Introduction
Cancer cachexia is a common syndrome, characterized
by skeletal muscle wasting, with or without loss of fat mass.
Systemic inflammation is essential for the pathogenesis of cancer
cachexia. C-reactive protein (CRP), a marker of systemic
inflammation, has been found to be elevated early in cancer
cachexia (1) and is associated
with decreased skeletal muscle mass (2). It has been reported (3,4) that
the levels of certain pro-inflammatory cytokines, including tumor
necrosis factor (TNF)-α and interleukin (IL)-6, are increased in
the serum of the mice with cancer cachexia and in patients with
cancer cachexia. Other studies (5,6) have
demonstrated that TNF-α is directly involved in cachexia by
inhibiting lipoprotein lipase and enhancing protein degradation,
and that IL-6 promotes skeletal muscle atrophy via signal
transducer and activator of transcription-3 (STAT3) signaling.
The liver is crucial for systemic inflammation in
cancer cachexia. A previous study (7) demonstrated that the number of IL-6
and IL-1 immunoreactive cells is significantly increased in the
locality of CD68-positive areas of the liver in cancer cachexia,
and that areas of CD68-positive macrophages in liver biopsies ares
increased in the patients with a more aggressive grades of tumor.
Further studies have demonstrated that Kupper cells and hepatocytes
act as a major source of circulating pro-inflammatory cytokines,
including TNF-α, IL-6 and proteolysis inducing factor, in cancer
cachexia, via the NF-κB- and STAT3-dependent signaling pathways
(8), or the cyclooxygenase-2
(Cox2)/prostaglandin E2 (PGE2) pathway (9). However, there remains controversy
regarding the effect of COX-2 on circulating levels of IL-6
(10).
L-carnitine is a key regulator of lipid metabolism
and exerts an anti-inflammatory effect in several disease settings.
For example, L-carnitine protects against carboplatin-mediated
renal injury by inhibiting renal tubular cell apoptosis (11) and, L-carnitine has been
demonstrated to prevent the progression of non-alcoholic
steatohepatitis in a mouse model by upregulating the mitochondrial
β-oxidation and redox system, accompanied by decreases in the
levels of IL-1 and TNF-α in the liver (12). In addition, L-carnitine has been
demonstrated to decrease the protein levels of TNF-α and IL-6 in
the fibrotic liver (13). Notably,
the levels of carnitine are markedly decreased in the serum of
patients with cancer cachexia (14). Oral supplementation of L-carnitine
prevent glutathione from decreasing further in tumor-bearing mice,
suggesting that it exerts a beneficial antioxidant effect in cancer
cachexia (15). However, the
effect of L-carnitine on the liver inflammatory response in cancer
cachexia remains to be elucidated.
It is understood that lipid metabolism disorders can
induce a pro-inflammatory response in the liver (16). A previous study (17) demonstrated that L-carnitine induces
the recovery of liver lipid metabolism dysfunction in cancer
cachexia, and is associated with the regulation of the expression
levels of carnitine palmitoyltransferase I and II (CPT I and II).
Our previous study (3)
demonstrated that L-carnitine ameliorates cachectic symptoms by
regulating the expression and activity of carnitine
palmitoyltransferase (CPT) in the liver, accompanied by a decrease
in the elevated serum levels of TNF-α and IL-6, suggesting that CPT
is involved in a certain aspect of the liver inflammatory response,
regulated by L-carnitine. Additionally, L-carnitine upregulates
peroxisome proliferator-activated receptor (PPAR)γ (18), a key regulator in the liver
inflammatory response and oxidative stress (19,20),
which has been found to be involved in regulating the expression of
CPT I (21). These findings led
the present study to hypothesize that L-carnitine may improve the
liver inflammatory response by regulating the CPT I-dependent PPARγ
signaling pathway. Therefore, the aim of the present study was to
investigate the role of the CPT I-dependent PPARγ signaling pathway
in the ameliorative effect of L-carnitine on the liver inflammatory
response in cancer cachexia in a colon-26 tumor-bearing mouse
model.
Materials and methods
Animals and cachexia model
The animal experiments performed in the present
study were approved by the Institute of Animal Use and Care
Committee of Tongji University (Shanghai, China). Adult male BALB/c
mice weighing 22–26 g were obtained from the Experimental Animal
Center of Tongji University (Shanghai, China) and housed at 24°C
with a 12-h light/dark cycle, and free access to water and mouse
chow. Cancer cachexia was induced in colon-26 tumor-bearing BALB/c
mice, as described in a previous study by our group (3). To establish the cachexia model, tumor
cells (1×106 cells in 0.1 ml of saline) were
subcutaneously inoculated into the right axillary fossa of BALB/c
mice.
Groups and experimental protocol
Based on the results obtained from our previous
study (3), cancer cachexia was
considered fully developed 11 days following tumor inoculation.
Therefore, subsequent interventions in the present study were
initiated on day 12.
