Introduction
Inflammatory bowel diseases (IBD), including
ulcerative colitis and Crohn's disease, are intractable disorders
of unknown etiology, which cause chronic inflammation of the
gastrointestinal tract (1). A
number of immune, genetic and environmental factors are known to
affect the onset and progression of colitis (2,3).
Several studies have suggested that the innate immune system, which
includes macrophages and dendritic cells, is involved in the
pathogenesis of colitis (4–6).
Macrophages are often pro-inflammatory; infiltrating
the injured mucosa and regulating the production of various
inflammatory cytokines, including interleukin (IL)-1β, IL-6, and
tumor necrosis factor (TNF)-α, as well as the neutrophil
chemoattractant, IL-8. Levels of these cytokines are elevated in
IBD (7,8) and animal models of colitis (9,10).
In addition, abnormally differentiated subsets of intestinal
macrophages are key in T-helper (Th)1 cell-dominant chronic
colitis, owing to elevated production of IL-12 and IL-23 (11). Consistent with thys, depletion of
macrophages and dendritic cells has been observed to ameliorate
disease symptoms in an IL-10 knockout mouse model of colitis
(12). Previous studies have also
demonstrated that alternatively activated M2 macrophages can
ameliorate experimental colitis through increased production of
IL-10 (13), and blood monocytes
can differentiate into either anti-inflammatory macrophages or
inflammatory dendritic cells in the colon, based on their
microenvironment (14). Therefore,
macrophages can adopt various phenotypes depending on the local
environment (15); however, the
precise mechanism underlying colitis remains to be fully
elucidated.
Originally described in melanoma cells, glycoprotein
nonmetastatic melanoma protein B (Gpnmb), also known as
osteoactivin and DC-HIL, is a heavily N-glycosylated type I
transmembrane domain protein, with a short cytoplasmic domain
containing an endosomal-sorting motif (16–18).
The gene encoding Gpnmb was isolated in our previous study, and was
differentially expressed in the livers of rats fed a
choline-deficient, L-amino acid-defined diet (19). In addition, our previous
investigations demonstrated that transgenic Gpnmb expression
attenuates the development of hepatic fibrosis (20). Previous studies have demonstrated
that the expression of Gpnmb in macrophages functions as a feedback
regulator of pro-inflammatory responses (21), and that the binding of Gpnmb on
antigen-presenting cells to syndecan-4 on activated T cells
inhibits T-cell activation (22).
Thus, the expression of Gpnmb on antigen-presenting cells may
negatively regulate inflammation. However, the potential
pathophysiological roles of Gpnmb in various disorders, including
IBD, remain largely unknown.
The present study aimed to characterize
Gpnmb-positive and -negative macrophages, and investigate their
roles in injured colonic mucosa. In a murine model of experimental
colitis, the effects of macrophages expressing and lacking Gpnmb on
the injured colonic mucosa were examined. In addition, the effects
on the expression of pro-inflammatory cytokines were examined in
vivo and in vitro to determine whether Gpnmb-positive
macrophages contribute to the severity of experimental colitis in
mice.
Materials and methods
Animals
BALB/c mice (n=30; 7–9 weeks-old; male) were
obtained from Kyudo Co., Ltd. (Tosu, Japan). DBA/2J Gpnmb mutant
(D2) mice (7–9 weeks-old; 11 male and 14 female) and DBA/2J-gpnmb+
(D2-gpnmb+) mice (7–9 weeks-old, 11 male and 14 female), in which
the mutated Gpnmb allele was replaced with a wild-type allele
(23), were purchased from The
Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed in a
24°C-controlled environment with a 12 h light/dark cycle, and two
to three mice were housed together. The mice were allowed ad
libitum access to standard mouse chow and tap water. All animal
experiments were approved by the Institutional Animal Care and Use
Committee of Kagoshima University (Kagoshima, Japan).
Dextran sulfate sodium (DSS)-induced
colitis
To induce experimental colitis, the mice received 2
or 3% DSS (molecular weight, 50 kDa; Ensuiko Sugar Refining Co.,
Tokyo, Japan) in their drinking water for 7 or 5 days,
respectively. The control mice received sterile filter-purified
distilled water.
To evaluate the severity of colitis, the mice were
weighed daily, and disease activity index (DAI) scores were
calculated. DAI scores ranged between 0 and 4, based on weight
loss, stool consistency and the presence or absence of fecal blood
(24). All mice were sacrificed
with an overdose of sodium pentobarbital (65–95 mg/kg; Kyoritsu
Seiyaku Co., Ltd., Tokyo, Japan) on days 0, 2, 5, 7, 10, or 14,,
and the large intestines were removed between the ileocecal
junction and the anal verge. The large intestines were assessed for
weight and length, incised longitudinally and fixed in 10% buffered
formalin (Wako Pure Chemical Industries, Ltd., Osaka, Japan).
Intestinal inflammation was scored, as described previously
(25). Briefly, grade 0 was
afforded to normal colonic mucosa; grade 1 to colonic mucosa with
loss of one-third of the crypts; grade 2 to colonic mucosa with
loss of two-thirds of the crypts; grade 3 to colonic mucosa in
which the lamina propria was covered with a single layer of
epithelium and mild inflammatory cell infiltration was present; and
grade 4 for colonic mucosa presenting erosions and marked
inflammatory cell infiltration.
Cell culture
RAW264.7 murine macrophage cells were obtained from
the Human Science Research Resources Bank (Osaka, Japan) and
cultured (2×106 cells/ml) in Dulbecco's modified Eagle's
medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with
10% fetal bovine serum (FBS; Invitrogen Life Technologies,
Carlsbad, CA, USA). The RAW264.7 cells were treated with 100
µg/ml lipopolysaccharide (LPS; Sigma-Aldrich) or
phosphate-buffered saline (PBS) for 24 h at 37°C.
In order to inhibit the expression of Gpnmb in the
RAW264.7 cells, small interfering (si)RNA specific for Gpnmb
(5′-UUAGCAUCUUCCUUCUGGCAUCUGG-3′; Invitrogen Life Technologies) was
transfected into the cells using RNAiMAX (Invitrogen Life
Technologies). Stealth RNAi Negative Control Medium GC Duplex
(Invitrogen Life Technologies) was used as a negative control siRNA
(NC-siRNA). One day prior to transfection, the cells were plated in
500 µl DMEM without antibiotics. The cells were 30–50%
confluent at the point of transfection. The macrophages transfected
with siRNA were cultured in RPMI-1640 medium (Invitrogen Life
Technologies) supplemented with 10% FBS for 24 h at 37°C in a
CO2 incubator.
