1. Introduction
According to the Heart Disease and Stroke Statistics
for 2014, ~620,000 US citizens present with acute myocardial
infarction for the first time and ~295,000 experience recurrent
acute myocardial infarction per year (1). It was estimated that every 34 sec,
one coronary event occurs and every 1 min and 23 sec, one patient
succumbs to coronary heart disease (CHD) in the USA (1). Acute coronary syndrome (ACS) is
treated using effective methods including percutaneous coronary
intervention, coronary artery bypass grafting and thrombolytic
agents, which are, however, constrained by a series of
pathophysiological changes, including myocardial necrosis,
arrhythmia and myocardial stunning following myocardial
ischemia-reperfusion injury (MIRI). Hence, identification of
endogenous negative regulatory factors of MIRI is urgently required
and has become a hot spot of cardiovascular research. In the last
decade, activating transcription factor 3 (ATF3) has been found to
negatively and directly regulate Toll-like receptor 4 (TLR4) signal
transduction (2), which represents
the main pathway mediating the MIRI-associated inflammatory
response.
ATF3 is a member of the ATF/cyclic adenosine
mono-phosphate response element binding (CREB) family, which is
rich in basic-region leucine zipper (bZIP) (3). The ATF3 gene contains four exons
encoding a 181 amino-acid protein with a molecular weight of 22 kDa
(3). ATF3 forms a homodimer with
connection via the bZIP structure and also combines with other
ATF/CERB family members or CCAAT/enhancer binding protein (C/EBP)
family members to form heterodimers (4). The formation of the ATF3 homodimer
and ATF3 heterodimer have numerous functions in the activation of
gene-transcription in the cell nucleus (5): The homodimers exert suppressive
effects on target gene transcription, while the heterodimers exert
suppressive or activating effects based on the status of cell and
promoter. Therefore, the transcription of target genes containing a
promoter with ability to bind to the homodimer is restrained by
ATF3.
2. ATF3 expression in MIRI
Induction factors of ATF3 expression
ATF3 expression is usually ubiquitous at low levels
in normal quiescent cells in the absence of cellular stressors.
ATF3 is a stress-response gene of the early phase and is induced to
activate transcription under several conditions of cellular stress,
including oxidative stress, genetic toxicity and inflammatory
response, which leads to a large increase in cytokines,
growth-stimulating factors (6,7).
MIRI is caused by inflammatory responses (8), activation of the complement system
(9), calcium overload (10), oxidative stress (8), cell apoptosis (11,12)
and autophagy (11), which
promotes the stimulation of ATF3 expression in the early stage.
Yamamoto et al (13) subjected Sprague Dawley rats to IR
after lung transplantation and discovered that the expression of
ATF3 was markedly elevated at 30 min and even more at 180 min.
Furthermore, Wang et al (14) reported that in ischemic brains of
adult mice, the mRNA expression of ATF3 was 60-fold increased at 6
h, followed by a three-fold reduction at 24 h after ischemic onset.
Song et al (15) reported
that following middle cerebral artery occlusion and reperfusion
injury, the expression of ATF3 was markedly increased in the
ipsilateral peri-infarct cortex at 1–2 days, but was declined at 3
days. These studies implied that ATF3 expression is transiently and
not persistently upregulated after IR injury. When the myocardium
is subjected to ischemia or hypoxia, an adaptive upregulation in
the expression of ATF3 occurrs. A bioinformatics analysis of gene
expression the in left ventricle following acute myocardial
infarction performed by Zhang et al (16) revealed that a series of genes,
including ATF3, were key transcription factors associated with the
immune response. Krivoruchko and Storey (17) found that under anoxia, the
expression of ATF3 in the heart was increased at the protein and
mRNA level.
Signaling pathways stimulating ATF3
expression in MIRI
Upon MIRI, certain endogenous molecules are
released, including heat-shock protein 60, fibronectin, hyaluronic
acid, defensin 3 and cell debris, which are referred to as
danger-associated molecular patterns (DAMPs) (18–20).