Experiment 1
A total of 18 tumor-bearing mice were equally
randomized into a vehicle control group, which received oral
administration of 2 ml saline daily; an L-carnitine group, which
received oral administration of 9 mg/kg daily (cat. no. C0158;
Sigma-Aldrich, St. Louis, MO, USA); and an L-carnitine+etomoxir
group, which received oral administration of 9 mg/kg L-carnitine
daily and intraperitoneal administration of 20 mg/kg etomoxir, an
inhibitor of CPT I (cat. no. E1905; Sigma-Aldrich) daily for 7
days.
Experiment 2
At the same time, a separate group of 30
tumor-bearing mice were equally randomized into a pioglitazone
group, GW9662 group, L-carnitine+pioglitazone group,
L-carnitine+GW9662 group and L-carnitine+GW9662+ pyrrolidine
dithiocarbamate (PDTC) group. The treatment administration was as
follows: Pioglitazone hydrochloride, a specific agonist of PPARγ
(10 mg/kg orally daily; cat. no. E6910; Sigma-Aldrich); GW9662, a
selective inhibitor of PPARγ (1 mg/kg daily intaperitoneally; cat.
no. M6191; Sigma-Aldrich); L-carnitine (9 mg/kg orally) +
pioglitazone (10 mg/kg per day orally); L-carnitine (9 mg/kg
orally) + GW9662 (1 mg/kg per day intraperitoneally); and
L-carnitine (9 mg/kg, orally) + GW9662 (1 mg/kg per day
intraperitoneally) and PDTC (120 mg/kg per day intraperitoneally),
a selective inhibitor of nuclear factor (NF)-κB, (cat. no. P8765;
Sigma-Aldrich;), respectively. In addition, six healthy mice
received no treatment, and were used as a normal control group.
Following intervention for 7 days, all mice in each
group in experiments 1 and 2 were anesthetized with 2%
intraperitoneal pentobarbital (40 mg/kg i.p.; Beyotime Institute of
Biotechnology, Haimen, China) and weighed. Blood (1.5 ml per mouse)
was collected from the inferior vena cava, close to the entrance of
the hepatic vein. Peripheral blood mononuclear cells (PBMCs) were
isolated for the measurement of NF-κB p65 using Ficoll-Isopaque
Plus density-gradient centrifugation at 800 × g for 20 min at 20°C
(cat. no. 10771; Sigma-Aldrich). The levels of serum inflammatory
agents (IL-6, TNF-α, PGE2 and CRP) and the oxidative stress
markers, malondialdehyde, (MDA) and superoxide dismutase (SOD) and
glutathione peroxidase(GSH-Px) were measured. The mice were
sacrificed by cervical dislocation. The intact liver was isolated
and stored in liquid nitrogen (Sinopharm Chemical Regent Co., Ltd.,
Shanghai, China).
Measurement of pro-inflammatory markers
and oxidative stress markers
The serum levels of TNF-α, IL-6, PGE2 and CRP were
detected using a enzyme linked immunosorbent assay (ELISA) kits
(TNF-α, cat. no. MTA00B; IL-6, cat. no. M6000B; PGE2, cat. no.
KGE004B; CRP, cat. no. MCRP00; R&D systems, Inc., Minneapolis,
MN, USA), according to the manufacturer's protocol. A total of 50
µl serum per well was added to a 96-well plate, followed by
incubation at 37°C for 2 h and subsequent determination of the
color intensity at 450 nm. MDA was measured using a thiobarbituric
acid reactive substance assay method, as described previously
(22). The reaction products were
obtained by isolating the organic layer and read at 532 nm. Serum
levels of SOD and GSH-Px were detected using kits (cat. nos. A001-1
and A006-1, respectively; Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) according to the manufacturer's
protocol. The optical density value was read on a spectrophotometer
(F96PRO; Shanghai Lengguang Industrial Co., Ltd, Shanghai,
China).
Histological analysis
The liver tissues were paraffin-embedded, sliced
into 5 µm sections and stained with hematoxylin and eosin
(Beyotime Institute of Biotechnology) for assessment of the degree
of liver inflammation, according to previously published criteria
(23). The scores (0–8) were used
for the assessment of steatosis, lobular inflammation and
hepatocyte ballooning. The liver sections were observed under the
light microscope equipped with a 10× objective (BM-600B; Ningbo
Barride Optics Co., Ltd., Ningbo, China).
Immunohistochemical analysis
The liver sections were incubated with 0.3% (v/v)
hydrogen peroxide (Beyotime Institute of Biotechnology) for 30 min
at room temperature to quench endogenous peroxidase activity, and
were then blocked for 2 h in phosphate-buffered saline (PBS;
Beyotime Institute of Biotechnology) containing 5% normal goat
serum and 2% bovine serum albumin (Beyotime Institute of
Biotechnology). Monoclonal antibodies (diluted 1:200) against PPARγ
(Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA; cat. no.
sc-7273) or PPARα (Abcam, Cambridge, MA; cat. no. ab2779) were
incubated with the fixed sections for 2 h, followed by five rinses
with PBS. The sections then were incubated with horseradish
peroxidase-conjugated goat anti-mouse antibody (diluted 1:500) for
1 h at room temperature. The relative expression levels of PPARα
and PPARγ were semi-quantitated as integrated optical density/area,
as described previously (24).