Isolation of thioglycollate-elicited
peritoneal macrophages (TEPMs)
The D2 and D2-gpnmb+ mice were intraperitone-ally
injected with 1 ml 10% thioglycollate (Sigma-Aldrich). The TEPMs
were harvested via peritoneal lavage with PBS 3 days
post-injection, and were cultured in RPMI-1640 medium supplemented
with 10% FBS. Following a 24 h incubation at 37°C, the medium was
replaced, and the TEPMs were treated with 10 µg/ml LPS or
PBS for an additional 24 h.
RNA extraction, and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was extracted from the cells or tissues
following homogenization using TRIzol® regent
(Invitrogen Life Technologies). Total RNA (0.5 µg) was
reverse transcribed using a PrimeScript™ RT Reagent kit (Takara Bio
Inc., Otsu, Japan). Each reaction mixture for RT (total volume 20
µl) consisted of total RNA (adjusted to 500 ng, 2.5
µl), oligo dT PCR primer (50 µM, 1 µl), 5X
PrimeScript buffer (4 µl), PrimeScript RT enzyme mix (1
µl), random 6 mers (100 µM, 1 µl) and RNase
free distilled H2O (10.5 µl). The synthesized
cDNA was amplified by RT-qPCR using SYBR Premix Ex Taq II
(Takara Bio Inc.). Each reaction mixture for PCR (total volume 20
µl) consisted of cDNA (adjusted to 500 ng, 2 µl),
forward and reverse PCR primer (10 µM, 0.8 µl each),
SYBR Premix Ex Taq II (10X, 10 µl), ROX reference dye (50X,
0.4 µl) and distilled H2O (6 µl). The
synthesized cDNA was quantified using an ABI prism 7700 Sequence
Detector (Applied Biosystems Life Technologies, Foster City, CA,
USA). The cycling conditions were as follows: One cycle at 95°C for
30 sec followed by 35 cycles each at 95°C for 5 sec and 60°C for 34
sec. The PCR primer sequences used are listed in Table I (Sigma-Aldrich). Gene expression
levels were normalized to those of β-actin, which was used as an
internal control.
 | Table IPolymerase chain reaction primer
sets. |
Table I
Polymerase chain reaction primer
sets.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Gpnmb |
ATCCCTGGCAAAGACCCAGAC |
CCTATTGGCTTGTACGCCTTGTG |
| IL-1β |
TCCAGGATGAGGACATGAGCAC |
GAACGTCACACACCAGCAGGTTA |
| IL-6 |
CCACTTCACAAGTCGGAGGCTTA |
GCAAGTGCATCATCGTTGTTCATAC |
| TNF-α |
AAGCCTGTAGCCCACGTCGTA |
GGCACCACTAGTTGGTTGTCTTTG |
| MCP-1 |
GCATCCACGTGTTGGCTCA |
CTCCAGCCTACTCATTGGGATCA |
| β-actin |
CATCCGTAAAGACCTCTATGCCAAC |
ATGGAGCCACCGATCCACA |
Western blot analysis
The RAW264.7 cells and TEPM from the D2 or D2-gpnmb+
were also washed PBS and removed from the dishes using a cell
scraper with 0.5% SDS (Promega Corporation, Madison, WI, USA). The
cell lysates were heated to 100°C for 5 min and cooled on ice.
Following centrifugation at 450 x g for 5 min at 20°C, the cell
lysates were analyzed for protein content using a Lowry Assay
(Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal quantities of
protein (1 µg/µl) were separated on 10% SDS-PAGE
(Wako Pure Chemical Industries, Ltd.) and transferred onto
polyvinylidene fluoride membranes (Bio-Rad Laboratories, Inc.).
Following blocking overnight at 4°C with 5% non-fat milk, the blots
were probed with primary antibodies for 1 h at room temperature.
The membranes were then incubated with the following primary
antibodies: Polyclonal goat anti-mouse Gpnmb (1:1,000; cat. no.
AF2330) purchased from R&D systems, Inc. (Minneapolis, MN,
USA). Polyclonal rabbit anti-mouse phosphorylated (p)-ERK1/2
(1:1,000, cat. no. 9101), ERK1/2 (1:1,000, cat. no. 3372),
phosphorylated c-jun N-terminal kinase (p-JNK; 1:1,000; cat. no.
9251), p-p mitogen activated kinase (MAPK; 1:1,000; cat. no. 4511),
p38 MAPL (1:1,000, cat. no. 9212) and IκB (1:1,000; cat. no. 9242),
as well as monoclonal rabbit anti-mouse JNK (1:1,000; cat. no.
9258) were purchased from Cell Signaling Technology Inc. (Danvers,
MA, USA). Monoclonal mouse anti-β-actin (1:1,000; cat. no. A5441)
was purchased from Sigma-Aldrich. The membranes were washed with
Tris-buffered saline (Bio-Rad Laboratories, Inc.) with Tween 20
(Wako Pure Chemical Industries) three times. Following incubation
of the membranes with the appropriate peroxidase-conjugated
secondary antibodies [polyclonal donkey anti-goat immunoglobulin
G-horseradish peroxidase (IgG-HRP), 1:2,000, cat. no. sc-2033;
polyclonal goat anti-rabbit IgG-HRP, 1:2,000, cat. no. sc-2004]
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) for
1 h at room temperature, the membranes were visualized using an
Enhanced Chemiluminescence Detection kit (cat. no. RPN2232 GE
Heathcare, Buckinghamshire, UK). Secondary antibodies were diluted
using Can Get Signal® (cat. no. NKB-301; Toyobo Co.,
Ltd., Osaka, Japan) Expression level quantification was performed
using Image J version 1.48 (National Institutes of Health,
Bethesda, MD, USA), and β-actin was used as an internal
control.
Immunohistochemistry
For immunohistochemical analysis, the colon tissues
were fixed in 10% buffered formalin and incubated with goat
anti-mouse Gpnmb antibodies (R&D Systems, Inc.) or rat
anti-mouse F4/80 antibodies (AbD Serotec, Kidlington, UK). For
immunofluorescence analysis, paraffin-embedded (Sigma-Aldrich) and
2 µm frozen tissue sections were incubated with goat
anti-mouse Gpnmb antibodies (R&D Systems, Inc.) or rat
anti-mouse CD68 antibodies (AbD Serotec), followed by imaging under
a fluorescence microscope (BZ-9000; Keyence Corporation, Osaka,
Japan).
