 and Bcl-2/Bax, suggesting that miR-146b may exerts its
influence by targeting RNase L.
Several questions remain unanswered by the present
study. Only the downregulation, and not the upregulation, of
miR-146b was investigated in this study. The expression levels of
routine miR-146 targets, including IRAK1 and TRAF6, were not
investigated, since their roles under hypoxic conditions are well
established.
Taken together, the present study indicated that the
inhibition of miR-146b increased hypoxia-induced apoptosis, which
may be partly caused by its target, RNase L. miR-146b has a
protective role in cardiac chronic hypoxia.
Acknowledgments
This study was supported by the National Science
Foundation of China (no. 81270228) The authors would like to thank
Miss Fu-Qing Tang for her technical assistance. (Department of
Cardiovascular Surgery, PLA, Xinqiao Hospital, Third Military
Medical University)
References
|
1
|
Jian Z, Li JB, Ma RY, Chen L, Zhong QJ,
Wang XF, Wang W, Hong Y and Xiao YB: Increase of macrophage
migration inhibitory factor (MIF) expression in cardiomyocytes
during chronic hypoxia. Clin Chim Acta. 405:132–138. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Waskova-Arnostova P, Kasparova D,
Elsnicova B, Novotny J, Neckar J, Kolar F and Zurmanova J: Chronic
hypoxia enhances expression and activity of mitochondrial creatine
kinase and hexokinase in the rat ventricular myocardium. Cell
Physiol Biochem. 33:310–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausser J and Zavolan M: Identification
and consequences of miRNA-target interactions - beyond repression
of gene expression. Nat Rev Genet. 15:599–612. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Condorelli G, Latronico MV and Cavarretta
E: microRNAs in cardiovascular diseases: Current knowledge and the
road ahead. J Am Coll Cardiol. 63:2177–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z
and Xiao Y: miR-138 protects cardiomyocytes from hypoxia-induced
apoptosis via MLK3/JNK/c-jun pathway. Biochem Biophys Res Commun.
441:763–769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang J, Chang JJ, Xu F, Ma XJ, Wu Y, Li
WC, Wang HJ, Huang GY and Ma D: MicroRNA deregulation in right
ventricular outflow tract myocardium in nonsyndromic tetralogy of
fallot. Can J Cardiol. 29:1695–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan SY, Zhang YY, Hemann C, Mahoney CE,
Zweier JL and Loscalzo J: MicroRNA-210 controls mitochondrial
metabolism during hypoxia by repressing the iron-sulfur cluster
assembly proteins ISCU1/2. Cell Metab. 10:273–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang D, Liu YL, Lü XD, Ling F, Liu AJ, Du
J and Han L: Lung microRNA profile in chronic cyanotic piglets with
decreased pulmonary blood flow. Chin Med J (Engl). 126:2260–2264.
2013.
|
|
9
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Curtale G, Mirolo M, Renzi TA, Rossato M,
Bazzoni F and Locati M: Negative regulation of Toll-like receptor 4
signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci USA.
110:11499–11504. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiang M, Birkbak NJ, Vafaizadeh V, Walker
SR, Yeh JE, Liu S, Kroll Y, Boldin M, Taganov K, Groner B, et al:
STAT3 induction of miR-146b forms a feedback loop to inhibit the
NF-kappaB to IL-6 signaling axis and STAT3-driven cancer
phenotypes. Sci Signal. 7:ra112014. View Article : Google Scholar
|
|
12
|
Mehmet H and Edwards AD: Hypoxia,
ischaemia, and apoptosis. Arch Dis Child Fetal Neonatal Ed.
75:F73–F75. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perry MM, Williams AE, Tsitsiou E,
Larner-Svensson HM and Lindsay MA: Divergent intracellular pathways
regulate interleukin-1beta-induced miR-146a and miR-146b expression
and chemokine release in human alveolar epithelial cells. FEBS
Lett. 583:3349–3355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hamid T, Gu Y, Ortines RV, Bhattacharya C,
Wang G, Xuan YT and Prabhu SD: Divergent tumor necrosis factor
receptor-related remodeling responses in heart failure: Role of
nuclear factor-kappaB and inflammatory activation. Circulation.
