Introduction
Traumatic brain injury (TBI) is one of the leading
causes of mortality and morbidity in adults and children worldwide,
which results in immediate and delayed motor and cognitive
deficiency (1). TBI-induced tissue
loss and cell death occur following primary injury (direct physical
disruption of the tissue) and secondary injury (delayed molecular
pathophysiological changes) (2).
Numerous pharmacological interventions are currently being
investigated to attempt to reduce the severity of secondary injury
following primary injury; however, current therapies are limited in
their utility and efficacy (1).
Excitotoxicity is widely recognized as an important
process in nervous cell death, produced by excessive release of
glutamate following acute brain injury. In order to protect neurons
from glutamate-mediated excitotoxicity, astrocytes transmit
excessive glutamate from the extracellular space (3,4).
Previous data confirmed that excitatory amino acid transporters are
important in maintaining extracellular glutamate concentrations
below excitotoxic levels (3). In
total, five subtypes of sodium-dependent glutamate transporters
have been identified: Glutamate aspartate transporter (GLAST;
EAAT1), glutamate transporter-1 (GLT-1; EAAT2), EAAT3, EAAT4 and
EAAT5 (5). Of these transporters
GLT-1 has a principal role, and accounts for >90% of glutamate
uptake in the brain (6). Previous
studies have demonstrated that GLT-1 dysfunction is associated with
the pathogenesis of multiple neurological disorders, including
Alzheimer's disease, amyotrophic lateral sclerosis, stroke, several
forms of epilepsy and TBI (7–11).
Therefore, drugs and agents that increase glutamate transport
activity may be significant in the treatment of neurological
diseases.
Harmine is a β-carboline alkaloid, which was
isolated >100 years ago (12).
Harmine exerts a series of pharmacological actions, including
antioxidative, antigenotoxic, antidepressant-like and
antiplasmodial actions (13–16).
Previously, harmine was observed to exert neuroprotective actions
in an in vivo investigation, and the beneficial effects were
specifically due to the elevation of GLT-1 (17). However, no experiments have been
performed to determine whether harmine provides neuroprotection
following TBI.
In the present study, intraperitoneal (i.p)
injection of harmine (30 mg/kg per day, for up to 5 days) into rats
was performed in order to investigate whether harmine attenuates
brain edema and improves functional recovery in a rat model of TBI.
Furthermore, to determine whether the mechanism underlying the
neuroprotective effects of harmine are associated with glutamate
transport and inflammatory factors, the expression levels of GLT-1
and inflammatory cytokines were examined in the hippocampus of the
rat brain. These investigations aimed to investigate a potential
novel therapeutic modality for the treatment of TBI.
Materials and methods
Animals and a controlled TBI model
All experimental procedures were approved by The
Third Military Medical University Committee (Chongqing, China) for
the use and care of animals in research, and were performed in
accordance with the The Third Military Medical University's
guidelines for the care and use of laboratory animals. A total of
150 male Sprague-Dawley rats (age, 10–12 weeks; weighing, 280–320
g; obtained from The Third Military Medical University Animal
Center) were used in the present study. All the rats had free
access to food and water, and were housed under a standard 12-h
light/dark cycle. The housing temperature was 25–28° and the rats
were housed in groups.
A previously described TBI procedure (18) was performed in the present study.
Briefly, the rats were anesthetized with sodium pentobarbital [i.p,
50 mg/kg; Bio-Rad Laboratories (Shanghai) Co., Ltd., Shanghai,
China], and a 5-mm craniotomy was performed over the left parietal
cortex, which was centered on the coronal suture [Bio-Rad
Laboratories (Shanghai) Co., Ltd.] and 3 mm lateral to the sagittal
suture. The TBI was performed using a pneumatic piston [Bio-Rad
Laboratories (Shanghai) Co., Ltd.] with a rounded metal tip (2.5 mm
diameter), which was angled 22.5° to vertical to ensure that the
tip was perpendicular with the brain surface at the center of the
craniotomy. A velocity of 4 m/s and a deformation depth of 2 mm
below the dura were used to generate the TBI in the procedure. The
bone flap was immediately replaced and sealed, and the scalp was
closed using sutures. Body temperature was monitored throughout the
surgery, to ensure it was maintained at 37.0±0.5°C. Whilst the rats
were recovering from anesthesia, they were placed in a heated cage
to maintain body temperature.
Group and drug administration
The rats were randomly divided into three groups:
Sham-operated group (sham; n=15); the TBI group (TBI; n=35) and the
TBI + harmine (Beijing Aoboxing Biotechnology Co., Ltd., Beijing,
China)-treated group (Harmine; n=35). Harmine was administered
immediately following TBI (i.p, 30 mg/kg per day) for up to 5 days.
The sham and TBI groups received equal volumes of 0.9% saline
solution (i.p.).
Measurement of brain edema
Brain edema was evaluated by analyzing the brain
water content, as described previously (19). Following anesthetization, as
described above, and sacrifice by exsanguination, the rat brains
were separated and weighed immediately using a chemical balance, in
order to obtain the wet weight (WW). Following drying in a
desiccating oven for 24 h at 100°C, the dry tissues were weighed to
obtain the dry weight (DW). The water percentage in the brain was
calculated according to the following formula Brain water % = (WW −
DW) / WW) × 100.
Behavioral recovery
The neurobehavioral status of the rats was evaluated
using a set of 10 tasks, collectively termed the neurologic
severity score (NSS) (20), which
assesses reflexes, alertness, coordination and motor abilities. A
score of one is awarded for failure to perform a particular task;
therefore, a score of 10 reflects maximal impairment, whereas a
normal rat scores 0. The rats were grouped as follows for
examination of behavioral recovery: Sham, n=3; TBI, n=7; and
Harmine, n=7. Following TBI, the NSS was evaluated at 1, 3 and 5
days. Each rat was assessed by an observer who was blinded to the
animal treatment. The difference between the initial NSS and that
at any later time-point was calculated for each rat, and this value
(ΔNSS) reflects the spontaneous or treatment-induced recovery of
motor function. Furthermore, the spatial learning ability of the
rats was assessed using a Morris water maze, as previously
described (21). Briefly, the
Morris water maze consisted of a black circular pool (diameter, 180
cm; height, 45 cm) filled with water (depth, 30 cm; temperature,
26°C) and divided into four equal quadrants: North (N), west (W),
south (S) and east (E). An escape platform (diameter, 12 cm;
height, 28 cm, made opaque with paint and submerged 2 cm) was
placed in the center of one of the quadrants, equidistant from the
sidewall and the center of the pool. Rats were trained to locate
the platform prior to TBI or undergoing the sham surgery. For each
trial, a rat was randomly placed into a quadrant start point (N, S,
E or W) facing the wall of the pool and was allowed ≤60 sec to
escape to the platform. Rats that failed to escape within 90 sec
were placed on the platform for ≤20 sec and returned to the cage
for a new trial (intertrial interval, 20 sec). The maze performance
was recorded using a video camera suspended above the maze and
interfaced with a video tracking system (HVS Imaging; Hampton, UK).