ELISA
The concentrations of IL-1β, IL-6, TNF-α, MCP-1, and
IL-10 were measured in the culture supernatants using the following
cytokine ELISA kits; IL-β mouse ELISA (Invitrogen Life
Technologies), mouse IL-6 quantikine ELISA (R&D Systems, Inc.),
mouse IL-10 quantikine ELISA (R&D Systems, Inc.), mouse TNF-α
quantikine ELISA (R&D Systems, Inc.), and MCP-1 mouse ELISA
(Invitrogen Life Technologies). Samples were analyzed in duplicate
using a microplate reader model 680 (Bio-Rad Laboratories, Inc.).
The concentrations of cytokines in the samples were calculated
using standard curves. Protein concentrations in each sample were
determined using an RC DC protein assay (Bio-Rad Laboratories,
Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
software version 17.0 (SPSS, Inc., Chicago, IL, USA). Data were
analyzed using the Mann-Whitney U test or Tukey's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Gpnmb is expressed in macrophages
infiltrating into injured colonic mucosa
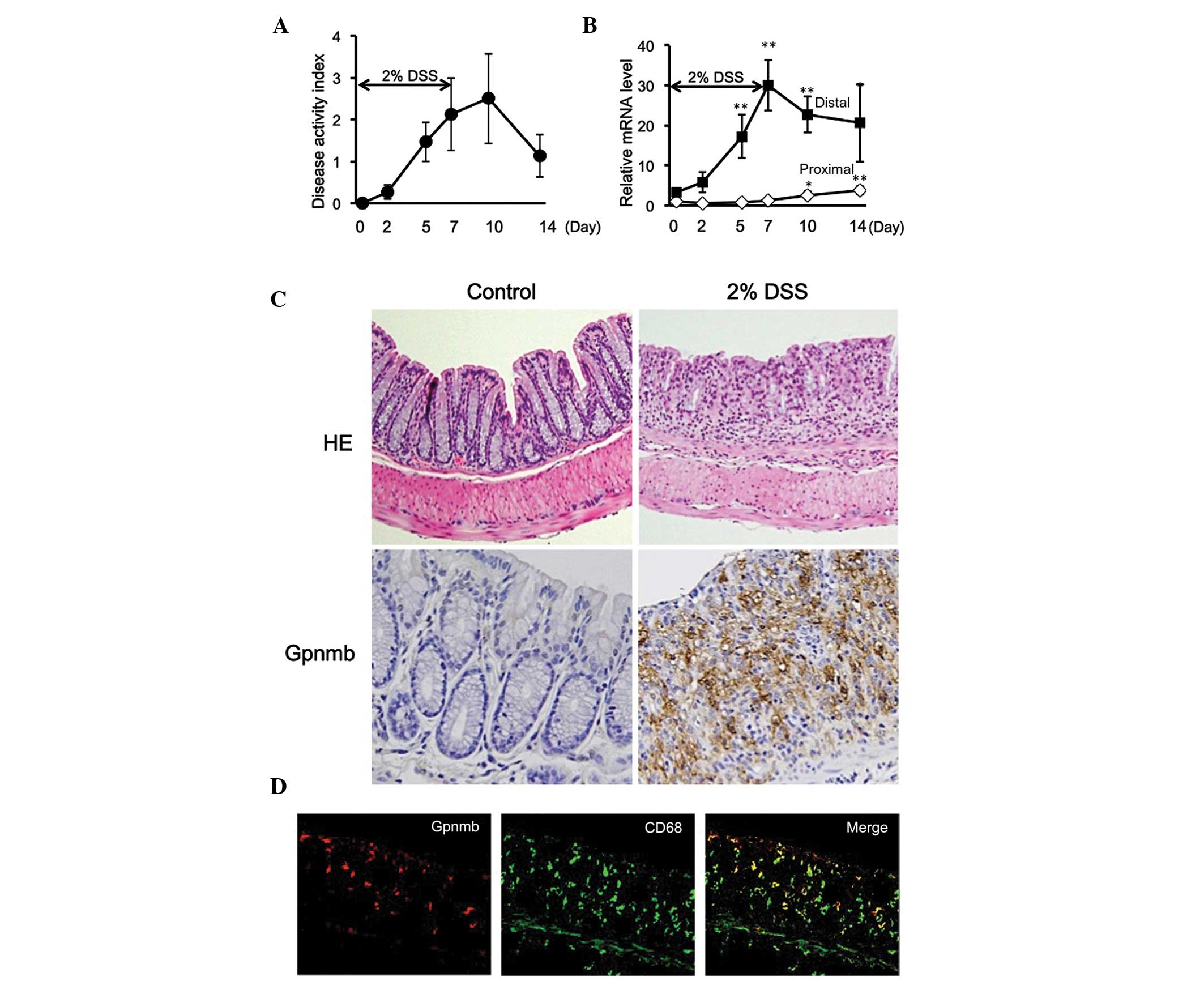
Following administration of the mice with 2% DSS for
7 days, DAI scores gradually increased until day 10 (3 days
following removal of DSS), and remained elevated above baseline
until day 14 (Fig. 1A). mRNA
expression levels of Gpnmb were increased in parallel with the
development of colitis in the distal large intestine (Fig. 1B), which was the primary site of
DSS-induced mucosal injury. The mRNA expression levels peaked on
day 7 and remained elevated even following the cessation of DSS
treatment (Fig. 1B). By contrast,
the mRNA expression levels of Gpnmb remained low in the proximal
large intestine throughout the experimental period (14 days;
Fig. 1B). Furthermore, protein
expression of Gpnmb was confirmed on day 7 using western blot
analysis, which was observed in the distal large intestine, but not
in the proximal large intestine (data not shown). Histologically,
the mice treated with 2% DSS exhibited destruction of epithelial
tissue, infiltration of inflammatory cells into the mucosa and
submucosa, edema and mucosal thickening by day 7 (Fig. 1C). Gpnmb was also observed in the
infiltrating inflammatory cells, but not in epithelial cells,
determined using immunohistochemistry (Fig. 1C). In order to detect the cells
positively expressing Gpnmb, double immunostaining was performed on
the samples. Gpnmb was expressed in a subset of cells positive for
the macrophage marker CD68 (Fig.
1D), and also in a subset of cells positive for F4/80 (data not
shown). These results indicated that Gpnmb is expressed in
macrophages infiltrating into the injured colonic mucosa.