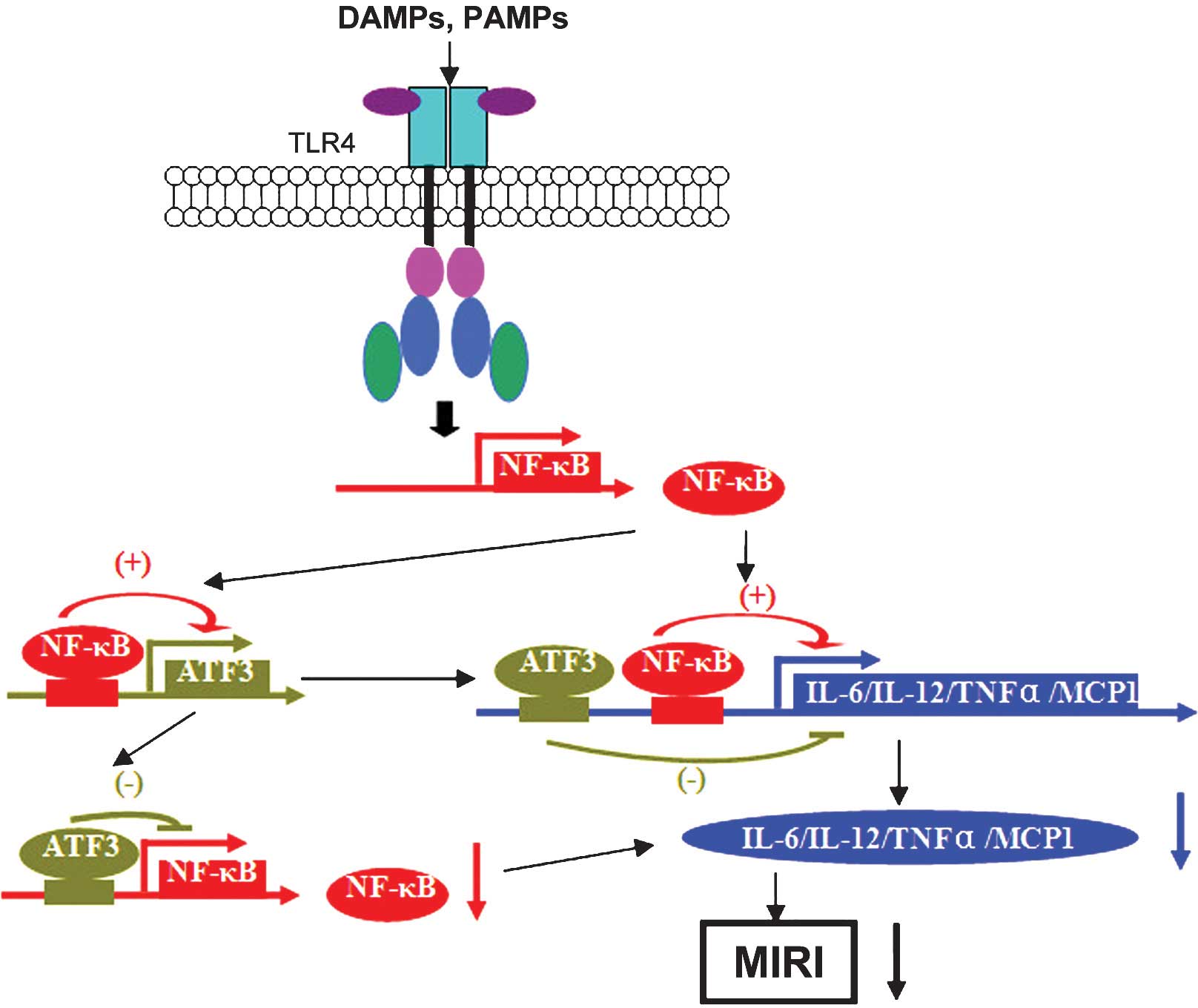
The recognition of DAMP by TLR4, which is located on the myocardial
cell surface, activates the myeloid differentiation factor 88
(MyD88)-dependent pathway as well as the Toll-like receptor adaptor
molecule 1 (TRIF)-dependent pathway, the so-called
MyD88-independent pathway, to promote the intranuclear
translocation of nuclear factor-κB (NF-κB). MIRI induces the
production of numerous inflammatory factors and chemotactic factors
and activation of immune cells (21,22)
as a result of myocardial damage.
