Introduction
Peripheral arterial disease (PAD), also known as
peripheral vascular disease, is involved in the atherosclerotic
occlusion of the arterial circulation to the lower extremities. The
disease may manifest symptoms of intermittent claudication or
severe chronic leg ischemia (1).
The major risk factors for PAD are older age (>40 years),
cigarette smoking and diabetes mellitus (2). Patients diagnosed with PAD suffer
from a limitation in exercise capacity and reduced quality of life,
and are at increased risk of cardiovascular-associated morbidity
and mortality (3,4).
Due to poor prognosis and the increasing number of
patients with PAD, an increasing number of investigations are
focussed on the identifications of biomarkers for PAD, and aim to
uncover the underlying mechanism of PAD. β2-microglobulin has been
considered to be a biomarker for PAD (5). In addition, Smadja et al found
that thrombospondin 1 is significantly up-regulated in patients
with PAD and may act as a plasmatic marker for PAD (6). Busti et al also reported that
the plasma levels of certain matrix metalloproteinases (MMPs),
including MMP-2 and MMP-9, are correlated with the development and
severity of PAD development and severity (7). Previously, Masud et al
screened 87 differentially expressed genes (DEGs) in PAD, and
examined the functions and pathways of the DEGs by performing gene
expression analysis of peripheral blood mononuclear cells (PBMCs)
in patients with PAD and normal controls (8). However, the interactions among these
genes and the exact molecular mechanism underlying PAD remain to be
elucidated.
Gene co-expression network-based approaches have
been widely used in analyzing microarray data, particularly for the
screening of functional modules (9,10).
One of the most useful gene co-expression network-based approaches
is Weighted Gene Co-expression Network Analysis (WGCNA), which has
been used to identify significant modules in a network and to
screen candidate targets or biomarkers for human diseases or cancer
(11–13). In the present study, the mRNA
expression profiles were analyzed in peripheral blood mononuclear
cell (PBMC) samples in patients diagnosed with PAD and in normal
controls, to identify the DEGs associated with PAD. In addition,
the significant modules were screened and a co-expression network
was constructed using WGCNA. The distinct Gene Ontology (GO)
functions and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathways were also obtained to further examine the associations
between the genes and PAD. These investigations aimed to provide
evidence to further elucidated the mechanism underlying PAD.
Materials and methods
Gene expression datasets
The microarray expression data (accession. no.
GSE27034) for PBMC samples in patients with PAD and controls
without PAD (8) were obtained from
the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database (14). Gene expression analysis was
performed based on a platform of [HG-U133_Plus_2] Affymetrix Human
Genome U133 Plus 2.0 Array (GPL570; Affymetrix, Inc., Santa Clara,
CA, USA). The datasets contained 37 samples, which included 19
samples from patients with PAD and 18 samples from normal
controls.
Data preprocessing and screening of
DEGs
The raw data were preprocessed via background
correction, quantile normalization and probe summarization using
the Affy package in R (Affymetrix, Inc.) (15). Subsequently, the expression
profiling probes were transformed to corresponding gene symbols. In
cases where there were more than one probe set in a single gene,
the average expression values of all probes for the specific gene
were defined as the gene expression value. In addition, when
several mRNAs were mapped by one probe, this probe was considered
to lack specificity and was excluded. Finally, the linear models
for microarray data (Limma) package in R language (version 3.22.7;
http://www.bioconductor.org/packages/3.0/bioc/html/limma.html)
(16) was used to normalize the
expression profiles and identify the DEGs between the PAD and
normal samples. The genes identified with the cutoff criteria of
fold change (FC)>1.5 and P-value<0.05 were reserved as DEGs
for subsequent analysis.
WGCNA
Initially, Pearson's correlation matrices for all
gene pairs were calculated using SPSS software, version 16.0 (SPSS,
Inc., Tokyo, Japan), and the correlation between gene m and
gene n was defined as Smn=|cor(m,n)|.