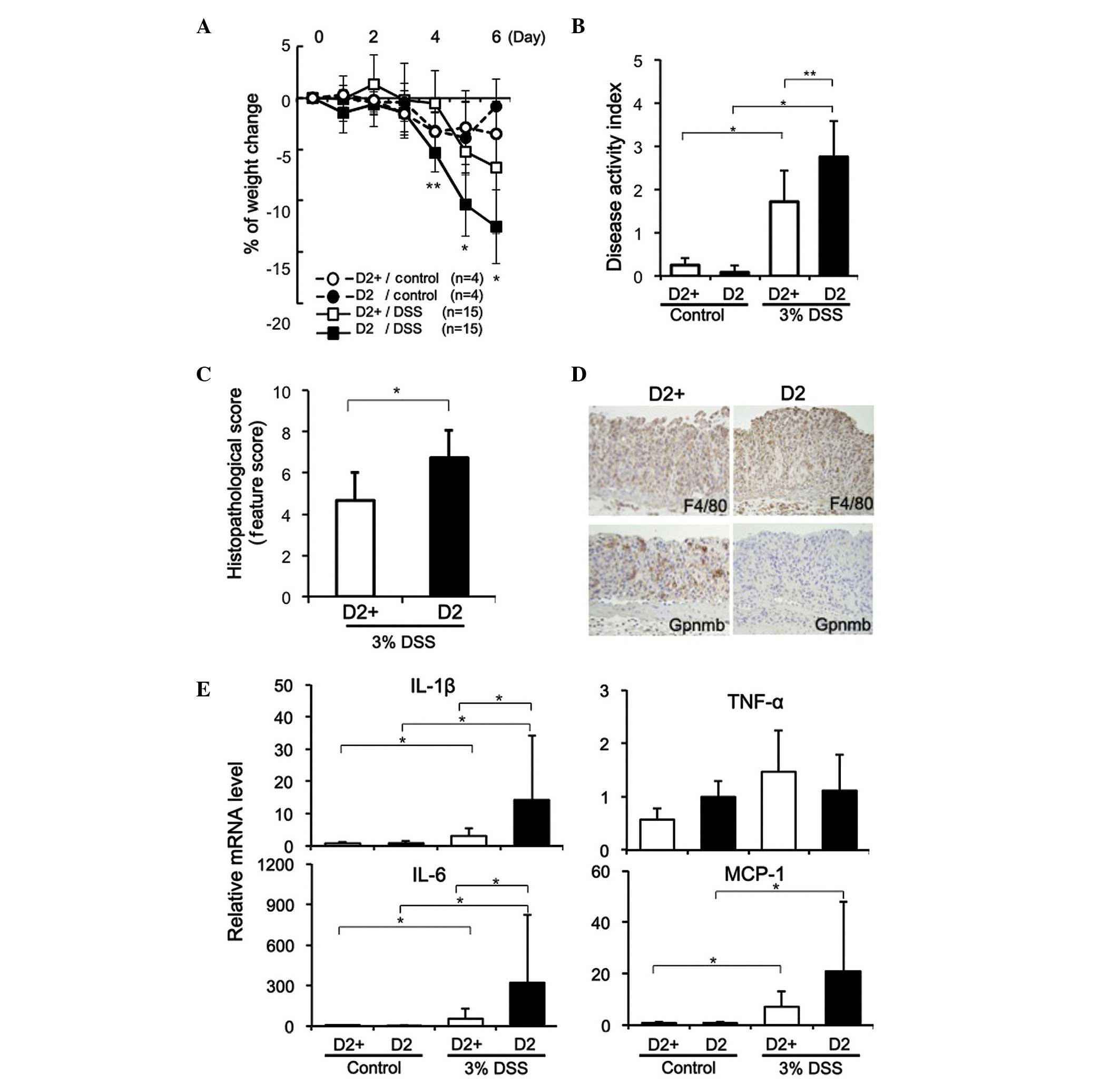
DSS-induced mucosal injury is exacerbated
in mice lacking Gpnmb
DSS (3%) was administered for 5 days to D2 mice,
which exhibit a naturally occurring mutation in Gpnmb (R150X), and
to D2-gpnmb+ mice, in which the mutated Gpnmb allele was replaced
with a wild-type allele (21). The
body weight of the D2 mice decreased significantly as colitis
developed, compared with the D2-gpnmb+ mice treated with DSS
(P<0.001 on day 4; P<0.01 on days 5 and 6), as shown in
Fig. 2A, and the D2 mice had
significantly higher DAI scores on day 5 (P<0.001; Fig. 2B). The colonic weight following
administration of DSS for 5 days was similar in the D2 and
D2-gpnmb+ mice; however, the colon lengths were significantly
shorter in the DSS-treated D2 mice, compared with the D2-gpnmb+
mice (7.1±0.8, vs. 8.0±1.1 cm; P<0.01). Histological analyses
revealed that colon tissues from the DSS-treated D2-gpnmb+ mice
were characterized by shorter crypts and mild inflammatory cell
infiltration, with no epithelial cell destruction. By contrast,
DSS-induced colitis in the D2 mice resulted in complete loss of
crypts, destruction of the epithelium and severe inflammatory cell
infiltration in the colon tissues (data not shown). In addition,
histopathological scoring revealed that DSS-induced colitis in the
D2 mice was significantly exacerbated, compared with that in the
D2-gpnmb+ mice (P<0.05; Fig.
2C).
The expression of Gpnmb was observed in the distal
colonic tissues from the DSS-treated D2-gpnmb+ mice (Fig. 2D), however, no Gpnmb was observed
in the colon tissues isolated from the DSS-treated D2 mice. In
addition, marked infiltration of F4/80-positive macrophages into
the injured colonic mucosa was observed in the D2-gpnmb+ and D2
mice, and a number of macrophages from the D2-gpnmb+ mice expressed
Gpnmb (Fig. 2D). As expected, the
expression of Gpnmb was not detected in macrophages infiltrating
the injured colonic mucosa of the D2 mice, regardless of whether
DSS was administered.
When the D2 and D2-gpnmb+ mice were treated with 3%
DSS for 5 days, the mRNA expression levels of IL-1β, IL-6, and
MCP-1 were significantly elevated, and significantly higher levels
of IL-1β and IL-6 were observed in the injured colon tissues from
the D2 mice, compared with in those from the D2-gpnmb+ mice
(Fig. 2E). The D2 mice treated
with 3% DSS also exhibited elevated expression levels of MCP-1 in
injured colon tissues; however, no significant difference was
observed in the expression of MCP-1 between the D2 and D2-gpnmb+
mice (P=0.075; Fig. 2E).