A number of studies indicated that the TLR4/NF-κB
signaling pathway participates in the expression of ATF3 (2,23,24).
Whitmore et al (23) showed
various TLRs that rapidly increased ATF3 expression in macrophages
and plasmacytoid dendritic cells in mice and humans. Gilchrist et
al (2) performed cDNA microarrays
on macrophages following their activation with bacterial
lipopolysaccharide (LPS), a TLR4 agonist, revealing that ATF3 mRNA
expression peaked at 1 h. These studies suggested that ATF3
expression was induced by TLR4/NF-κB signaling. A study by Suganami
et al (24) further
verified this result in vitro and in vivo by
demonstrating that in RAW264 macrophages, the saturated fatty acids
LPS, stearate and palmitate markedly increased the expression of
ATF3 at the protein and mRNA level, while palmitate did not affect
the mRNA expression of ATF3 in peritoneal macrophages from
TLR4-knockdown C3H/HeJ mice. Furthermore, treatment of RAW264
macrophages with the NF-κB inhibitor BAY11-7085 significantly
suppressed the palmitate-induced mRNA expression of ATF3, while
ATF3 promoter activity was significantly enhanced in HEK293 cells
following selective NF-κB activation via transient overexpression
of its p65 and p50 sub-units (24). These observations suggested that
the expression of ATF3 is induced via the TLR4/NF-κB pathway.
3. Protective effects of ATF3 against
MIRI
Role of ATF3 in IRI
While ATF3 expression is induced by activation of
the TLR4/NF-κB signaling pathway, ATF3 also negatively regulates
the TLR4 signal transduction pathway (2,23),
thereby exerting a protective effect against IRI. Of note, the
protective effects of ATF3 have been documented with regard to IRI
in the kidney (25–27), cerebrum (14) and liver (28). Li et al (25) observed that ATF3-deficient mice
exhibited increased rates of mortality, kidney dysfunction,
inflammation and apoptosis compared with those of wild-type mice
following renal IR, while gene transfer-mediated restoration of
ATF3 in the kidney protected these ATF3-deficient mice from
IR-induced injury. Yoshida et al (26) utilized adenovirus-mediated gene
transfer to overexpress ATF3 in mice, which had a protective effect
against renal IRI. Furthermore, Chen et al (27) reported that in ATF3-deficient mice,
the induction of adhesion molecules P- and E-selectin,
interleukin-6, intercellular adhesion molecule, vascular cellular
adhesion molecule and monocyte chemotactic protein-1 was enhanced
compared with that in wild-type mice during renal IR-induced
inflammation. Their in vitro study showed that MCP-1
expression in epithelial cells as well as macrophage migration was
inhibited by epithelium-derived exosomal ATF3 RNA and that
IR-induced kidney injury was attenuated following administration of
exosome containing ATF3 RNA derived from epithelial cells (27). In the research of brain injury
after transient focal cerebral ischemia, knockout of ATF3
significantly exacerbated the infarct volume and worsened
neurological function and up regulated neural apoptosis,
inflammatory gene expression and cellular inflammatory response
(14). In mouse models of warm and
cold liver IRI, Rao et al (28) showed that ATF3 deficiency
significantly aggravated IR-induced liver injury and demonstrated
that ATF3 mediated local cytoprotection against TLR4-driven
inflammation in liver IRI (28).
While only few studies have investigated the role of
ATF3 in MIRI, certain protective effects have been indicated
(29). Furthermore, Brooks et
al (30) observed an obvious
increase of inflammatory cells and neutrophils in ATF3-null hearts
subjected to myocardial IR after ischemic pre-conditioning
(30). While genetic deletion of
ATF3 did not decrease the inflammatory response and attenuated
monocyte and neutrophil infiltration in non-pre-conditioned hearts,
it abolished the cardioprotective effects of ischemic
pre-conditioning (30). These
results suggested that ATF3 may negatively regulate the
inflammatory response in MIRI.