Subsequently, the Pearson's correlation matrices were transformed
into matrices of connection strengths using a power function, as
follows amn = power (Smn, β) =
|Smn|β. The β value is set as the weighting
coefficient only when the correlation coefficient between log (k)
and log [p(k)] reaches 0.9, where p(k) represents the proportion of
nodes with connectivity (k). Thus, the β value (β=16) was
determined, so that the initial correlation coefficient reached
0.9. Following determination of the adjacency parameter, the
correlation matrix was transformed into an adjacency matrix, which
was subsequently transformed into a topological overlap matrix
(TOM). The TOM (17) was computed
using the following formula:
where lmn indicates the sum of the products of
the adjacency coefficients of the nodes connected to m and
n. km indicates the sum of the adjacency
coefficients of the nodes only connected to m. kn
indicates the sum of the adjacency coefficient of the nodes only
connected to n. If two nodes are not connected to each other
and share no neighbors, =0.
Identification of significant modules and
construction of the co-expression network
The obtained DEGs were hierarchically clustered
using the dissimilarity coefficient as the distance measure, with
each branch corresponding to a module. The modules containing at
least 30 genes were assigned using the software of dynamic tree cut
R package (version 1.62; Bioconductor, Los Angeles, CA, USA), which
applies a novel dynamic branch cutting method for the detection of
clusters in a dendrogram according to their shape (18). Following module detection, the
eigengenes for each module were calculated, and the merged close
modules were clustered into new modules. The module, which
exhibited the maximal absolute correlation coefficient was
considered to be the module that is most significantly associated
with PAD. The network significance approach determines the module
associated with PAD based on module significance (MS) (19). The MS indicates the average gene
significance (GS) of all the genes in a module, and the GS of a
gene is defined as -lg p, where p indicates the P-values obtained
with Student's t-test using SPSS software, version 16.0. High MS
values indicate a high association with PAD. In addition, the gene
pairs, which were identified with the correlation coefficient
>0.7 in the selected significant module were used to construct
the co-expression network using Cytoscape software (version: 3.2.0;
http://www.cytoscape.org/) (20).
Functional enrichment analyses
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) (21)
was used to identify the enriched functions in the DEGs, with the
cutoff criterion of a false discovery rate (FDR) <0.05. In