TEPMs lacking Gpnmb expression exhibit
enhanced expression of pro-inflammatory cytokines
The mRNA expression of Gpnmb in the TEPMs from the
D2 mice was low or undetectable, regardless of LPS stimulation;
whereas the mRNA expression of Gpnmb was reduced in the D2-gpnmb+
mice following LPS stimulation (Fig.
3A). In the absence of LPS, TEPMs from the D2-gpnmb+ and D2
mice expressed low mRNA levels of IL-1β, IL-6, TNF-α, MCP-1 and
IL-10. Conversely, LPS stimulated the expression of these mRNAs in
the TEPMs isolated from the D2-gpnmb+ and D2 mice, and the mRNA
expression levels of IL-1β, IL-6, TNF-α and MCP-1 were
significantly higher in the TEPMs from D2 mice, compared with those
from the D2-gpnmb+ mice. In addition, the expression of IL-10 was
markedly lower following LPS stimulation in the D2 mice, compared
with the D2-gpnmb+ mice, although this difference was not
statistically significant. The protein expression levels of IL-1β
and TNF-α were significantly higher, and the expression of IL-10
was significantly lower in the supernatants of TEPMs from the D2
mice, compared with those from the D2-gpnmb+ mice (Fig. 3B). These results suggested that the
inhibition of inflammatory cytokines and the stimulation of
anti-inflammatory cytokines by Gpnmb in macrophages may reduce
inflammation in the injured colonic mucosa.
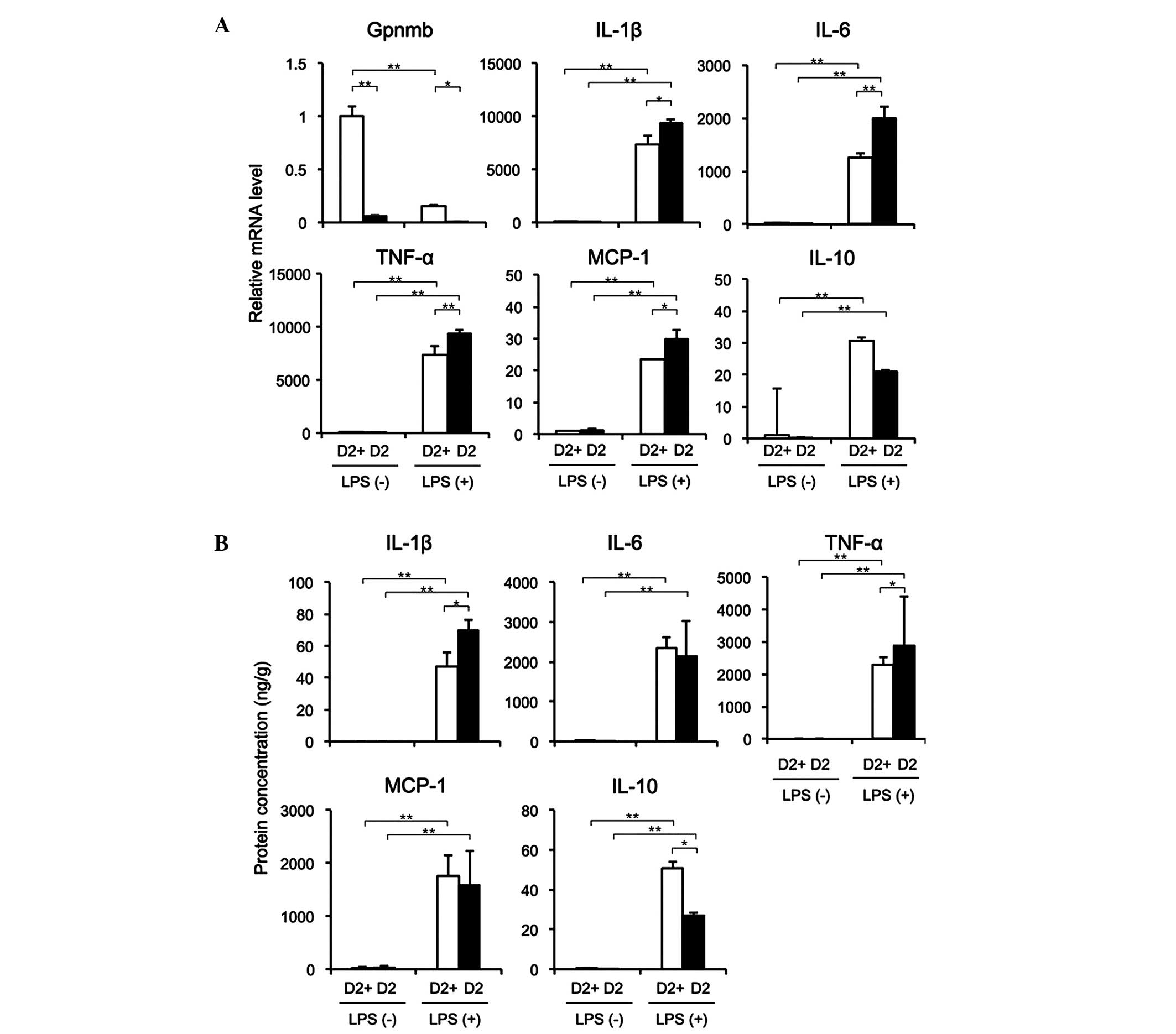 | Figure 3TEPMs lacking Gpnmb expression
exhibit enhanced expression of pro-inflammatory cytokines. TEPMs
were isolated from D2 and D2-gpnmb+ (D2+) mice. (A) mRNA expression
levels of Gpnmb, IL-1β, IL-6, TNF-α, MCP-1 and IL-10 in the absence
or presence of LPS were detected using reverse
transcription-quantitative polymerase chain reaction. mRNA
expression levels in TEPMs isolated from D2+ mice without LPS
treatment were set as 1. (B) Protein levels of IL-1β, IL-6, TNF-α,
MCP-1 and IL-10 in the supernatants of TEPMs in the absence or
presence of LPS, examined using ELISA. The protein levels were
adjusted by the total protein concentration in the supernatant.
Data are presented as the mean ± standard deviation (n=3).