The mean escape latency of a total of five trials was calculated.
This assessment was performed 1, 3 and 5 days following TBI or sham
surgery.
Hematoxylin and eosin (H&E)
staining
At 24 h post-surgery, the rats (sham, n=3; TBI, n=7;
and Harmine, n=7) were anesthetized, as described above, and
perfused intracardially with isotonic sodium chloride solution,
followed by 4% (w/v) paraformaldehyde [Bio-Rad Laboratories
(Shanghai) Co., Ltd.] in 0.1 M sodium phosphate buffer [pH 7.4;
Bio-Rad Laboratories (Shanghai) Co., Ltd.]. The rats were
sacrificed via exsanguination. The brains were subsequently removed
and fixed for 48 h in 4% (w/v) paraformaldehyde. Following
fixation, the brains were embedded in paraffin [Bio-Rad
Laboratories (Shanghai) Co., Ltd.], sliced into 4 µm coronal
sections at the bregma, and stained with H&E [Bio-Rad
Laboratories (Shanghai) Co., Ltd.]. The surviving and dying neurons
in the CA1 area per 1 mm were subsequently quantified under an
Olympus BX51 microscope (Olympus Corporation, Tokyo, Japan).
Immunofluorescence
At 24 h post-TBI, coronal sections were incubated
with 10% normal donkey serum [Bio-Rad Laboratories (Shanghai) Co.,
Ltd. for 30 min at room temperature in phosphate-buffered saline
(PBS) containing 0.1% Triton X-100 [Bio-Rad Laboratories (Shanghai)
Co., Ltd.]. The sections were then incubated at 4°C overnight with
the following primary antibodies: Goat anti-rat neuron-specific
nuclear protein [NeuN; 1:200 (cat. no. sc-31154); Santa Cruz
Biotechnology, Inc., Dallas, TX, USA] and rabbit anti-rat caspase 3
[1:50; (cat. no. sc-7148); Santa Cruz Biotechnology, Inc.]. The
sections were then washed with PBS four times at room temperature,
followed by an incubation with appropriate fluorescent-labeled
secondary antibodies [1:200 (cat. nos. sc-2342 and sc-2341) Santa
Cruz Biotechnology, Inc.] for 1 h at room temperature. The sections
were incubated with DAPI (1 ng/Nl; Beijing Aoboxing Biotechnology
Co., Ltd.) to counterstain the nucleus. Subsequently, the sections
were washed with PBS and mounted using water-based mounting medium
containing anti-fading agents (Biomeda; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Confocal images were captured using a
laser scanning confocal microscope (Olympus FV1000; Olympus
Corporation, Tokyo, Japan) and digital imaging software (FV10-ASW
1.5 Viewer; Olympus Corporation).
Western blot analysis
The rats (sham, n=3; TBI, n=7; and Harmine, n=7)
were anesthetized and underwent intracardiac perfusion with 0.1
mol/l PBS (pH 7.4). The cortex region of the brain was rapidly
isolated and the brain tissues were homogenized using a
homogenizer, total proteins were extracted using protein extraction
reagent [both Bio-Rad Laboratories (Shanghai) Co., Ltd.] and
protein concentration was determined using a bicinchoninic acid
reagent (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) method. The protein samples (5 mg) were separated
by 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
[Bio-Rad Laboratories (Shanghai) Co., Ltd.] and were subsequently
transferred onto polyvinylidene fluoride membranes (Roche
Diagnostics Deutschland GmbH, Mannheim, Germany). The blots were
then blocked with 5% fat-free dry milk for 1 h at room temperature.
Following blocking, the membranes were incubated with the following
primary antibodies overnight at 4°C: Rabbit anti-GLT-1 polyclonal
antibody (cat. no. sc-15317), rabbit anti-interleukin (IL)-1β
polyclonal antibody (cat. no. sc-7884), rabbit anti-tumor necrosis
factor (TNF)-α polyclonal antibody (cat. no. sc-7895), rabbit
anti-caspase 3 polyclonal antibody (cat. no. sc-7148) and mouse
anti-β-actin monoclonal antibody [(cat. no. sc-7210) all 1:500;
Santa Cruz Biotechnology, Inc.). The membranes were then incubated
with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G
(IgG; cat. no. sc-2027) and anti-mouse IgG (cat. no. sc-2025) for 2
h at room temperature (1:5,000; Santa Cruz Biotechnology, Inc.).
Following incubation with the secondary antibody, the immunoblots
were then visualized following development with an enhanced
chemiluminescence detection system [Bio-Rad Laboratories (Shanghai)
Co., Ltd.], and densitometric signals were quantified using an
enhanced chemiluminescence detection system [Bio-Rad Laboratories
(Shanghai) Co., Ltd.]. The western blotting results were analyzed
using ImageJ 1.41 software (National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. SPSS 16.0 (SPSS, Inc., Chicago, IL, USA) was used to
analyze the data. Statistical analysis was performed using one-way
analysis of variance followed by the Student-Newman-Keuls post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Treatment with harmine attenuates
cerebral edema
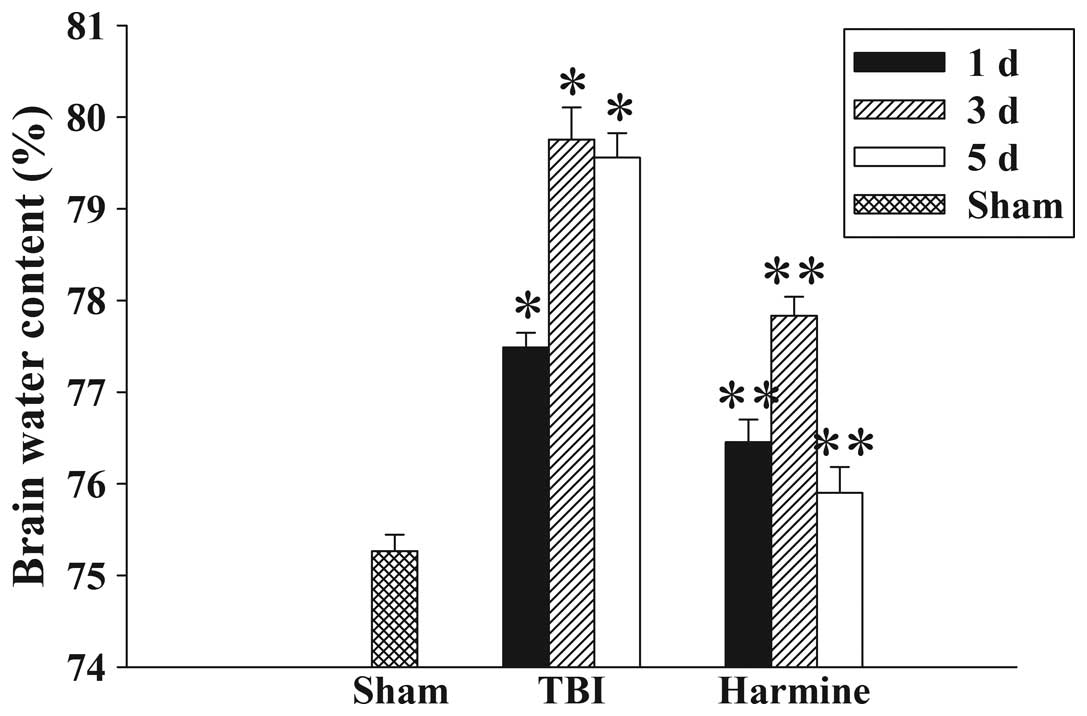
The wet-dry weight method was used to evaluate brain
edema. As shown in Fig. 1, brain
water content was significantly increased in the TBI group,
compared with the sham group at 1, 3 and 5 days following trauma.