Mechanisms of ATF3-mediated attenuation
of MIRI Anti-inflammatory effects of ATF3
The TLR4 downstream signaling cascade of the
inflammatory pathway promotes the release of interleukin(IL)-6 and
IL-12b, whose promoter regions contain binding sites for CREB/ATF
and NF-κB (2). ATF3 binds to the
ATF/CREB sites in the promoter regions of the target genes and
thereby inhibits the binding of NF-κB, which suppresses the
transcription of IL-6 and IL-12b (2). Furthermore, transcription of the
inflammatory factor tumor necrosis factor (TNF)-α was shown to be
restrained by ATF3 by blocking the AP-1 binding site in its
promoter to interfere with the transcriptional activation by NF-κB
(24,31). The effect of ATF3 binding to the
CREB/ATF or AP-1 recognition site of target genes is linked with
ATF3-associated histone deacetylase (HDAC), which performs
chromosomal remodeling, which has an important role in the
regulation of transcriptional activity (32,33).
While histone acetyltransferases mediate the acetylation of
histones to unwind the chromatin structure, which provides access
by positive transcriptional regulators and thereby promotes
transcriptional activation, HDACs have the opposite effect and
mediate chromatin condensation, which represses transcriptional
activation due to the resulting inaccessibility of gene promoters.
Numerous studies have confirmed that ATF3-associated HDAC caused
condensation of the chromatin structure and blocked the binding of
NF-κB to the promoter regions of IL-6, IL-12, TNF-α and MCP-1,
which inhibited the transcription of these inflammatory factors and
attained the suppression of TLR4-associated inflammatory signaling
pathways (23,25,34).
Furthermore, it has been evidenced that HDAC in had protective
effects against MIRI (35,36). In conclusion, ATF3 inhibits
TLR4/NF-κB inflammatory signaling to reduce MIRI (Fig. 1).
Anti-apoptotic effects of ATF3
Yoshida et al (26) revealed that adenovirus-mediated
overexpression of ATF3 in HK2 cells reduced
H2O2-induced cell death and this protective
effect was associated with a decrease of p53 mRNA and an increase
of p21 mRNA. ATF3 was also shown to prevent reactive oxygen
species-induced renal injury via upregulation of p21 and
downregulation of p53 (33). It is
well known that protein kinase inhibitor p21 has a vital role in
cell apoptosis by regulating cell-cycle progression, while p53 is
able to induce apoptosis and suppress cell proliferation;
therefore, enhancement of p21 and reduction of p53 expression
following overexpression of ATF3 leads to cell-cycle arrest and
prevents apoptosis. Krivoruchko and Storey (17) demonstrated that ATF3 prevented
adriamycin-induced myocardial-cell apoptosis by blocking the
transcription of p53 through binding to the PF-1 site of its gene
promoter, thereby exerting protective effects on the myocardium.
Thus, the prevention of apoptosis is another mechanism of the
protective effects of ATF3 against MIRI.
4. Conclusion
ATF/CERB family member ATF3 is considered to be a
regulatory factor of gene transcription. While ATF3 expression is
maintained at low levels in normal quiescent cells, it is induced
by several cellular stressors, including IRI, which triggers the
activation of the TLR4/NF-κB signaling pathway as an early stage of
the molecular stress response. A large number of studies have
demonstrated that ATF3 homodimer binds to the promoter regions of
its target genes and recruits HDAC, which in turn inhibits the
transcription of these genes to interrupt the synthesis of the
associated inflammatory and chemotactic factors involved in the
TLR4/NF-κB pathway, which ultimately exerts protective effects
against MIRI. Although few studies have reported the protective
effects of endogenous regulatory factor ATF3 in MIRI, the
anti-inflammatory and anti-apoptotic effects of ATF3 are
well-documented; there fore overexpression of ATF3 may represent a
promising therapeutic strategy for the prevention of MIRI.
Acknowledgments
The present study work was supported by the National
Natural Science Foundation of China (grant nos. 81170133, 81200088
and 81470387) and the Outstanding Medical Academic Leader Program
of Hubei Province (grant no. 201304).
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Executive summary: Heart disease and stroke statistics-2014 update:
A report from the American Heart Association. Circulation.