*P<0.01; **P<0.001. TEMPs,
thioglycollate-elicited peritoneal macrophages; Gpnmb, glycoprotein
nonmetastatic melanoma protein B; DSS, dextran sulfate sodium; IL,
interleukin; TNF, tumor necrosis factor; MCP, monocyte
chemoattractant protein; LPS, lipopolysaccharide. |
Expression of Gpnmb in response to LPS,
and its effects on cytokine and chemokine production in murine
macrophages
The siRNA-mediated knockdown of Gpnmb in RAW264.7
macrophages in the present study was effective. The Gpnmb-specific
siRNA (Gp-siRNA) reduced the mRNA and protein expression levels of
Gpnmb to ~1/3 of the levels observed in the cells transfected with
NC-siRNA (Fig. 4A and B).
Furthermore, inhibition of the expression of Gpnmb led to
significant increases in the expression levels of IL-1β, IL-6,
TNF-α and MCP-1 (Fig. 4C). In
addition, when the expression of Gpnmb was knocked down by
Gp-siRNA, p38 and ERK1/2 were phosphorylated at significantly
higher levels; whereas the phosphorylation of JNK and the
expression of IκB were not significantly affected (Fig. 4C). These results indicated that
Gpnmb negatively regulated the production of pro-inflammatory
cytokines and chemokines by inhibiting signaling pathways involving
p38 and ERK1/2.
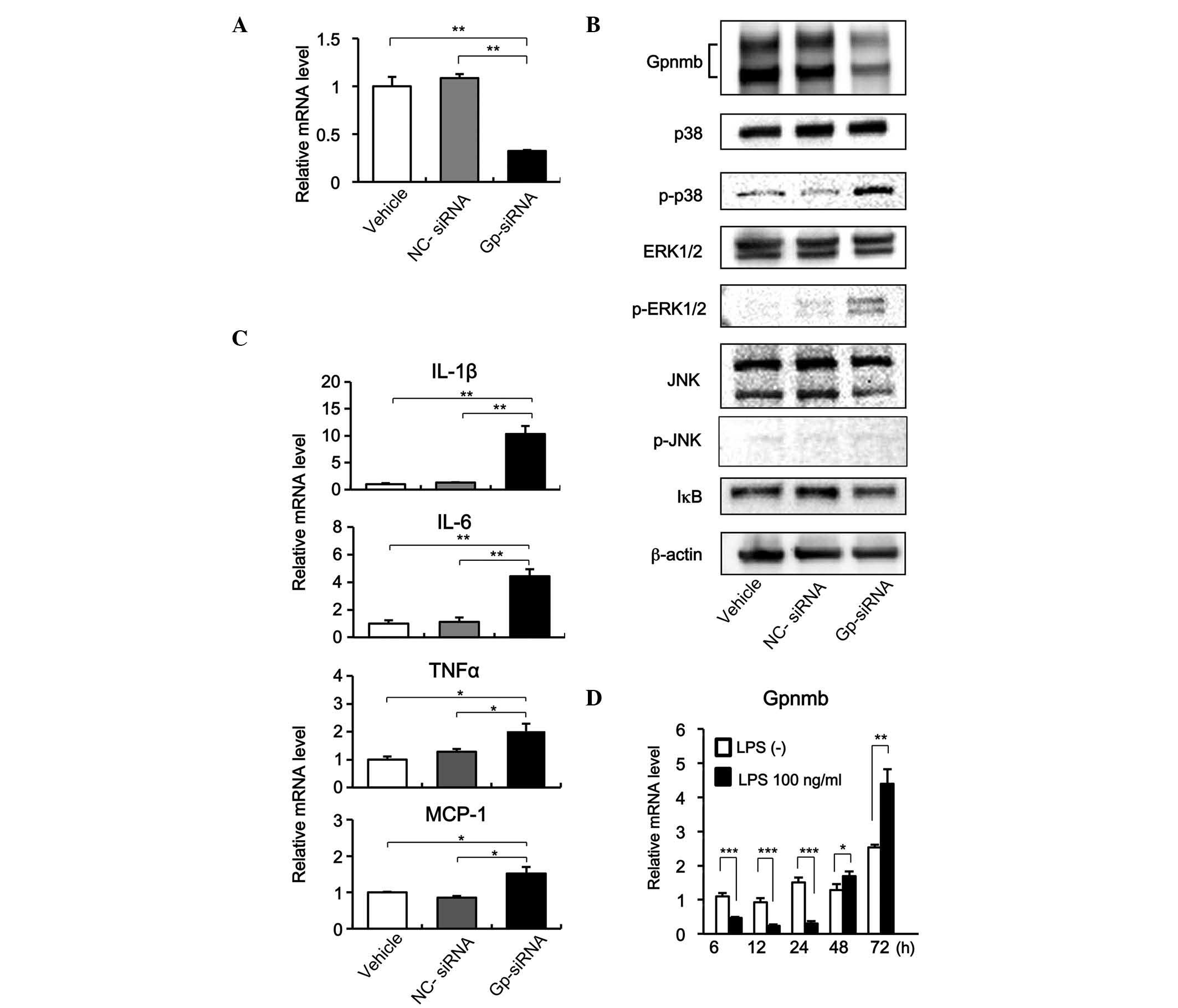 | Figure 4Reduced Gpnmb expression increases
cytokine and chemokine production and activates p38 and ERK1/2
signaling. (A) RAW264.7 murine macrophage cells were transfected
with NC-siRNA, Gp-siRNA, or vehicle alone, and the expression of
Gpnmb was examined using RT-qPCR. Data are presented as the mean ±
SD (n=3; *P<0.001). (B) Effects of Gpnmb knockdown on
levels of mRNAs encoding IL-1β, IL-6, TNF-α and MCP-1 were examined
using RT-PCR. Data are presented as the mean ± SD (n=3;
*P<0.01; **P<0.001. (C) Effects of
Gpnmb knockdown on p38, ERK1/2, and JNK signaling and IκB
activation were examined using western blotting. (D) Expression of
Gpnmb in the RAW264.7 cells was decreased in response to LPS and
increased following the removal of LPS. RAW264.7 macrophage cells
were exposed to LPS (100 µg/ml) for 24 h and changes in
Gpnmb were examined using RT-qPCR. mRNA expression levels in the
cells without LPS for 6 h were set as 1. Data are presented as the
mean ± SD (n=3). *P<0.05; **P<0.01;
***P<0.001. Gpnmb, glycoprotein nonmetastatic
melanoma protein B; IL, interleukin; TNF, tumor necrosis factor;
MCP, monocyte chemoattractant protein; LPS, lipopolysaccharide;
ERK, extracellular signal-regulated kinase; NC, negative control;
Gp, Gpnmb; siRNA, small interfering RNA; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; JNK, c-Jun
N-terminal kinase; p-, phosphorylated. |
Macrophages infiltrating into injured tissues are
have been suggested to alter phenotypes in response to the myriad
of stimuli present during wound healing (26). The present study investigated
changes in the expression of Gpnmb in murine RAW264.7 cells in
response to LPS. The RAW264.7 cells expressed Gpnmb in the absence
of LPS (Fig. 4B). The cells
exposed to LPS for 24 h expressed significantly lower levels of
Gpnmb during the treatment period, compared with the control cells
treated with PBS alone. However, the expression of Gpnmb increased
significantly over the 2 days following the removal of LPS
(Fig. 4D).