119:1386–1397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng HS, Sivachandran N, Lau A, Boudreau
E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI and Fish JE:
MicroRNA-146 represses endothelial activation by inhibiting
pro-inflammatory pathways. EMBO Mol Med. 5:949–966. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Zuo X, Wang Q, Boudreau E, Zhao
JL, Baltimore D, Delgado-Olguin P, Cybulsky MI and Fish JE:
Nicorandil inhibits hypoxia-induced apoptosis in human pulmonary
artery endothelial cells through activation of mitoKATP and
regulation of eNOS and the NF-kappaB pathway. Int J Mol Med.
32:187–194. 2013.PubMed/NCBI
|
|
17
|
Hurst DR, Edmonds MD, Scott GK, Benz CC,
Vaidya KS and Welch DR: Breast cancer metastasis suppressor 1
up-regulates miR-146, which suppresses breast cancer metastasis.
Cancer Res. 69:1279–1283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang X, Ha T, Liu L, Zou J, Zhang X,
Kalbfleisch J, Gao X, Williams D and Li C: Increased expression of
microRNA-146a decreases myocardial ischaemia/reperfusion injury.
Cardiovasc Res. 97:432–442. 2013. View Article : Google Scholar :
|
|
19
|
Halkein J, Tabruyn SP, Ricke-Hoch M,
Haghikia A, Nguyen NQ, Scherr M, Castermans K, Malvaux L, Lambert
V, Thiry M, et al: MicroRNA-146a is a therapeutic target and
biomarker for peripartum cardiomyopathy. J Clin Invest.
123:2143–2154. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Walker SR, Xiang M and Frank DA: STAT3
Activity and Function in Cancer: Modulation by STAT5 and miR-146b.
Cancers (Basel). 6:958–968. 2014. View Article : Google Scholar
|
|
21
|
Gu Q, Kong Y, Yu ZB, Bai L and Xiao YB:
Hypoxia-induced SOCS3 is limiting STAT3 phosphorylation and NF-κB
activation in congenital heart disease. Biochimie. 93:909–920.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hilfiker-Kleiner D, Hilfiker A, Fuchs M,
Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z,
Podewski E, et al: Signal transducer and activator of transcription
3 is required for myocardial capillary growth, control of
interstitial matrix deposition, and heart protection from ischemic
injury. Circ Res. 95:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Domingo-Gil E and Esteban M: Role of
mitochondria in apoptosis induced by the 2-5A system and mechanisms
involved. Apoptosis. 11:725–738. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malathi K, Paranjape JM, Ganapathi R and
Silverman RH: HPC1/RNASEL mediates apoptosis of prostate cancer
cells treated with 2′,5′-oligoadenylates, topoisomerase I
inhibitors, and tumor necrosis factor-related apoptosis-inducing
ligand. Cancer Res. 64:9144–9151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chakrabarti A, Jha BK and Silverman RH:
New insights into the role of RNase L in innate immunity. J
Interferon Cytokine Res. 31:49–57. 211PubMed/NCBI
|
|
26
|
Siddiqui WA, Ahad A and Ahsan H: The
mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update.
Arch Toxicol. 89:289–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: Bcl-2/Bax: A rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
28
|
Shalini S, Dorstyn L, Dawar S and Kumar S:
Old, new and emerging functions of caspases. Cell Death Differ.
22:526–539. 2015. View Article : Google Scholar
|
|
29
|
Li G, Xiang Y, Sabapathy K and Silverman
RH: An apoptotic signaling pathway in the interferon antiviral
response mediated by RNase L and c-Jun NH2-terminal kinase. J Biol
Chem. 279:1123–1131. 2004. View Article : Google Scholar
|