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the PBMCs using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA; cat. no. 15596-018), according to the manufacturer's
instructions. The cDNA was generated using a MultiScribe Reverse
Transcriptase kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.; cat. no. 4368814). RNA (2 µg) was reverse-transcribed
to cDNA with 1 µl oligo(dT) (0.1 µg/µl) and 5
µl 0.1% diethylpyrocarbonate-treated H2O at 70°C
for 5 min. The primers used were as follows: Forward 5′-ACA GAC CCA
GGA GTG ACA A-3′ and reverse 5′-CAT GGA CAC ACC CTG GTT CAG-3′ for
NF-κB p65; and forward 5′-TGG TGG ACC TCA TGG CCT AC-3′ and reverse
5′-GCA ACT GAG GGC CTC TCT-3′ for GAPDH. All primers were
synthesized by Sangon Biotech Co., Ltd (Shanghai, China). The qPCR
reactions were performed using SYBR green PCR master mix (Qiagen,
Shanghai, China; cat. no. 204141) in a 50-µl PCR reaction
containing 1 µl cDNA using an iCycler thermocycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) with the following
thermocycling conditions: 50°C for 2 min and 95°C for 15 min,
followed by 40 cycles of 94°C for 15 sec, 55°C for 30 sec and 72°C
for 30 sec. PCR products were detected using an ABI7500 Real-Time
PCR Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The house-keeping gene, GAPDH, was used as an internal
control. Data were normalized to GAPDH, and the relative expression
levels were calculated using the 2−ΔΔCq method as
described previously (3).
Experiments were performed in triplicate samples.
Western blot analysis
Total protein extracts were obtained by
homogenization of tissues using protein sample buffer [100 mM
Tris-HCl (pH 6.8), 200 mM dithiothreitol, 4% sodium dodecyl sulfate
(SDS), 0.2% bromphenol blue and 20% glycerol] and a classic
protease inhibitor cocktail (Beyotime Institute of Biotechnology).
Protein concentrations were measured by the bicinchoninic acid
method (Pierce Biotechnology, Inc., Rockford, IL, USA). Protein
samples were heated at 100°C for 10 min, and 40 µg was
applied to a 10% SDS-polyacrylamide gel. Following electrophoresis,
the proteins were electrophoretically transferred onto
polyvinylidene difluoride membranes (Millipore Co., Billerica, MA,
USA). The membranes were stained with 0.5% Ponceau S (Beyotime
Institute of Biotechnology) to assure equal protein loading,
blocked for 1 h with 5% powdered non-fat dry milk in 25 mM Tris-HCl
(pH 8.0), 144 mM NaCl and 0.1% Tween 20 (TBS-T), and incubated
overnight at 4°C with the following primary antibodies: PPAR-α,
PPAR-γ, NF-κB P65 (cat. no. ab16502; Abcam), Cox-2 (catalog no.
sc-166475; Santa Cruz Biotechnology) and β-actin (cat. no. ab8227;
Abcam). Following incubation with the goat anti-rabbit (cat. no.
A0277) or mouse (cat. no. A0286) secondary antibodies (Beyotime
Institute of Biotechnology; 1:2,000 dilution), the membranes were
briefly washed twice and then three times for 10 min each with
TBS-T. Immunodetected proteins were visualized in a
FluorChem® HD2 analysis system (Protein Simple Co.,
Shanghai, China) using the enhanced chemiluminescent ECL assay kit
(Santa Cruz Biotechnology, Inc.) according to the manufacturer's
recommended protocol.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using analysis of variance, followed by
a least significant difference t-test for post-hoc comparison. SPSS
13.0 software (SPSS, Inc., Chicago, IL, USA) was used for all
statistical analyses. P≤0.05 was considered to indicate a
statistically significant difference.