Treatment with harmine significantly reduced the tissue water
content at 1, 3 and 5 days, a compared with the TBI group.
Treatment with harmine attenuates
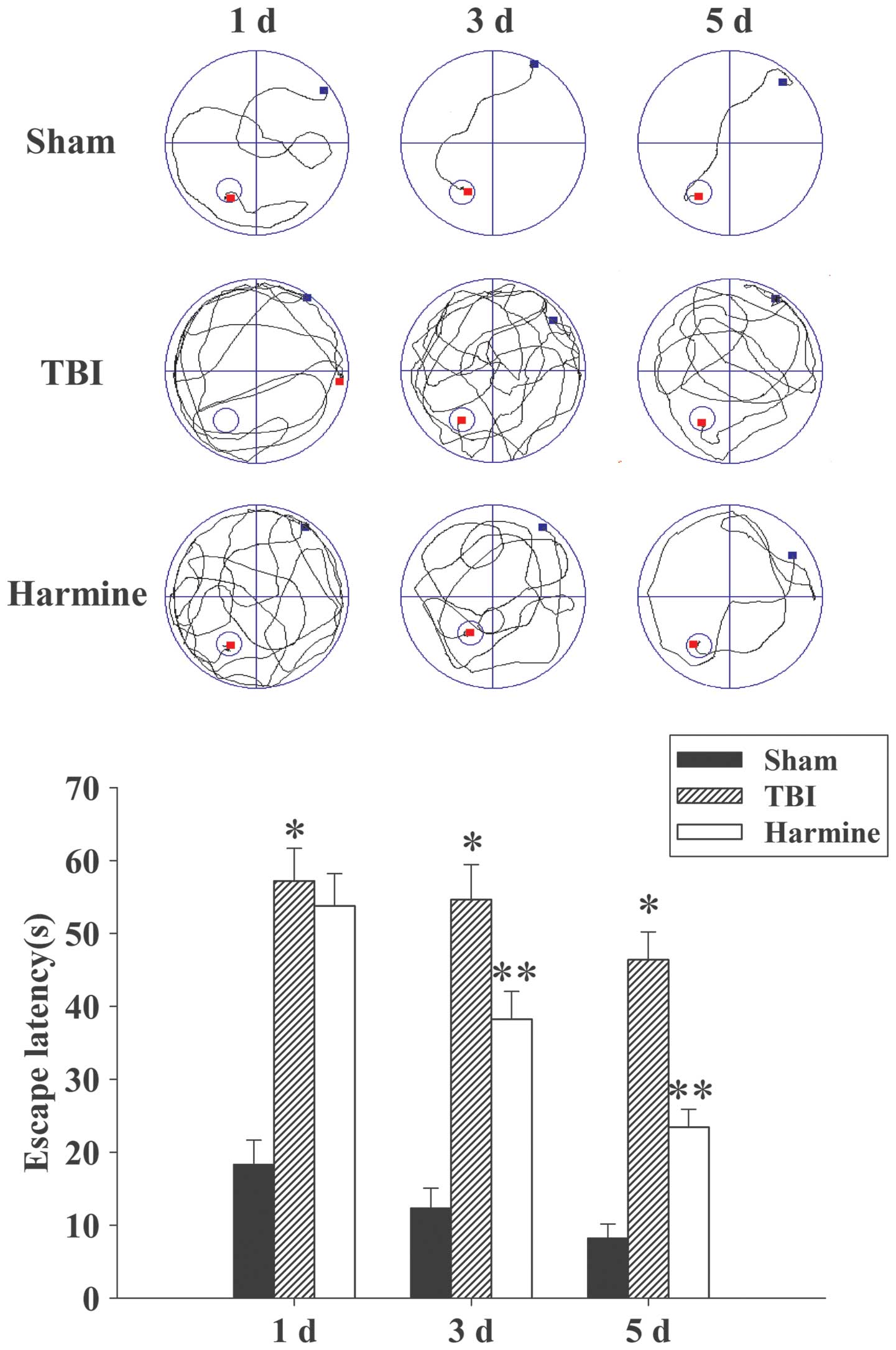
TBI-induced learning and spatial memory dysfunction
Since treatment with harmine successfully attenuated
brain edema, the present study examined whether harmine also
improved spatial learning function, which was assessed using a
Morris water maze at 1, 3 and 5 days post-TBI or sham surgery. As
shown in Fig. 2, TBI resulted in a
significant spatial learning deficit at 1, 3 and 5 days, compared
with the sham group, and harmine treatment significantly reduced
the escape latency at 3 and 5 days, compared with the TBI
group.