addition, the KEGG Orthology-Based Annotation System (KOBAS) was
applied to identify the significantly enriched pathways using a
hypergeometric test (22) with the
cutoff criterion of P<0.05
Results
Screening of DEGs
Under the cutoff criteria of FC>1.5 and
P<0.05, a total of 148 DEGs were obtained, which included 91
upregulated and 57 downregulated genes.
WGCNA analysis and identification of key
modules
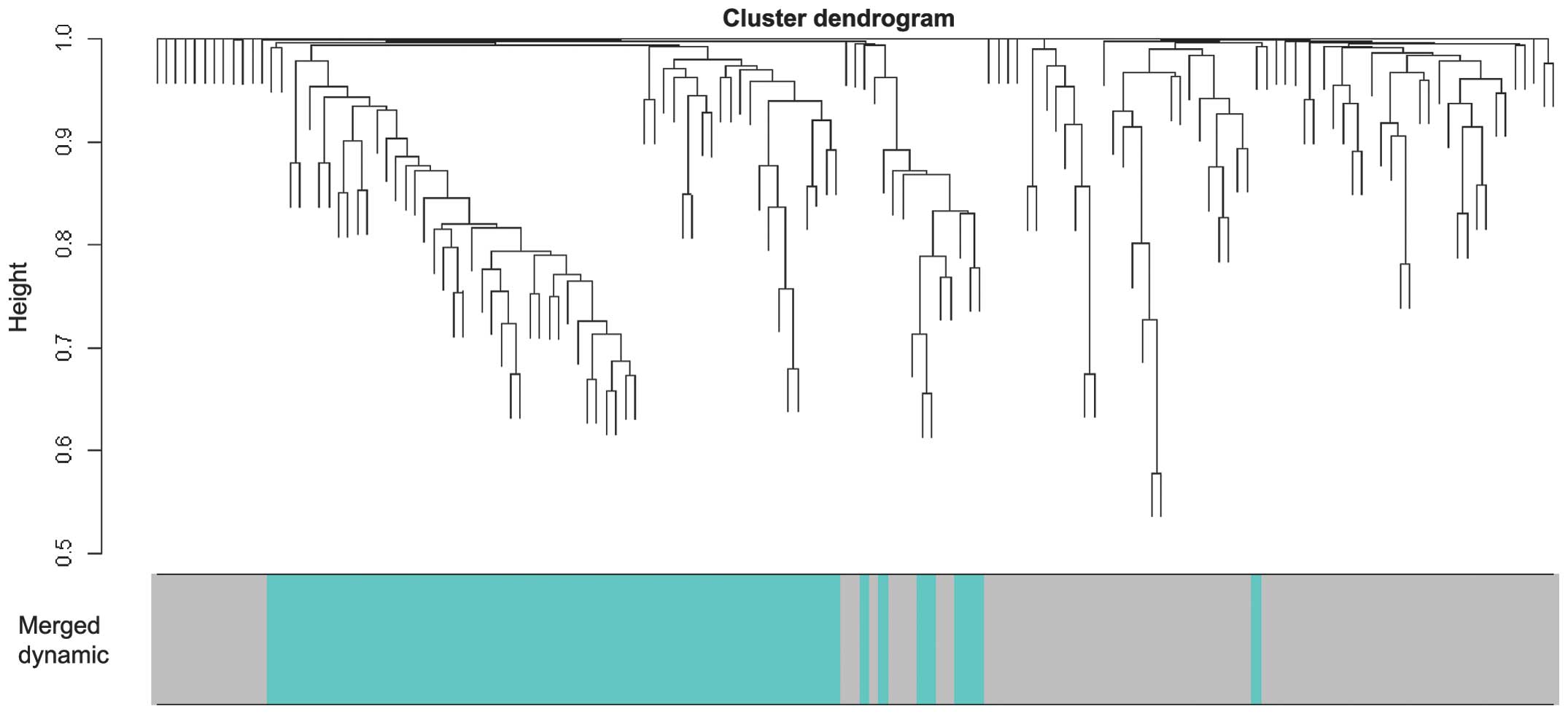
The 148 genes were subjected to WGCNA analysis,
where the genes exhibiting similar patterns of expression were
grouped into modules via hierarchical average linkage clustering
(Fig. 1). The present study used
two parameters (MS and P-value) to measure the relevance between
the modules and PAD. Initially, the MS value of each module was
calculated, and the higher the MS value, the more relevant the
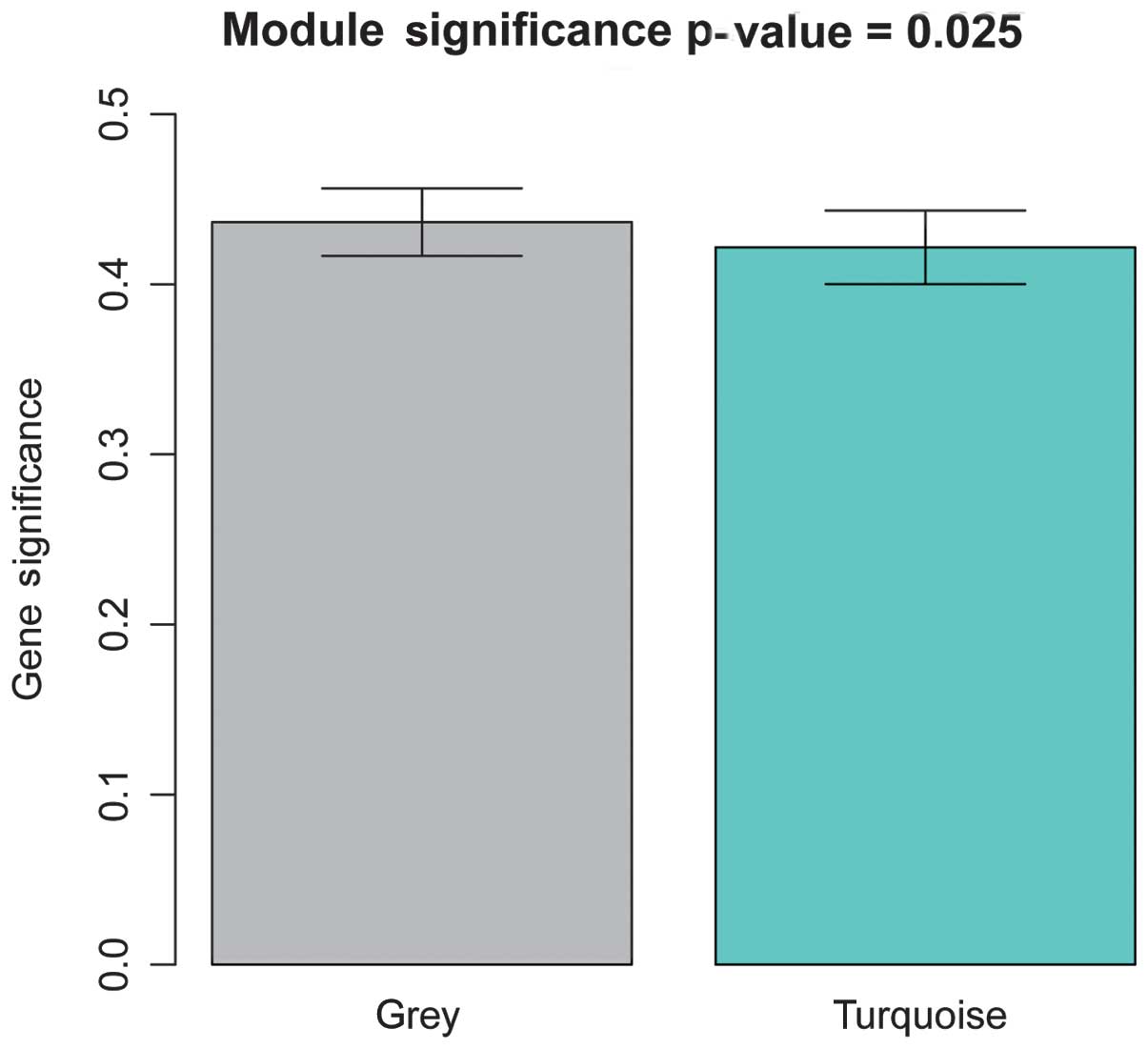
correlation between the module and PAD was. In total, two modules
(Fig. 2) were identified with a
threshold of P<0.05, which were termed the gray and turquoise
modules. Furthermore, the gray module was found to exhibit a higher
GS value, compared with the turquoise module, which suggested that
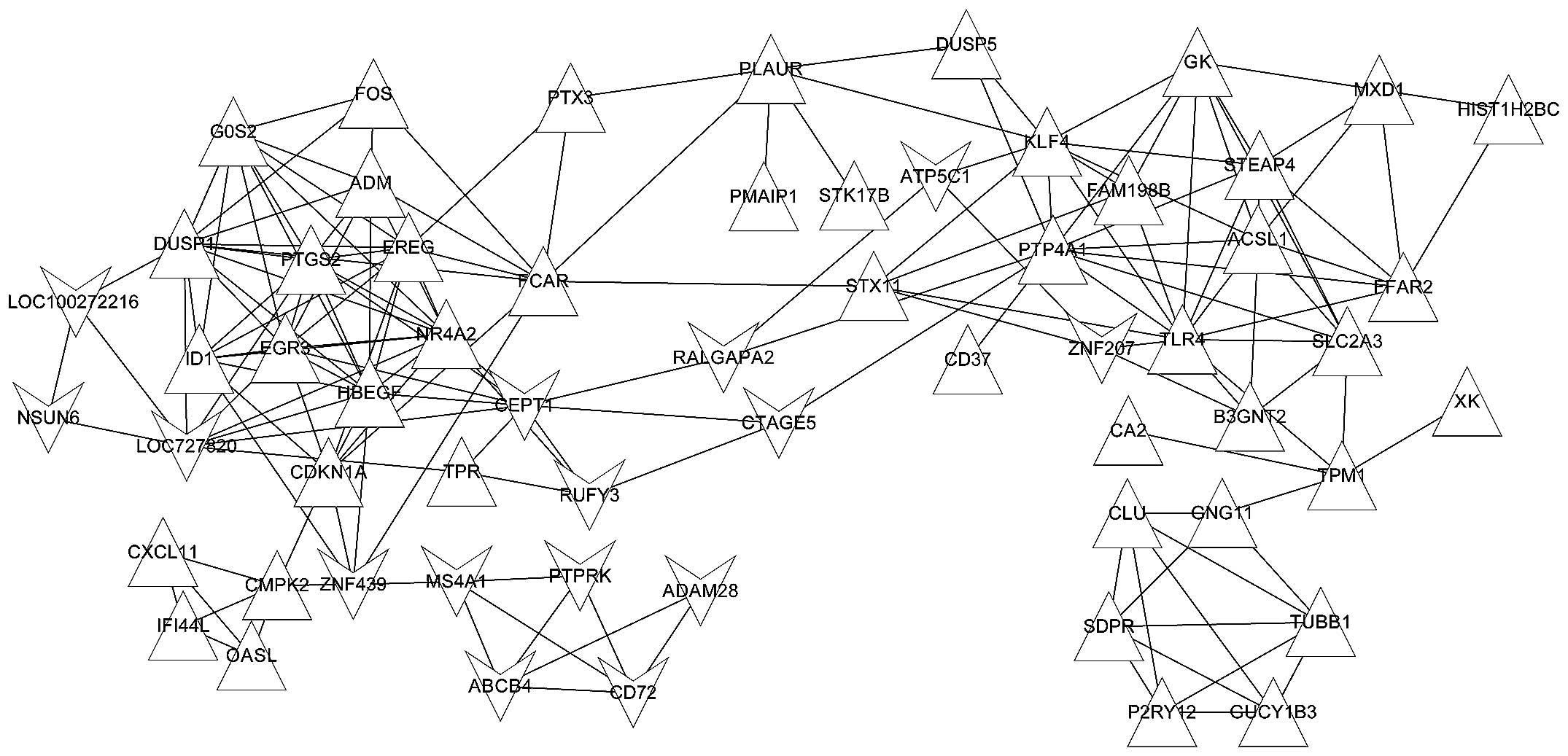
the gray module was most significant. Finally, a co-expression
network was constructed for the DEGs in the gray module with the