Results
L-carnitine relieves the liver
inflammatory response in mice with cancer cachexia
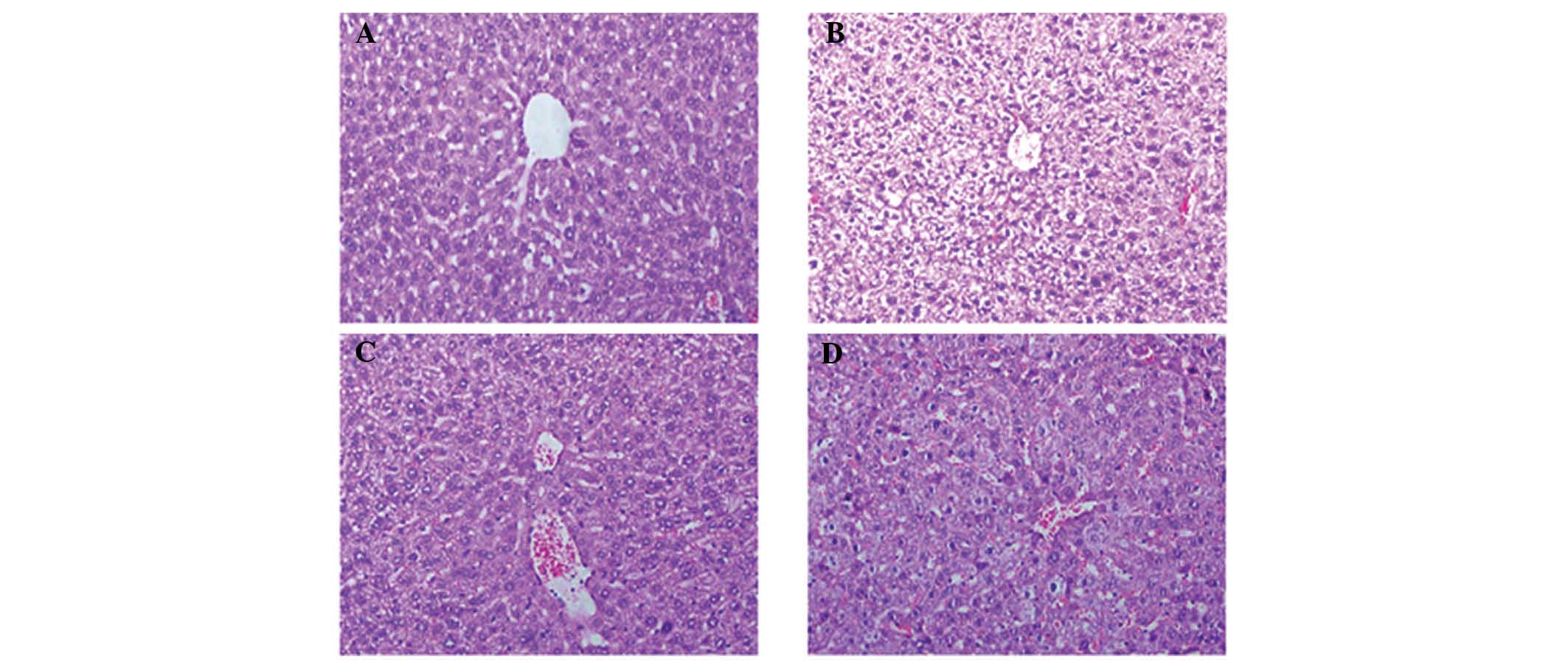
Compared with the normal control mice (Fig. 1A), histological analysis of the
liver tissue obtained from the mice with cancer cachexia receiving
saline showed hepa-tocyte necrosis, liver cell cord derangement and
hydropic or fatty degeneration of liver cells (Fig. 1B), which were relieved markedly by
L-carnitine (Fig. 1C). The effects
of L-carnitine on the liver inflammatory response were reversed
notably by etomoxir, the inhibitor of CPT I (Fig. 1D).
Effects of L-carnitine on serum levels of
MDA, SOD and GSH-Px
Compared with the healthy mice, there was a
significant increase in the serum levels of MDA, and a significant
decrease in the serum levels of SOD and GSH-Px in the mice with
cachexia receiving saline. However, L-carnitine markedly increased
the serum levels of SOD and GSH-Px, and significantly reduced the
serum levels of MDA, compared with the mice with cachexia receiving
saline, and these effects of L-carnitine were impaired markedly by
treatment with etomoxir (Table
I).
 | Table IL-carnitine decreases serum levels of
MDA, SOD and GSH-Px oxidative-stress markers. |
Table I
L-carnitine decreases serum levels of
MDA, SOD and GSH-Px oxidative-stress markers.
| Marker | Normal control | Vehicle
control | L-carnitine | L-carnitine +
etomoxir |
|---|
| MDA (nmol/ml) | 7.60±1.01 | 10.35±0.40a | 9.37±0.65b | 10.2±0.33c |
| SOD (U/ml) | 90.08±1.67 | 55.81±8.64a | 75.77±3.54b | 56.4±7.51d |
| GSH-Px (U/ml) | 222.43±11.7 | 180.6±6.22a | 204.03±6.06e |
179.39±11.77d |
Effects of L-carnitine on the protein
expression levels of PPAR-α/PPAR-γ in the liver of mice with
cachexia
In the normal control mice, the expression levels of
PPARα and PPARγ were detected at basal level, which were decreased
markedly at the protein level in the mice with cachexia receiving
saline. However, these changes were reversed following treatment of
animals with L-carnitine alone. This reversal effect of L-carnitine
on the decreased expression levels of PPAR-α and PPARγ in the mice
with cachexia receiving saline was almost eradicated following
etomoxir treatment (Fig. 2A and
B).