Discussion
The results of the present study demonstrated that
DSS induced mucosal injury between the rectum and the distal colon,
whereas the proximal colon remained unaffected. In this murine
model, infiltrating macrophages expressed Gpnmb in injured colonic
tissues, whereas the expression of Gpnmb was almost undetectable in
the proximal colon tissues, even when the mice were treated with
DSS for 7 days. These results indicated that macrophages recruited
to the injured mucosal tissue express Gpnmb, whereas the resident
macrophages of the uninjured intestine do not. In addition, the
results suggested a potential role for Gpnmb in suppressing
inflammatory responses in colitis by attenuating pro-inflammatory
cytokine production mediated by p38 and ERK signaling.
An inflammatory environment causes recruited
macrophages to assume heterogeneous phenotypes, which together
assist in balancing the damaging and healing mechanisms within the
tissue (14,27). The present study demonstrated that
the expression of Gpnmb gradually increased during 7 days of DSS
administration, and remained elevated, even following the removal
of DSS from the drinking water. In addition, when RAW264. 7 cells
were treated with LPS for 24 h, the expression of Gpnmb was reduced
in response to inflammatory stimuli in vitro; however, the
expression of Gpnmb gradually increased following removal of the
LPS, ultimately rising above the baseline level at 72 h. These
results suggested that macrophages infiltrating the injured mucosa
expressed Gpnmb in response to the inflammatory environment, and
that these Gpnmb-positive macrophages were involved in a delayed
phase of inflammation and/or tissue repair processes.
Macrophages involved in tissue repair can assume the
alternatively activated phenotype (26), whereas macrophages, which are
isolated from wounds, are not alternatively activated. The
phenotype of macrophages in injury changes over time, and does not
reflect the classic classification schemes (28). In the present study, the expression
of Gpnmb in the macrophage cell line was downregulated in the
presence of LPS, but was upregulated following the removal of LPS.
In vivo experiments demonstrated that elevated levels of
Gpnmb persisted following the removal of DSS from the drinking
water. These results suggested that Gpnmb-expressing macrophages
are involved in the wound-healing process and ameliorate mucosal
inflammation by modulating the expression of pro-inflammatory
cytokines.
In the identification of genes, which are
preferentially expressed in macrophages, Rippoll et al
(21) identified Gpnmb as a
macrophage-specific gene, reporting that the expression of Gpnmb is
inhibited in RAW264.7 cells exposed to LPS for 21 h. Conversely,
the expression of Gpnmb in RAW264.7 cells reduces the
LPS/interferon (IFN)-γ-induced secretion of IL-6 and IL-12p40
(21). The results of the present
study were consistent with these findings; in which the expression
of Gpnmb in RAW264.7 cells decreased during 24 h treatment with
LPS. Furthermore, siRNA-mediated knockdown of Gpnmb in RAW264.7
cells increased the expression of IL-1β, IL-6, TNF-α and MCP-1, in
the absence of LPS and IFN-γ. These results indicated that Gpnmb
suppressed pro-inflammatory cytokine production in macrophages,
suggesting that Gpnmb-positive macrophages negatively regulates
inflammation in the injured intestinal mucosa. In addition, the
present study demonstrated that inhibition of the expression of
Gpnmb in RAW264.7 cells resulted in the enhanced phosphorylation of
p38 and ERK1/2; however, no significant difference was observed in
the expression of IκB, which is directly associated with nuclear
factor (NF)-κB activation. Zhou et al (29) previously demonstrated that the
absence of Gpnmb increased the expression of IL-18 in the
iris/ciliary body by increasing the activation of NF-κB and the
phosphorylation of MAPKs, predominantly in CD69-positive cells.
These results suggest that Gpnmb suppresses pro-inflammatory
cytokine production in inflammatory cells via the attenuation of
MAPK signaling. Furthermore, p38/MAPK phosphatase-1 regulates AKT,
which coordinates phenotypic transitions of macrophages, and
resolution of inflammation during tissue repair (30). Akt1 and Akt2 also contribute
differentially to macrophage polarization (31). Therefore, Gpnmb may be associated
with macrophage polarization.
In conclusion, the present study demonstrated that
macrophages infiltrating the injured intestinal mucosa express
Gpnmb. These cells are anti-inflammatory, due to their decreased
production of pro-inflammatory cytokines, and elevated levels of
Gpnmb persisted following removal of the inflammatory stimulus.
These results suggested that Gpnmb-positive macrophages may be
important in ameliorating mucosal injury. Although further
investigations are required to clarify the roles of Gpnmb-positive
macrophages in the injured mucosa, these findings provide novel
information regarding the roles of innate immunity in tissue
injury, inflammation and healing of the intestinal mucosa.
Acknowledgments
The authors would like to thank Ms. Yuko
Morinaga-Nakamura for her technical assistance. This study was
supported by grants from the Ministry of Education, Culture,
Sports, Science and Technology of Japan (grant nos. 19590763 and
22590742 to Dr Akio Ido), and from the Ministry of Health, Labour
and Welfare of Japan as an official project of the Intractable
Inflammatory Bowel Diseases Study Group of Japan (to Dr Hirohito
Tsubouchi). Drs Akihiro Moriuchi, Hirohito Tsubouchi and Dr Akio
Ido hold endowed faculty positions in research for HGF tissue
repair and regenerative medicine, and received funds from Eisai
Co., Ltd (Tokyo, Japan).