cutoff criterion of a correlation coefficient >0.7 between two
genes (Fig. 3). The resulting
co-expression network contained a total of 60 genes and 167
interactions.
Functional enrichment analysis for DEGs
in the gray module
Functional annotations and pathway enrichment
analysis of the DEGs in gray module were performed on the basis of
their gene composition. The GO functional analysis revealed that
the gray module was involved in responses to stimuli (Table I). For example, six DEGs, including
cyclin-dependent kinase inhibitor 1A (CDKN1A), FBJ murine
osteosarcoma viral oncogene homolog (FOS) and
prostaglandin-endo-peroxide synthase 2 (PTGS2), were
enriched in response to glucocorticoid stimulus; and eight DEGs,
including PTPRK, FOS and CDKN1A, were enriched
in response to inorganic substance. In addition, the pathway
analysis indicated that the gray module was significantly involved
in four pathways, including the ErbB signaling pathway
(HBEGF, CDKN1A and EREG), oxytocin signaling
pathway (FOS, CDKN1A and PTGS2), circadian
entrainment (GUCY1B3, FOS, GNG11) and
Toll-like receptor signaling pathway (FOS, CXCL11,
TLR4), as presented in Table
II.
 | Table IEnriched GO terms for the
differentially expressed genes in the gray module. |
Table I
Enriched GO terms for the
differentially expressed genes in the gray module.