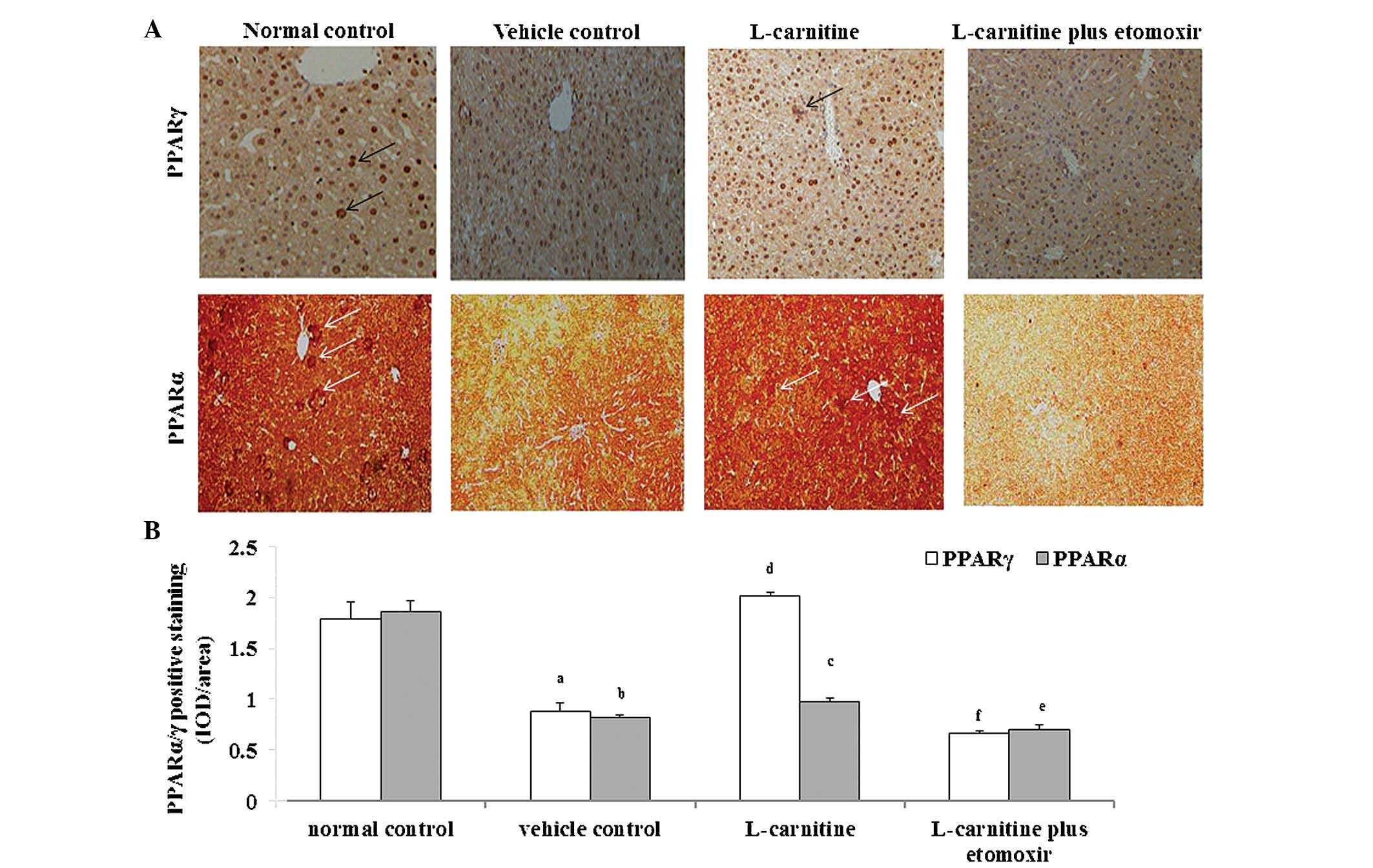 | Figure 2Effects of L-carnitine on protein
expression levels of PPARα and PPARγ in the liver of cachectic
mice. (A) Cancer cachectic mice were administered with saline
(vehicle control), L-carnitine (9 mg/kg per day), and CPT I
inhibitor etomoxir (20 mg/kg per day) + L-carnitine for 8 days (n=6
in each group), following which then the protein levels of PPARα
and PPARγ in the liver were assayed using immunohistochemistry.
Healthy untreated mice were used as normal controls (n=6). The
positive staining for PPARα and PPARγ is indicated by the white and
black arrows, respectively (magnification, ×100). (B) The relative
expression levels of PPARα and PPARγ were semi-quantitated as the
IOD/area. Data are expressed as the mean ± standard deviation.
aP<0.05 and bP<0.01, vs. normal
control; cP<0.05 and dP<0.01, vs.
vehicle control; eP<0.05 and fP<0.01,
vs. L-carnitine. PPAR, peroxisome proliferator-activated receptor;
CPT I, carnitine palmitoyltransferase I; IOD, integrated optical
density. |
L-carnitine decreases the expression of
NF-κB p65 in the PBMCs of mice with cancer cachexia in a
PPARγ-dependent manner
Compared with the normal control mice, the mRNA and
protein expression levels of NF-κB p65 in the PBMCs were markedly
elevated in the mice with cancer cachexia receiving saline.