129:399–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gilchrist M, Thorsson V, Li B, Rust AG,
Korb M, Roach JC, Kennedy K, Hai T, Bolouri H and Aderem A: Systems
biology approaches identify ATF3 as a negative regulator of
Toll-like receptor 4. Nature. 441:173–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hai TW, Liu F, Coukos WJ and Green MR:
Transcription factor ATF cDNA clones: An extensive family of
leucine zipper proteins able to selectively form DNA-binding
heterodimers. Genes Dev. 3:2083–2090. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen BP, Liang G, Whelan J and Hai T: ATF3
and ATF3 delta Zip: Transcriptional repression versus activation by
alternatively spliced isoforms. J Biol Chem. 269:15819–15826.
1994.PubMed/NCBI
|
|
5
|
Hai T and Curran T: Cross-family
dimerization of transcription factors Fos/Jun and ATF/CREB alters
DNA binding specificity. Proc Natl Acad Sci USA. 88:3720–23724.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang HY, Wek SA, McGrath BC, Lu D, Hai T,
Harding HP, Wang X, Ron D, Cavener DR and Wek RC: Activating
transcription factor 3 is integral to the eukaryotic initiation
factor 2 kinase stress response. Mol Cell Biol. 24:1365–1377. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hai T, Wolford CC and Chang YS: ATF3, a
hub of the cellular adaptive-response network, in the pathogenesis
of diseases: Is modulation of inflammation a unifying component?
Gene Expr. 15:1–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang M, Chen J, Zhao J and Meng M:
Etanercept attenuates myocardial ischemia/reperfusion injury by
decreasing inflammation and oxidative stress. PLoS One.
9:e1080242014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ilczuk T, Wasiutynski A, Wilczek E and
Gornicka B: The study of the protein complement in myocardial
infarction. Immunol Lett. 162:262–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma HJ, Li Q, Ma HJ, Guan Y, Shi M, Yang J,
Li DP and Zhang Y: Chronic intermittent hypobaric hypoxia
ameliorates ischemia/reperfusion-induced calcium overload in heart
via Na/Ca2+ exchanger in developing rats. Cell Physiol Biochem.
34:313–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J,
Zhang L, Tang Y, Liu J, Zhang X and Gao F: Mitochondrial JNK
activation triggers autophagy and apoptosis and aggravates
myocardial injury following ischemia/reperfusion. Biochim Biophys
Acta. 1852:262–270. 2015. View Article : Google Scholar
|
|
12
|
Kim MY, Lim SH and Lee J: Intake of hot
water-extracted apple protects against myocardial injury by
inhibiting apoptosis in an ischemia/reperfusion rat model. Nutr
Res. 34:951–960. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamamoto S, Yamane M, Yoshida O, Okazaki
M, Waki N, Toyooka S, Oto T and Miyoshi S: Activations of
mitogen-activated protein kinases and regulation of their
downstream molecules after rat lung transplantation from donors
after cardiac death. Transplant Proc. 43:3628–3633. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang L, Deng S, Lu Y, Zhang Y, Yang L,
Guan Y, Jiang H and Li H: Increased inflammation and brain injury
after transient focal cerebral ischemia in activating transcription
factor 3 knockout mice. Neuroscience. 220:100–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song DY, Oh KM, Yu HN, Park CR, Woo RS,
Jung SS and Baik TK: Role of activating transcription factor 3 in
ischemic penumbra region following transient middle cerebral artery
occlusion and reperfusion injury. Neurosci Res. 70:428–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang T, Zhao LL, Cao X, Qi LC, Wei GQ,
Liu JY, Yan SJ, Liu JG and Li XQ: Bioinformatics analysis of time
series gene expression in left ventricle (LV) with acute myocardial
infarction (AMI). Gene. 543:259–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krivoruchko A and Storey KB: Activation of
the unfolded protein response during anoxia exposure in the turtle
Trachemys scripta elegans. Mol Cell Biochem. 374:91–103. 2013.
View Article : Google Scholar
|
|
18
|
Arslan F, de Kleijn DP and Pasterkam G:
Innate immune signaling in cardiac ischemia. Nat Rev Cardiol.