Treatment with harmine attenuates
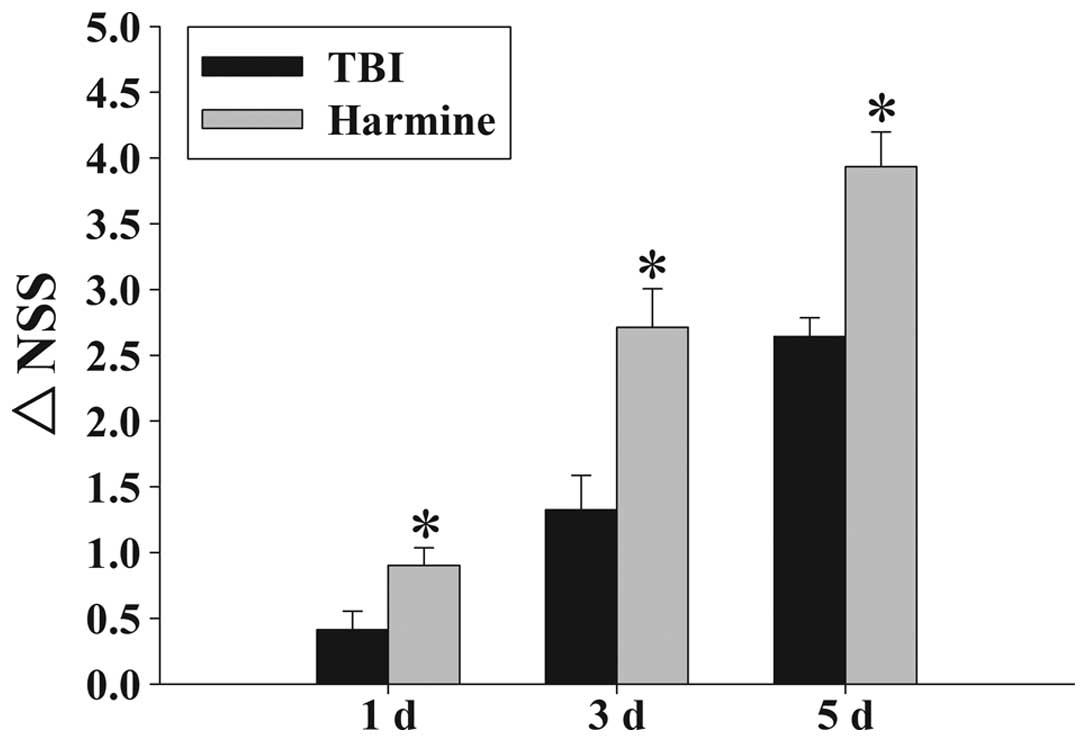
TBI-induced motor deficits
Temporal variations in the functional recovery of
the TBI rats are demonstrated in Fig.
3 and expressed as ΔNSS. Post-TBI administration of harmine
significantly improved the motor function recovery of the rats at
1, 3 and 5 days following TBI, compared with the TBI group without
harmine treatment.
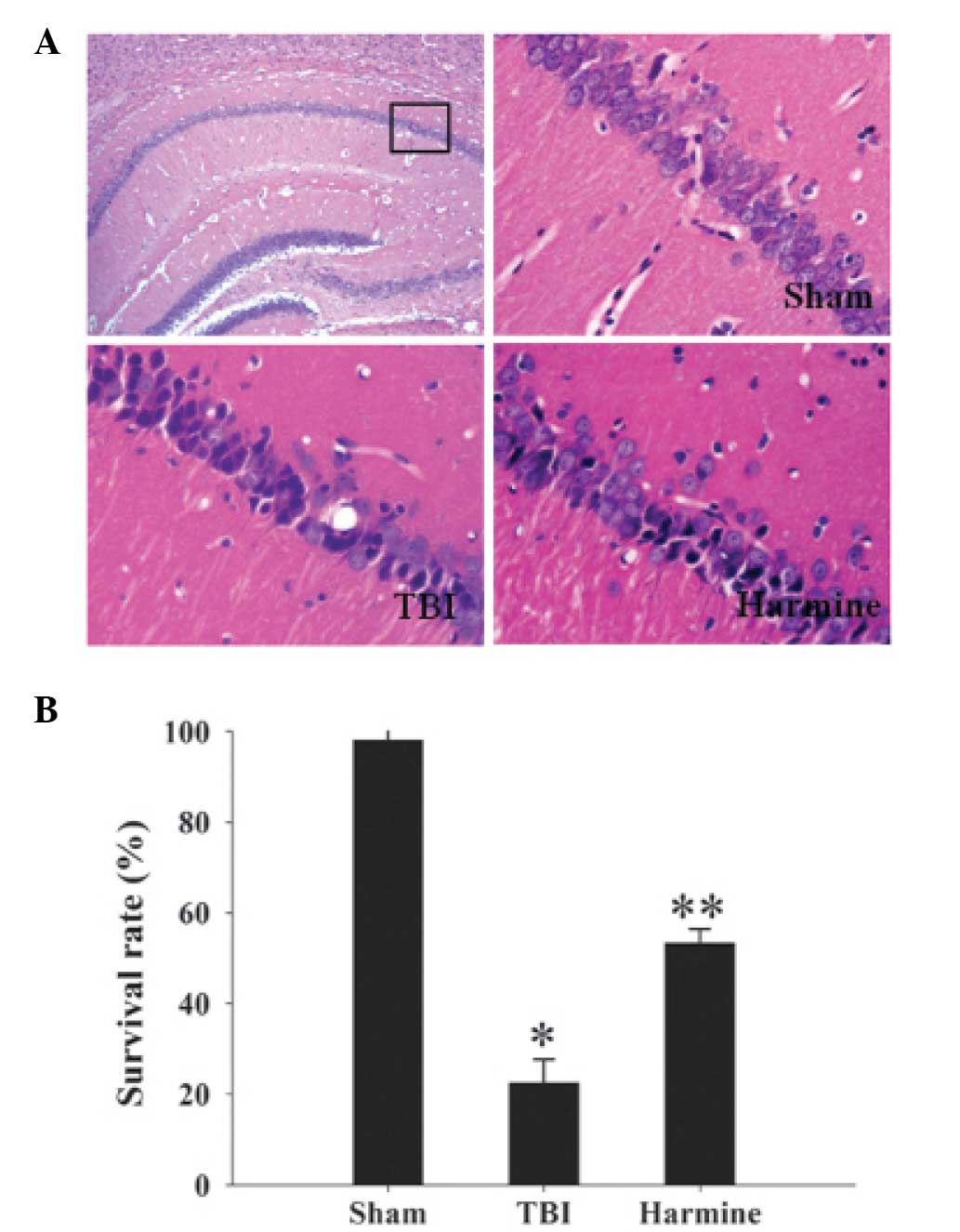
Treatment with harmine suppresses
neuronal death in the hippocampal region following TBI
Neuronal survival was assessed at 24 h using H&E
staining. Morphologically, the nuclei of the normal neurons were
round and poorly stained, whereas the nuclei of the dying neurons
were pyknotic and darkly stained (Fig.
4A). As shown in Fig. 4B,
compared with the sham group, TBI resulted in a marked decrease in
the survival rate of the neurons. However, the neuronal survival
rate in the harmine-treated group was significantly increased,
compared with the TBI group.