However, the increased expression of NF-κB p65 in the mice of the
vehicle control group was decreased significantly by L-carnitine or
pioglitazone (a specific agonist of PPARγ). The effects of
L-carnitine on NF-κB p65 at the mRNA (Fig. 3A) and protein (Fig. 3B) levels were significantly
weakened by GW9662, a selective inhibitor of PPAR-γ.
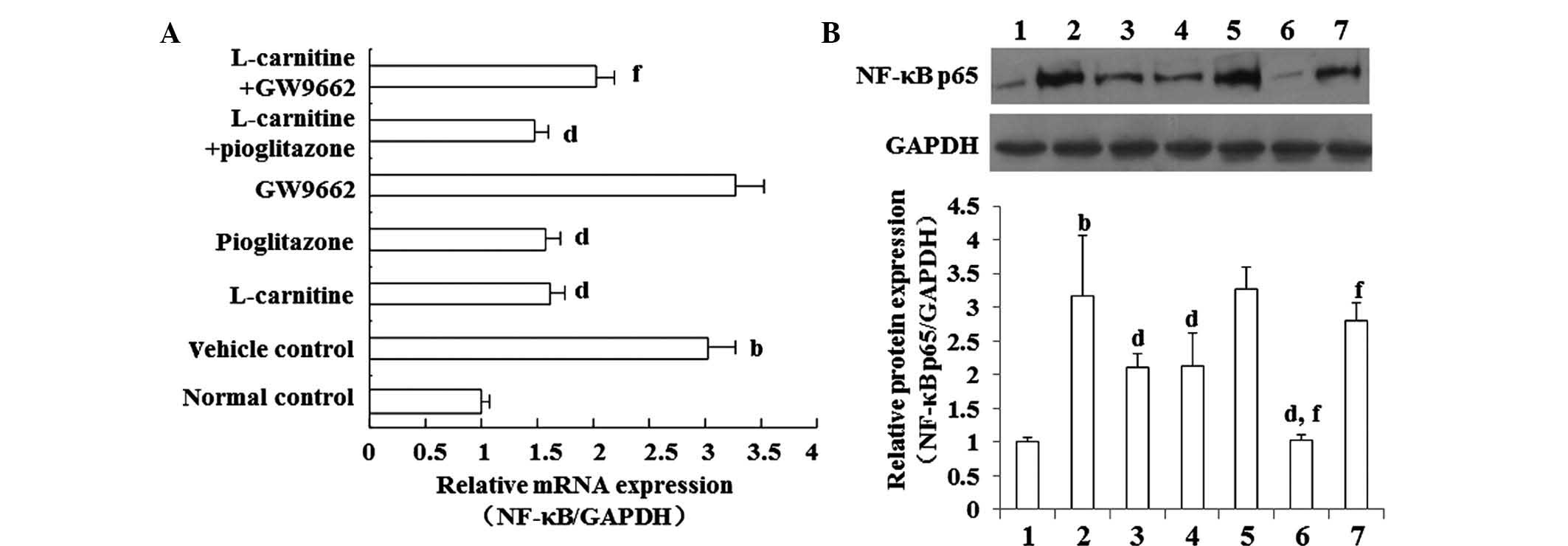 | Figure 3L-carnitine decreases the serum
expression levels of NF-κB p65 in cachectic mice. Cancer cachectic
mice were administered saline (vehicle control; Lane 2), oral
L-carnitine (9 mg/kg per day; Lane 3), oral pioglitazone
hydrochloride (10 mg/kg daily; Lane 4), intraperitoneal GW9662 (1
mg/kg daily; Lane 5), oral L-carnitine ± pioglitazone hydrochloride
(Lane 6), and L-carnitine + GW9662 for 8 days (n=6 in each group;
Lane 7), following which the serum concentration of NF-κB p65 were
determined at the (A) mRNA and (B) protein levels in peripheral
blood mononuclear cells using reverse transcription-quantitative
polymerase chain reaction and western blot analysis, respectively.
Healthy untreated mice were used as normal controls (n=6; Lane 1).
Data are expressed as the mean ± standard deviation.
bP<0.01, vs. normal control; dP<0.01,
vs. vehicle control; fP<0.01,. vs. L-carnitine.