| Accession no. | Term | Count | P-value | FDR | Gene |
|---|
| GO: 0051384 | Response to
glucocorticoid stimulus | 6 | 1.29E-05 | 0.020401798 | CDKN1A, FOS,
PTGS2, DUSP1, ADM, ABCB4 |
| GO: 0010035 | Response to
inorganic substance | 8 | 1.41E-05 | 0.022301137 | PTPRK, FOS,
CDKN1A, PTGS2, DUSP1, NR4A2, CA2, TPM1 |
| GO: 0010033 | response to organic
substance | 13 | 1.70E-05 | 0.026762768 | PTGS2, NR4A2,
TLR4, GNG11, PMAIP1, ABCB4, FOS, CDKN1A, ACSL1, ADM, DUSP1, ID1,
CA2 |
| GO: 0031960 | Response to
corticosteroid stimulus | 6 | 1.97E-05 | 0.031048526 | FOS, CDKN1A,
PTGS2, DUSP1, ADM, ABCB4 |
| GO: 0009991 | Response to
extracellular stimulus | 8 | 2.23E-05 | 0.035204556 | FOS, CDKN1A,
ACSL1, PTGS2, DUSP1, ADM, NR4A2, KLF4 |
| Table IIEnriched Kyoto Encyclopedia of Genes
and Genomes database pathways for the differentially expressed
genes in the gray module. |
Table II
Enriched Kyoto Encyclopedia of Genes
and Genomes database pathways for the differentially expressed
genes in the gray module.
| Term | Description | Input | Gene |
|---|
| hsa04012 | ErbB signaling
pathway | 1839|1026|2069 | HBEGF, CDKN1A,
EREG |
| hsa04921 | Oxytocin signaling
pathway |
2353|1026|5743|2983 | FOS, CDKN1A,
PTGS2, GUCY1B3 |
| hsa04713 | Circadian
entrainment | 2983|2353|2791 | GUCY1B3, FOS,
GNG11 |
| hsa04620 | Toll-like receptor
signaling pathway | 2353|6373|7099 | FOS, CXCL11,
TLR4 |
Discussion
PAD is among the common manifestations of systemic
atherosclerosis, which affects other major circulations involving
the cerebral and coronary circulations (23). However, the molecular mechanism
underlying the disease remains to be fully elucidated. In the
present study, two significant modules involved in PAD were
identified and screened, and the co-expressed network was
constructed using the WGCNA method by bioinformatics analysis. The
present study found that certain genes in the most significant
module were closely associated with the response to glucocorticoid
stimulus, determined using enrichment analysis. The present study
aimed to screen the important modules and genes, which were
identified to be associated with PAD and examine the molecular
mechanism of PAD.
The two modules were obtained using WGCNA, of which
the gray module was found to be the most significant module. The
genes in the gray module were associated with the responses to
stimulus. CDKN1A, FOS and PTGS2 were
significantly upregulated in the gray module. CDKN1A, also
known as p21/WAF1/CIP1, is a regulator of cell cycle progression at
the G1 and S phase (24), which is required for the response
to DNA damage by inducing cell cycle arrest, inhibiting DNA
replication and regulating fundamental cellular processes,
including apoptosis and gene transcription (25). Furthermore, CDKN1A can
interaction with proliferating cell nuclear antigen, which is a DNA
polymerase accessory factor and has a regulatory role in DNA damage
repair and the S phase of DNA replication (25,26).
In addition, CDKN1A has also been found to be associated
with atherosclerosis and myocardial infarction (27). The results of the present study
also showed that CDKN1A was involved in the responses to
stimulus, including the response to glucocorticoid stimulus,
response to inorganic substance and response to organic substance.
Therefore, the present study suggested that CDKN1A may be
involved in the processes and pathways, which are associated with
the cell cycle in patients with PAD.