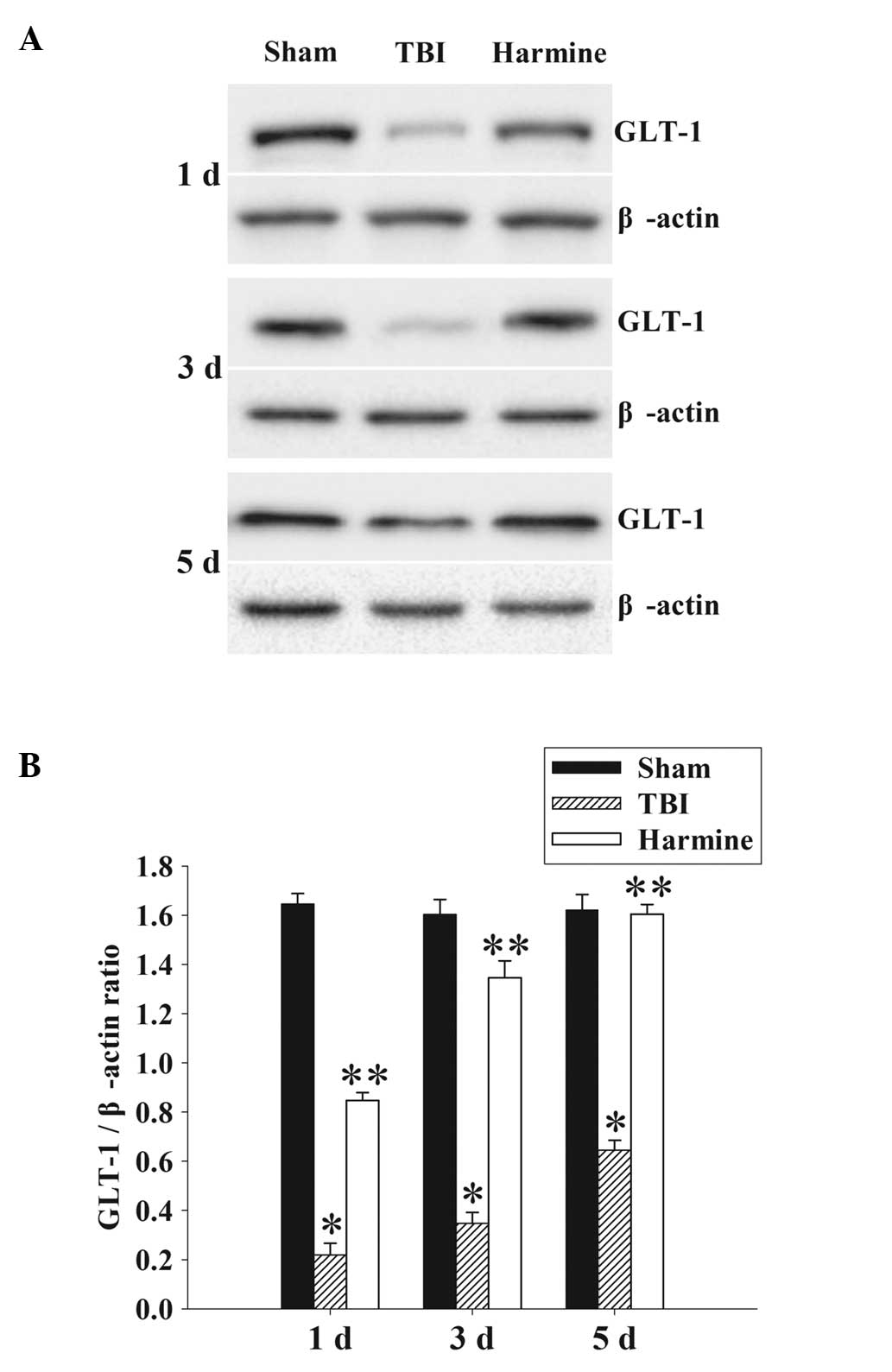
Treatment with harmine increases the
expression of GLT-1 in the hippocampus following TBI
The protein expression levels of GLT-1 in the
hippocampus were analyzed using western blot analysis. As shown in
Fig. 5, at 1, 3 and 5 days
post-surgery, there was a significant downregulation in the
expression of GLT-1 in the TBI group, compared with the sham group.
Administration of harmine resulted in marked elevation in the
expression of GLT-1, compared with the TBI group.
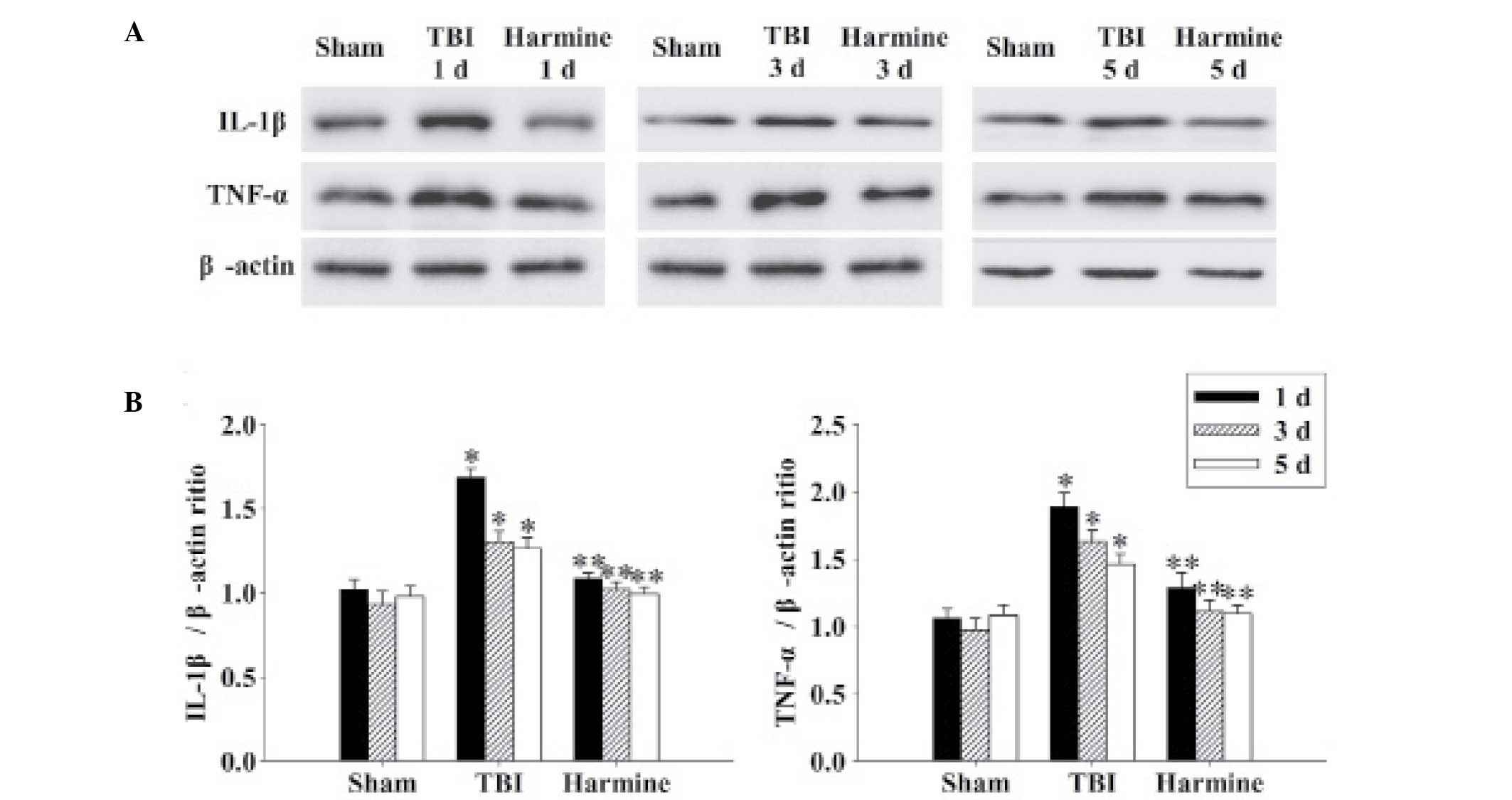
Treatment with harmine attenuates the
expression of inflammatory cytokines in the hippocampus following
TBI
The expression levels of IL-1β and TNF-α were
detected in the hippocampus 1, 3 and 5 days following TBI. As shown
in Fig. 6, the expression levels
of the inflammatory cytokines in the sham group were low; however,
the expression levels of IL-1β and TNF-α were markedly elevated at
each of the time-points in the TBI group. Furthermore, TBI-induced
expression of IL-1β and TNF-α was significantly reduced following
administration of harmine.