8:292–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng Y and Chao W: Toll-like receptors and
myocardial inflammation. Int J Inflam. 9:170352–170373. 2011.
|
|
20
|
Delarosa O, Dalemans W and Lombardo E:
Toll-like receptors as modulators of mesenchymal stem cells. Front
Immunol. 3:1822012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
DelaRosa O and Lombardo E: Modulation of
adult mesenchymal stem cells activity by toll-like receptors:
Implications on therapeutic potential. Mediators of Inflamm.
10:865601–865609. 2010.
|
|
22
|
Shimazu R, Akashi S, Ogata H, Nagai Y,
Fukudome K, Miyake K and Kimoto M: MD-2, a molecule that confers
lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp
Med. 189:1777–1782. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Whitmore MM, Iparraguirre A, Kubelka L,
Weninger W, Hai T and Williams BR: Negative regulation of
TLR-signaling pathways by activating transcription factor-3. J
Immunol. 179:3622–3630. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suganami T, Yuan X, Shimoda Y,
Uchio-Yamada K, Nakagawa N, Shirakawa I, Usami T, Tsukahara T,
Nakayama K, Miyamoto Y, et al: Activating transcription factor 3
constitutes a negative feedback mechanism that attenuates saturated
fatty acid/toll-like receptor 4 signaling and macrophage activation
in obese adipose tissue. Circ Res. 105:25–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li HF, Cheng CF, Liao WJ, Lin H and Yang
RB: ATF3-mediated epigenetic regulation protects against acute
kidney injury. J Am Soc Nephrol. 21:1003–1013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshida T, Sugiura H, Mitobe M, Tsuchiya
K, Shirota S, Nishimura S, Shiohira S, Ito H, Nobori K, Gullans SR,
et al: ATF3 protects against renal ischemia-reperfusion injury. J
Am Soc Nephrol. 19:217–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen HH, Lai PF, Lan YF, Cheng CF, Zhong
WB, Lin YF, Chen TW and Lin H: Exosomal ATF3 RNA attenuates
pro-inflammatory gene MCP-1 transcription in renal
ischemia-reperfusion. J Cell Physiol. 229:1202–1211. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rao J, Qian X, Li G, Pan X, Zhang C, Zhang
F, Zhai Y, Wang X and Lu L: ATF3-mediated NRF2/HO-1 signaling
regulates TLR4 innate immune responses in mouse liver
ischemia/reperfusion injury. Am J Transplant. 15:76–87. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
da Silva R, Lucchinetti E, Pasch T, Schaub
MC and Zaugg M: Ischemic but not pharmacological preconditioning
elicits a gene expression profile similar to unprotected
myocardium. Physiol Genomics. 20:117–130. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brooks AC, Guo Y, Singh M, McCracken J,
Xuan YT, Srivastava S, Bolli R and Bhatnagar A: Endoplasmic
reticulum stress-dependent activation of ATF3 mediates the late
phase of ischemic preconditioning. J Mol Cell Cardiol. 76:138–147.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim HB, Kong M, Kim TM, Suh YH, Kim WH,
Lim JH, Song JH and Jung MH: NFATc4 and ATF3 negatively regulate
adiponectin gene expression in 3T3-L1 adipocytes. Diabetes.
55:1342–1352. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kouzarides T: Acetylation: A regulatory
modification to rival phosphorylation? EMBO J. 19:1176–1179. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheung P, Allis CD and Sassone-Corsi P:
Signaling to chromatin through histone modifications. Cell.
103:263–271. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng CF and Lin H: Acute kidney injury
and the potential for ATF3-regulated epigenetic therapy. Toxicol
Mech Methods. 21:362–366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao TC, Cheng G, Zhang LX, Tseng YT and
Padbury JF: Inhibition of histone deacetylases triggers
pharmacologic preconditioning effects against myocardial ischemic
injury. Cardiovasc Res. 76:473–481. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Granger A, Abdullah I, Huebner F, Stout A,
Wang T, Huebner T, Epstein JA and Gruber PJ: Histone deacetylase
inhibition reduces myocardial ischemia-reperfusion injury in mice.
FASEB J. 22:3549–3560. 2008. View Article : Google Scholar : PubMed/NCBI
|