References
|
1
|
Podolsky DK: Inflammatory bowel disease
(1). N Engl J Med. 325:928–937. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiocchi C: Inflammatory bowel disease:
Etiology and pathogenesis. Gastroenterology. 115:182–205. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loftus EV Jr: Clinical epidemiology of
inflammatory bowel disease: Incidence, prevalence, and
environmental influences. Gastroenterology. 126:1504–1517. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rogler G, Andus T, Aschenbrenner E, Vogl
D, Falk W, Schölmerich J and Gross V: Alterations of the phenotype
of colonic macrophages in inflammatory bowel disease. Eur J
Gastroenterol Hepatol. 9:893–899. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xavier RJ and Podolsky DK: Unravelling the
pathogenesis of inflammatory bowel disease. Nature. 448:427–434.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mowat AM: Anatomical basis of tolerance
and immunity to intestinal antigens. Nat Rev Immunol. 3:331–341.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rogler G and Andus T: Cytokines in
inflammatory bowel disease. World J Surg. 22:382–389. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Banks C, Bateman A, Payne R, Johnson P and
Sheron N: Chemokine expression in IBD. Mucosal chemokine expression
is unselectively increased in both ulcerative colitis and Crohn's
disease. J Pathol. 199:28–35. 2003. View Article : Google Scholar
|
|
9
|
Jurjus AR, Khoury NN and Reimund JM:
Animal models of inflammatory bowel disease. J Pharmacol Toxicol
Methods. 50:81–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garside P: Cytokines in experimental
colitis. Clin Exp Immunol. 118:337–339. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamada N, Hisamatsu T, Okamoto S, Sato T,
Matsuoka K, Arai K, Nakai T, Hasegawa A, Inoue N, Watanabe N, et
al: Abnormally differentiated subsets of intestinal macrophage play
a key role in Th-1-dominat chronic colitis through excess
production of IL-12 and IL-23 in response to bacteria. J Immunol.
175:6900–6908. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watanabe N, Ikuta K, Okazaki K, Nakase H,
Tabata Y, Matsuura M, Tamaki H, Kawanami C, Honjo T and Chiba T:
Elimination of local macrophages in intestine prevents chronic
colitis in interleukin-10-deficient mice. Dig Dis Sci. 48:408–414.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hunter MM, Wang A, Parhar KS, Johnston MJ,
Van Rooijen N, Beck PL and McKay DM: In vitro-derived alternatively
activated macrophages reduce colonic inflammation in mice.
Gastroenterology. 138:1395–1405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rivollier A, He J, Kole A, Valatas V and
Kelsall BL: Inflammation switches the differentiation program of
Ly6Chi monocytes from antiinflammatory macrophages to inflammatory
dendritic cells in the colon. J Exp Med. 209:139–155. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weterman MA, Ajubi N, van Dinter IM, Degen
WG, van Muijen GN, Ruitter DJ and Bloemers HP: nmb, a novel gene,
is expressed in low-metastatic human melanoma cell lines and
xenografts. Int J Cancer. 60:73–81. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Safadi FF, Xu J, Smock SL, Rico MC, Owen
TA and Popoff SN: Cloning and characterization of osteoactivin, a
novel cDNA expressed in osteoblasts. J Cell Biochem. 84:12–26.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shikano S, Bonkobara M, Zukas PK and
Ariizumi K: Molecular cloning of a dendritic cell-associated
transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion
of endothelial cells through recognition of heparan sulfate
proteoglycans. J Biol Chem. 276:8125–8134. 2001. View Article : Google Scholar
|
|
19
|
Onaga M, Ido A, Hasuike S, Uto H, Moriuchi
A, Nagata K, Hori T, Hayash K and Tsubouchi H: Osteoactivin
expressed during cirrhosis development in rats fed a
choline-deficient, L-amino acid-defined diet, accelerates motility
of hepatoma cells. J Hepatol. 39:779–785. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abe H, Uto H, Takami Y, Takahama Y,
Hasuike S, Kodama M, Nagata K, Moriuchi A, Numata M, Ido A and
Tsubochi H: Transgenic expression of osteoactivin in the liver
attenuates hapatic fibrosis in mice. Biochem Biophys Res Commun.
356:610–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ripoll VM, Irvine KM, Ravasi T, Sweet MJ
and Hume DA: Gpnmb is induced in macrophages by IFN-gammma and
lipopolysaccharide and acts as a feedback regulator of
proinflammatory responses. J Immunol. 178:6557–6566. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chung JS, Dougherty I, Cruz PD Jr and
Ariizumi K: Syndecan-4 mediates the coinhibitory function of DC-HIL
on T cell activation. J Immunol. 179:5778–5784. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Howell GR, Libby RT, Marchant JK, Wilson
LA, Cosma IM, Smith RS, Anderson MG and John SW: Absence of
glaucoma in DBA/2J mice homozygous for wild-type versions of Gpnmb
and Tyrp1. BMC Genetics. 8:452007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cooper HS, Murthy SN, Shah RS and
Sedergran DJ: Clinicopathological study of dextran sulfate sodium
experimental murine colitis. Lab Invest. 69:238–249.
1993.PubMed/NCBI
|
|
25
|
Kihara N, de la Fuente SG, Fujino K,
Takahashi T, Pappas TN and Mantyh CR: Vanilloid receptor-1
containing primary sensory neurons mediate dextran sulphate sodium
induced colitis in rats. Gut. 52:713–719. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramachandran P, Pellicoro A, Vernon MA,
Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A,
Gordon-Walker TT, et al: Differential Ly-6C expression identifies
the recruited macrophage phenotype, which orchestrates the
regression of murine liver fibrosis. Proc Natl Acad Sci USA.
109:E3186–E3195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daley JM, Brancato SK, Thomay AA, Reichner
JS and Albina JE: The phenotype of murine wound macrophages. J
Leukoc Biol. 87:59–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou X, Li F, Kong L, Tomita H, Li C and
Cao W: Involvement of inflammation, degradation, and apoptosis in a
mouse model of glaucoma. J Biol Chem. 280:31240–31248. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perdiguero E, Sousa-Victor P, Ruiz-Bonilla
V, Jardí M, Caelles C, Serrano AL and Muñoz-Cánoves P:
p38/MKP-1-regulated AKT coordinates macrophage transitions and
resolution of inflammation during tissue repair. J Cell Biol.
195:307–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arranz A, Doxaki C, Vergadi E, Martinez de
la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E, Androulidaki A,
Venihaki M, Margioris AN, et al: Akt1 and Akt2 protein kinases
differentially contribute to macrophage polarization. Proc Natl
Acad Sci USA. 109:9517–9522. 2012. View Article : Google Scholar : PubMed/NCBI
|