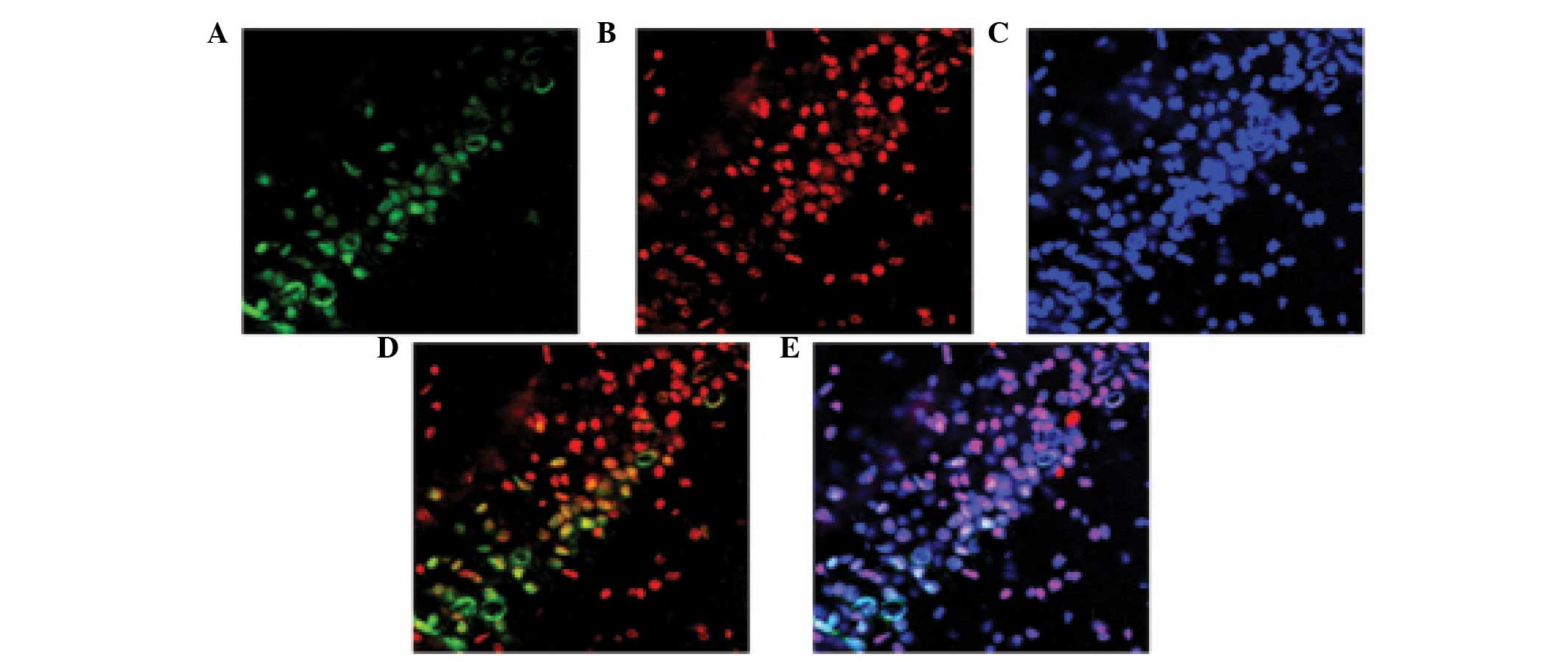
Caspase 3 and NeuN are co-localized in
the hippocampus following TBI
The co-localization of NeuN and caspase 3 was
detected using immunofluorescent staining 24 h post-TBI. As shown
in Fig. 7, the majority of
TBI-induced apoptosis occurred in the neurons.