FOS/c-FOS is a proto-oncogene, which is
induced by a variety of extracellular stimuli (28). FOS acts as a transcription
factor, which promotes the expression of specific cell cycle
regulatory genes (29) and leads
to early G1-phase cyclin accumulation, and enhances cyclin D- and
cyclin E-associated kinase activities (30). In addition, it has been reported
that FOS interacts with the c-jun proto-oncogene to form
transcription factor activating protein 1 (31), which is a crucial protein required
for cell adaptation to environmental changes (32). PTGS2, also termed
cyclooxygenase-2, is unexpressed under normal conditions in the
majority of cells. The inhibition of PTGS2 has been shown to
improve endothelial function in coronary artery disease (33). Additionally, PTGS2 has been
found to interact with caveolin 1 (34), which is a predominant component of
caveolae plasma membranes and is associated with the repair of DNA
damage through regulating the important molecules involved in
maintaining genomic integrity (35). Thus, FOS and PTGS2
are also cell cycle-associated genes and may be involved in the
pathogensis of PAD.
In conclusion, the present study used the WGCNA
approach to analyze the mRNA expression profile of PAD, and
obtained two key modules. The results suggested that the gray
module-associated genes may be associated with the development of
PAD, particularly CDKN1A, FOS and PTGS2. These
genes may be involved in the pathogenesis of PAD by regulating the
cell cycle, and offer potential for use as potential therapeutic
targets for PAD. However, further experiments are required to
verify these results.
References
|
1
|
Hiatt WR and Nehler MR: Peripheral
arterial disease. 2007
|
|
2
|
Hiatt WR: Medical treatment of peripheral
arterial disease and claudication. N Engl J Med. 344:1608–1621.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cronberg C, Sjöberg S, Albrechtsson U,
Leander P, Lindh M, Norgren L, Danielsson P, Sonesson B and Larsson
EM: Peripheral arterial disease. Contrast-enhanced 3D MR
angiography of the lower leg and foot compared with conventional
angiography. Acta Radiol. 44:59–66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ouriel K: Peripheral arterial disease.
Lancet. 358:1257–1264. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilson AM, Kimura E, Harada RK, Nair N,
Narasimhan B, Meng XY, Zhang F, Beck KR, Olin JW, Fung ET and Cooke
JP: Beta2-Microglobulin as a biomarker in peripheral arterial
disease: Proteomic profiling and clinical studies. Circulation.
116:1396–1403. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smadja DM, D'audigier C, Bièche I, Evrard
S, Mauge L, Dias JV, Labreuche J, Laurendeau I, Marsac B, Dizier B,
et al: Thrombospondin-1 is a plasmatic marker of peripheral
arterial disease that modulates endothelial progenitor cell
angiogenic properties. Arterioscler Thromb Vasc Biol. 31:551–559.
2011. View Article : Google Scholar
|
|
7
|
Busti C, Falcinelli E, Momi S and Gresele
P: Matrix metalloproteinases and peripheral arterial disease.
Intern Emerg Med. 5:13–25. 2010. View Article : Google Scholar
|
|
8
|
Masud R, Shameer K, Dhar A, Ding K and
Kullo IJ: Gene expression profiling of peripheral blood mononuclear
cells in the setting of peripheral arterial disease. J Clin
Bioinforma. 2:62012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Horvath S and Dong J: Geometric
interpretation of gene coexpression network analysis. PLoS Comput
Biol. 4:e10001172008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruan J, Dean AK and Zhang W: A general
co-expression network-based approach to gene expression analysis:
Comparison and applications. BMC Syst Biol. 4:82010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malki K, Tosto MG, Jumabhoy I, Lourdusamy
A, Sluyter F, Craig I, Uher R, McGuffin P and Schalkwyk LC:
Integrative mouse and human mRNA studies using WGCNA nominates
novel candidate genes involved in the pathogenesis of major
depressive disorder. Pharmacogenomics. 14:1979–1990. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Udyavar AR, Hoeksema MD, Clark JE, Zou Y,
Tang Z, Li Z, Li M, Chen H, Statnikov A, Shyr Y, et al:
Co-expression network analysis identifies spleen tyrosine kinase
(SYK) as a candidate oncogenic driver in a subset of small-cell
lung cancer. BMC Syst Biol. 7(Suppl 5): S12013. View Article : Google Scholar
|
|
13
|
Zhao H, Cai W, Su S, Zhi D, Lu J and Liu
S: Screening genes crucial for pediatric pilocytic astrocytoma
using weighted gene coexpression network analysis combined with
methylation data analysis. Cancer Gene Ther. 21:448–455. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995,
Database issue. 2013. View Article : Google Scholar
|
|
15
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of affymetrix genechip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics and Computational Biology Solutions
Using R and Bioconductor. Gentleman R, Carey V, Huber W, Irizarry R
and Dudoit S: Springer; New York: pp. 397–420. 2005, View Article : Google Scholar
|
|
17
|
Yip AM and Horvath S: Gene network
interconnectedness and the generalized topological overlap measure.