NF-κB, nuclear factor-κB. |
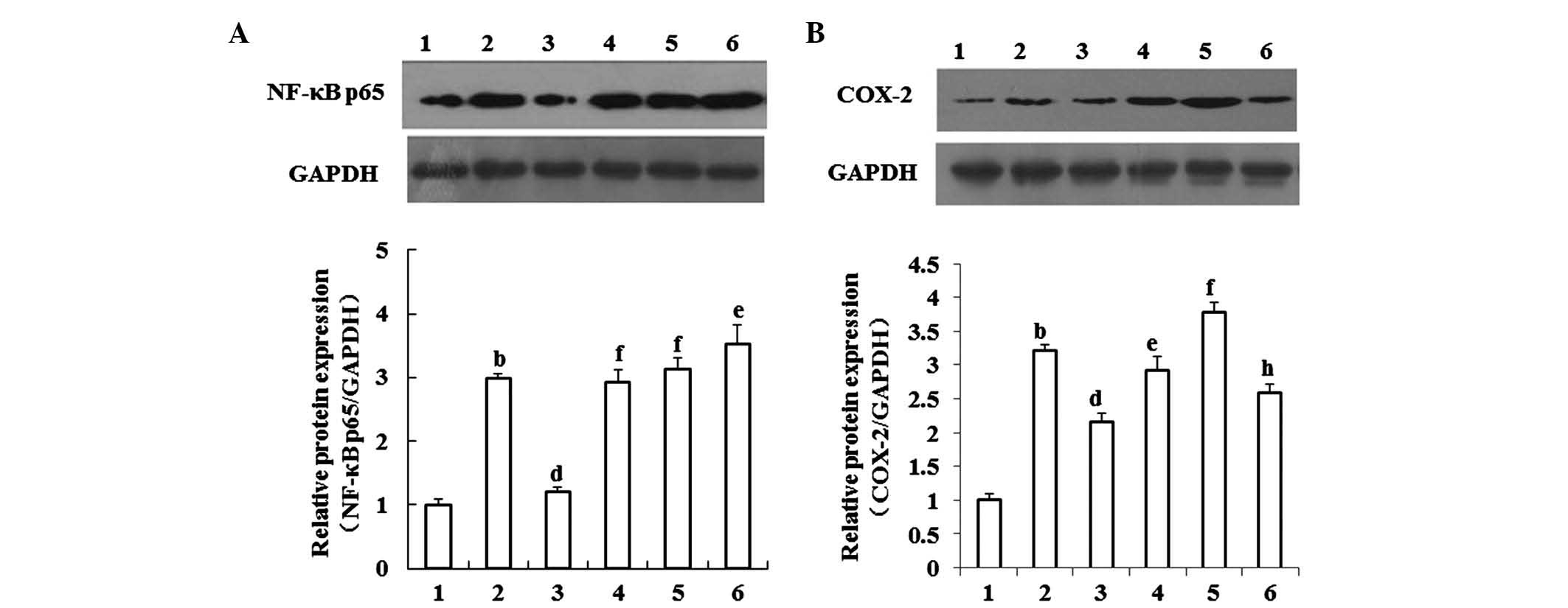
L-carnitine decreases the expression of
Cox-2 in the livers of mice with cachexia, partly by suppressing
NF-κB signaling
NF-κB p65 (Fig. 4A)
and Cox-2 (Fig. 4B) were expressed
at basal levels in the livers of the normal control mice, and were
elevated in the livers of the mice with cachexia. L-carnitine
decreased the elevated expression levels of NF-κB p65 and Cox-2 in
the livers of the mice with cachexia. This effect of L-carnitine
was reversed by GW9662, a selective inhibitor of PPARγ. The
inhibitory effect of GW9662 on L-carnitine on Cox-2 was impaired by
PDTC, a selective inhibitor of NF-κB signaling.
L-carnitine decreases the serum levels of
PGE2, CRP, TNF-α and IL-6 pro-inflammatory markers in mice with
cachexia, partly by suppressing PPARγ-NF-κB signaling
Compared with the healthy mice, there was a
significant increase in the serum levels of PGE2, CRP, TNF-α and
IL-6 in the mice with cachexia receiving saline. The serum
concentrations of these markers were decreased markedly by
L-carnitine. This effect of L-carnitine was impaired by GW9662, and
PDTC reversed the effect of GW9662 on the inhibition serum
pro-inflammatory markers by L-carnitine (Table II).
| Table IIL-carnitine decreases serum levels of
pro-inflammatory agents in cancer cachectic mice. |
Table II
L-carnitine decreases serum levels of
pro-inflammatory agents in cancer cachectic mice.
| Inflammatory agent
(pg/ml) | Normal control | Vehicle
control | L-carnitine | GW9662 | L-carnitine+
GW9662 | L-carnitine+
GW9662+PDTC |
|---|
| PGE2 | 40.01±1.43 | 122.83±4.13a | 108.00±1.08b | 131.82±4.84 | 121.17±4.35c | 103.01±6.62e |
| CRP | 7.19±0.57 | 16.98±1.48a | 10.52±1.01b | 17.71±0.97 | 15.64±0.83d | 9.67±0.53f |
| IL-6 | 2.69±0.31 | 28.11±5.20a | 16.44±2.58b | 28.22±5.56 | 28.81±3.85d | 17.53±2.46f |
| TNF-α | 1.68±0.38 | 35.25±3.00a | 19.98±2.78b | 32.22±2.89 | 29.48±3.68d | 22.81±1.76e |
Discussion
Cancer cachexia is a wasting syndrome, which is
character-ized by systemic inflammation, body weight loss, atrophy
of white adipose tissue and skeletal muscle, all of which are often
correlated with high mortality rates and poor quality of life in
patients with cancer (2). Liver
lipid metabolism disorders contribute to cancer cachexia symptoms
through inducing the pro-inflammatory response in the liver to
aggravate systemic inflammation (8,25). A
previous study (21) demonstrated
that L-carnitine, a key regulator of lipid metabolism, induces the
recovery of lipid metabolism disorders in the liver, and decrease
circulating pro-inflammatory cytokines to improve the symptoms of
cachexia in association with regulating the expression and activity
of CPT. This suggests that CPT is pivotal in the regulation of
L-carnitine in the liver inflammatory response. The results of the
present study demonstrated that L-carnitine attenuated the liver
inflammatory response and oxidative stress via CPT I-dependent
PPARγ-NF-κB signaling.