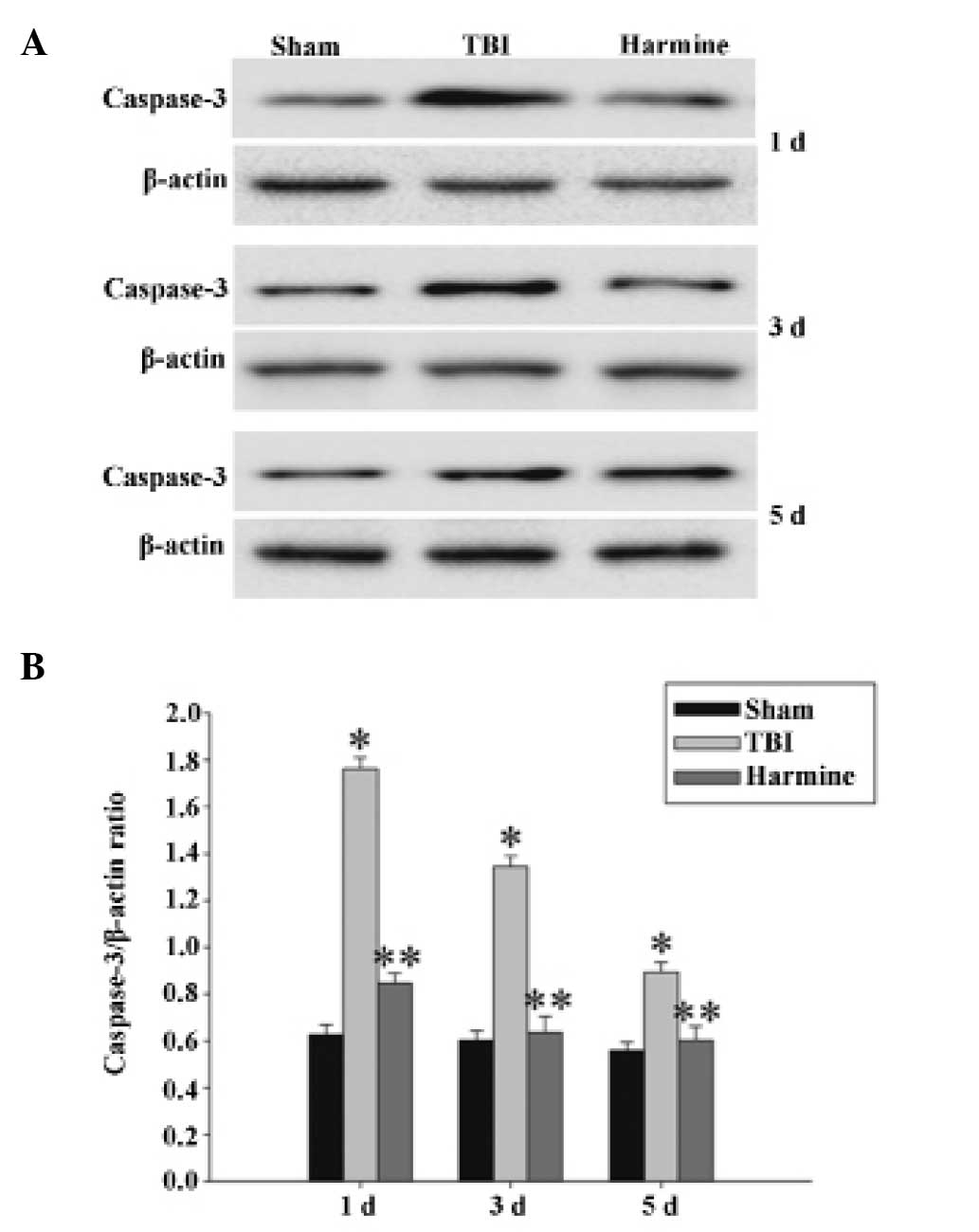
Treatment with harmine reduces the
expression of caspase 3 in the hippocampus following TBI
The expression of caspase 3 in the hippocampus were
detected using western blot analysis. As shown in Fig. 8, at 1, 3 and 5 days post-TBI, the
expression of caspase 3 was significantly increased in the TBI
group, compared with the sham group. However, the administration of
harmine significantly reduced the expression of caspase 3, compared
with the TBI group.
Discussion
The present study demonstrated that administration
of harmine (30 mg/kg per day) for up to 5 days immediately
following TBI had neuroprotective potential in the rats. Treatment
with harmine attenuated post-traumatic cerebral edema, and improved
learning and memory abilities. Furthermore, the present study
investigated the mechanisms underlying the protective effects of
harmine on TBI. At the molecular level, the protein expression
levels of GLT-1 and inflammatory cytokines in the hippocampus of
the rat brain were detected using western blot analysis. Treatment
with harmine resulted in a marked elevation in the expression of
GLT-1, as well as significant reductions in the expression levels
of IL-1β and TNF-α, suggesting that harmine attenuated apoptotic
neuronal death in the hippocampus. Previous studies have reported
that treatment with harmine significantly attenuate
glutamate-induced and oxidative stress-induced neuronal death in
neuronal cell cultures (15,17).
In addition, a previous in vivo study demonstrated that
harmine exerts neuroprotective effects against glutamate-mediated
excitotoxicity in a rat model of amyotrophic lateral sclerosis
disease (17). These findings,
together with the findings of the present study, improve current
understanding of harmine-mediated neuroprotection in neurological
disorders.
Previous in vivo studies have demonstrated
that GLT-1 dysfunction causes acute and chronic brain injury, and
that glutamate-mediated excitotoxicity is considered to be an
important process in neurological disorders (22,23).
The accumulation of excess extracellular glutamate and subsequent
overstimulation of glutamatergic receptors increases the production
of reactive and excitotoxic oxygen/nitrogen species, which may
induce oxidative stress leading to neuronal death (24). Previous studies have reported that
elevated activity and expression of GLT-1 induces demonstrated in
rat models of various acute and chronic neurologic diseases
(7–11), including a rat model of TBI, in
which the overexpression of GLT-1 was observed to significantly
reduce glutamate overflow, decrease cell death and improve
behavioral recovery (20). The
results of the present study confirmed that treatment with harmine
markedly elevated the expression of GLT-1 in the hippocampus
following trauma. These findings may demonstrate one of the
predominant mechanisms by which harmine exerts its neuroprotective
effects in TBI.
Treatment with harmine not only protects neurons
against glutamate insult via elevating the expression level of
GLT-1, but it also led to the exertion of anti-inflammatory effects
following TBI. Previous studies have demonstrated that inflammatory
cytokines can directly and indirectly attenuate the expression of
glutamate transporters in astrocytes, which has a significant role
in TBI-induced glutamate excitotoxicity, and it has been reported
that IL-1β and TNF-α regulate glutamate transmission directly
through elevation of the expression of
α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors
(25–27). In the present study, the expression
levels of inflammatory cytokines in the hippocampus of the rat
brain were suppressed following treatment with harmine. Although
the mechanism underlying the neuroprotective effects of harmine on
TBI remain to be fully elucidated, the data of the present study
suggested that attenuation of inflammation may synergistically
reduce extracellular glutamate. This suggests that the recruitment
of attenuated inflammation occurs a physiological action, which
acts synergically with the GLT-1 response to protect neurons from
TBI-induced insult and death, and further investigation of this is
required.
The present study performed only preliminarily
examination of the neuroprotective effects induced by harmine on
TBI rats; therefore, further investigations are required to
establish the appropriate routes of administration, the exact
time-frame, and whether these effects are observed following
different injury magnitudes.
The results of the present study demonstrated that
administration of harmine significantly attenuated cerebral edema,
and improved learning and memory abilities following experimental
TBI. Furthermore, treatment with harmine markedly elevated the
expression of GLT-1 and significantly attenuated the expression
levels of IL-1β and TNF-α, thereby attenuating apoptotic neuronal
death in the hippocampus. These findings indicated that
administration of harmine (30 mg/kg per day, up to 5 days)
following TBI can be neuroprotective, via anti-inflammatory and
anti-excitotoxicity effects following experimental TBI in rats.
These findings may represent a potential novel therapeutic modality
for the treatment of TBI.