BMC Bioinformatics. 8:222007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langfelder P, Zhang B and Horvath S:
Defining clusters from a hierarchical cluster tree: The dynamic
tree cut package for R. Bioinformatics. 24:719–720. 2008.
View Article : Google Scholar
|
|
19
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:559–571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Da W, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu J, Mao X, Cai T, Luo J and Wei L: KOBAS
server: A web-based platform for automated annotation and pathway
identification. Nucleic Acids Res. 34:W720–W724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
American Disease Association: Peripheral
arterial disease in people with diabetes. J Am Podiatr Med Assoc.
95:309–319. 2005. View
Article : Google Scholar
|
|
24
|
Gartel AL and Radhakrishnan SK: Lost in
transcription: p21 repression, mechanisms and consequences. Cancer
Res. 65:3980–3985. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cazzalini O, Scovassi AI, Savio M, Stivala
LA and Prosperi E: Multiple roles of the cell cycle inhibitor
p21(CDKN1A) in the DNA damage response. Mutat Res. 704:12–20. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gulbis JM, Kelman Z, Hurwitz J, O'donnell
M and Kuriyan J: Structure of the C-terminal region of
p21(WAF1/CIP1) complexed with human PCNA. Cell. 87:297–306. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rodriguez I, Coto E, Reguero JR, González
P, Andrés V, Lozano I, Martín M, Alvarez V and Morís C: Role of the
CDKN1A/p21, CDKN1C/p57 and CDKN2A/p16 genes in the risk of
atherosclerosis and myocardial infarction. Cell Cycle. 6:620–625.
2007. View Article : Google Scholar
|
|
28
|
Abate C, Luk D, Gagne E, Roeder RG and
Curran T: Fos and jun cooperate in transcriptional regulation via
heterologous activation domains. Mol Cell Biol. 10:5532–5535. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hunter T: Oncoprotein networks. Cell.
88:333–346. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Braun-Dullaeus RC, Mann MJ and Dzau VJ:
Cell cycle progression New therapeutic target for vascular
proliferative disease. Circulation. 98:82–89. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mahner S, Baasch C, Schwarz J, Hein S,
Wölber L, Jänicke F and Milde-Langosch K: C-Fos expression is a
molecular predictor of progression and survival in epithelial
ovarian carcinoma. Br J Cancer. 99:1269–1275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bossis G, Malnou CE, Farras R,
Andermarcher E, Hipskind R, Rodriguez M, Schmidt D, Muller S,
Jariel-Encontre I and Piechaczyk M: Down-regulation of c-Fos/c-Jun
AP-1 dimer activity by sumoylation. Mol Cell Biol. 25:6964–6979.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chenevard R, Hürlimann D, Béchir M,
Enseleit F, Spieker L, Hermann M, Riesen W, Gay S, Gay RE, Neidhart
M, et al: Selective COX-2 inhibition improves endothelial function
in coronary artery disease. Circulation. 107:405–409. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liou JY, Deng WG, Gilroy DW, Shyue SK and
Wu KK: Colocalization and interaction of cyclooxygenase-2 with
caveolin-1 in human fibroblasts. J Biol Chem. 276:34975–34982.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu H, Yue J, Pan Z, Wu H, Cheng Y, Lu H,
Ren X, Yao M, Shen Z and Yang JM: Involvement of Caveolin-1 in
repair of DNA damage through both homologous recombination and
non-homologous end joining. PloS One. 5:e120552010. View Article : Google Scholar : PubMed/NCBI
|