Abbreviations:
|
TBI
|
traumatic brain injury
|
|
GLT-1
|
glutamate transporter 1
|
|
NSS
|
neurologic severity score
|
|
IL-1β
|
interleukin-1β
|
|
TNF-α
|
tumor necrosis factor-α
|
|
NeuN
|
neuron-specific nuclear protein
|
References
|
1
|
Wu X, Hu J, Zhuo L, Fu C, Hui G, Wang Y,
Yang W, Teng L, Lu S and Xu G: Epidemiology of traumatic brain
injury in eastern China, 2004: A prospective large case study. J
Trauma. 64:1313–1319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Werner C and Engelhard K: Pathophysiology
of traumatic brain injury. Br J Anaesth. 99:4–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnston MV: Excitotoxicity in perinatal
brain injury. Brain Pathol. 15:234–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palmer AM, Marion DW, Botscheller ML,
Swedlow PE, Styren SD and DeKosky ST: Traumatic brain
injury-induced excitotoxicity assessed in a controlled cortical
impact model. J Neurochem. 61:2015–2024. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Amara SG and Fontana AC: Excitatory amino
acid transporters: Keeping up with glutamate. Neurochem Int.
41:313–318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanaka K, Watase K, Manabe T, Yamada K,
Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi
T, et al: Epilepsy and exacerbation of brain injury in mice lacking
the glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lauderback CM, Hackett JM, Huang FF,
Keller JN, Szweda LI, Markesbery WR and Butterfield DA: The glial
glutamate transporter, GLT-1, is oxidatively modified by
4-hydroxy-2-nonenal in the Alzheimer's disease brain: The role of
Abeta1-42. J Neurochem. 78:413–416. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rothstein JD, Van Kammen M, Levey AI,
Martin LJ and Kuncl RW: Selective loss of glial glutamate
transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol.
38:73–84. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rao VL, Dogan A, Todd KG, Bowen KK, Kim
BT, Rothstein JD and Dempsey RJ: Antisense knockdown of the glial
glutamate transporter GLT-1, but not the neuronal glutamate
transporter EAAC1, exacerbates transient focal cerebral
ischemia-induced neuronal damage in rat brain. J Neurosci.
21:1876–1883. 2001.PubMed/NCBI
|
|
10
|
Samuelsson C, Kumlien E, Flink R, Lindholm
D and Ronne-Engström E: Decreased cortical levels of astrocytic
glutamate transport protein GLT-1 in a rat model of posttraumatic
epilepsy. Neurosci Lett. 289:185–188. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rao VL, Dogan A, Bowen KK, Todd KG and
Dempsey RJ: Antisense knockdown of the glial glutamate transporter
GLT-1 exacerbates hippocampal neuronal damage following traumatic
injury to rat brain. Eur J Neurosci. 13:119–128. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao R, Peng W, Wang Z and Xu A:
beta-Carboline alkaloids: Biochemical and pharmacological
functions. Curr Med Chem. 14:479–500. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Réus GZ, Stringari RB, de Souza B,
Petronilho F, Dal-Pizzol F, Hallak JE, Zuardi AW, Crippa JA and
Quevedo J: Harmine and imipramine promote antioxidant activities in
prefrontal cortex and hippocampus. Oxid Med Cell Longev. 3:325–331.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moura DJ, Richter MF, Boeira JM, Pêgas
Henriques JA and Saffi J: Antioxidant properties of beta-carboline
alkaloids are related to their antimutagenic and antigenotoxic
activities. Mutagenesis. 22:293–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fortunato JJ, Réus GZ, Kirsch TR,
Stringari RB, Fries GR, Kapczinski F, Hallak JE, Zuardi AW, Crippa
JA and Quevedo J: Effects of beta-carboline harmine on behavioral
and physiological parameters observed in the chronic mild stress
model: Further evidence of antidepressant properties. Brain Res
Bull. 81:491–496. 2010. View Article : Google Scholar
|
|
16
|
Astulla A, Zaima K, Matsuno Y, Hirasawa Y,
Ekasari W, Widyawaruyanti A, Zaini NC and Morita H: Alkaloids from
the seeds of Peganum harmala showing antiplasmodial and
vasorelaxant activities. J Nat Med. 62:470–472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Sattler R, Yang EJ, Nunes A, Ayukawa
Y, Akhtar S, Ji G, Zhang PW and Rothstein JD: Harmine, a natural
beta-carboline alkaloid, upregulates astroglial glutamate
transporter expression. Neuropharmacology. 60:1168–1175. 2011.
View Article : Google Scholar :
|
|
18
|
Chen SF, Hsu CW, Huang WH and Wang JY:
Post-injury baicalein improves histological and functional outcomes
and reduces inflammatory cytokines after experimental traumatic
brain injury. Br J Pharmacol. 155:1279–1296. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Täuber MG, Khayam-Bashi H and Sande MA:
Effects of ampicillin and corticosteroids on brain water content,
cerebrospinal fluid pressure, and cerebrospinal fluid lactate
levels in experimental pneumococcal meningitis. J Infect Dis.
151:528–534. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu D, Sanberg PR, Mahmood A, Li Y, Wang L,
Sanchez-Ramos J and Chopp M: Intravenous administration of human
umbilical cord blood reduces neurological deficit in the rat after
traumatic brain injury. Cell Transplant. 11:275–281.
2002.PubMed/NCBI
|
|
21
|
Cui C, Cui Y, Gao J, Sun L, Wang Y, Wang
K, Li R, Tian Y, Song S and Cui J: Neuroprotective effect of
ceftriaxone in a rat model of traumatic brain injury. Neurol Sci.
35:695–700. 2014. View Article : Google Scholar
|
|
22
|
Namura S, Maeno H, Takami S, Jiang XF,
Kamichi S, Wada K and Nagata I: Inhibition of glial glutamate
transporter GLT-1 augments brain edema after transient focal
cerebral ischemia in mice. Neurosci Lett. 324:117–120. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han F, Shioda N, Moriguchi S, Qin ZH and
Fukunaga K: Downregulation of glutamate transporters is associated
with elevation in extracellular glutamate concentration following
rat microsphere embolism. Neurosci Lett. 430:275–280. 2008.
View Article : Google Scholar
|
|
24
|
Rauen T and Kanner BI: Localization of the
glutamate transporter GLT-1 in rat and macaque monkey retinae.
Neurosci Lett. 169:137–140. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim
JM, Kim SJ, Park DK, Jung KH, Song EC, et al: Pharmacological
induction of ischemic tolerance by glutamate transporter-1 (EAAT2)
upregulation. Stroke. 38:177–182. 2007. View Article : Google Scholar
|
|
26
|
Pickering M, Cumiskey D and O'Connor JJ:
Actions of TNF-alpha on glutamatergic synaptic transmission in the
central nervous system. Exp Physiol. 90:663–670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lai AY, Swayze RD, El-Husseini A and Song
C: Interleukin-1 beta modulates AMPA receptor expression and
phosphorylation in hippocampal neurons. J Neuroimmunol. 175:97–106.
2006. View Article : Google Scholar : PubMed/NCBI
|