The liver is the major site of lipid metabolism and
a predominant source of circulating pro-inflammatory factors in
cancer cachexia (12,16). Patients with hepatocellular
carcinoma which progresses to cancer cachexia often exhibit
accompanied chronic liver inflammation, which is one of the major
factors leading to poor prognosis (26). Our previous study showed that
L-carnitine ameliorates the symptoms of cancer cachexia (3). In the present study, it was found
that L-carnitine ameliorated the liver inflammatory response by
relieving hepatocyte necrosis, liver cell cord derangement and
hydropic or fatty degeneration of liver cells, suggesting that
L-carnitine ameliorated cancer cachexia by inducing the recovery
from liver inflammation. In addition, etomoxir, as an inhibitor of
CPT I almost eradicated the effect of L-carnitine on liver
inflammation, suggesting that CPT I was a mediator in the
improvement of liver inflammation by L-carnitine.
The β-oxidation of fatty acids in the mitochondria
is disrupted in the liver in cancer cachexia, resulting in
oxidative stress (27), which is
supported by the results of the present study that the serum levels
of oxidative stress markers were increased in the mice with cancer
cachexia. The dysfunction of mitochondria in β-oxidation finally
induces a pro-inflammatory response. In the present study, the
results showed that L-carnitine decreased the elevated levels of
oxidative stress markers, suggesting that L-carnitine relieved the
liver inflammatory response by inhibiting oxidative stress. Our
previous study demonstrated that the activity of CPT I, a key
mediator in the β-oxidation of fatty acids, is decreased in the
mitochondria of the livers of mice with cachexia liver, and is
increased by L-carnitine (3). In
the present study, it was demonstrated that etomoxir, as an
inhibitor of CPT I, reversed the amelioratory effect of L-carnitine
on oxidative stress. These results suggested that L-carnitine
inhibited oxidative stress and improved liver inflammation in a CPT
I-dependent manner.
PPARs are transcription factors belonging to a
superfamily of nuclear receptors, and three isoforms (α, δ and γ)
have been described, in which PPARα and γ are known to regulate
lipid metabolism and oxidative stress (28,29).
Furthermore, PPARα and γ have been previously demonstrated to exert
an inhibitory effect on tumor growth, muscle atrophy, and
pro-inflammatory cytokine secretion and signaling in cancer
cachexia (30–33). In the present study, the protein
expression levels of PPARα and γ in the liver were decreased in the
mice with cancer cachexia, which was accompanied by a notable liver
inflammatory response. These changes were restored by L-carnitine,
suggesting that L-carnitine improved the liver inflammatory
response by regulating the expression levels of PPARα and/or
PPARγ.
Notably, it has been demonstrated that PPARα and
PPARγ coactivators induce the expression of CPT I through different
regions of the CPT-1A gene (21).
In the present study, the promotion by L-carnitine on the
expression levels of PPARα and γ were reversed by etomoxir, an
inhibitor of CPT, suggesting that L-carnitine regulated the
expression of PPAR in a CPT I-dependent manner, and that CPT I may
have an indirect effect on regulating the expression of PPAR.
Although the present study was unable to provide direct evidence
that CPT I induces the expression of PPAR, the results indicated
that L-carnitine ameliorated the liver inflammatory response by
regulating CPT I-dependent PPAR signaling.
Of note, the present study demonstrated that the
increase in the expression of PPARγ in the liver induced by
L-carnitine was more significant, compared with that of PPARα,
suggesting that the amelioration effects of L-carnitine on the
liver inflammatory response may be dependent more on PPARγ and less
on PPARα signaling. These results are consistent with those of a
previous study in a cyclophosphamide-induced hepatotoxic model,
which reported that PPARγ signaling, but not PPARα signaling,
mediated antioxidant and anti-inflammatory effects in the liver
(19).
NF-κB is known to regulate liver inflammation and
oxidative stress (34). A previous
study (35) demonstrated that the
elevation in the expression levels of NF-κB p65 contributes
substantially to the progression of cancer cachexia, suggesting
that NF-κB signaling is essential in cancer cachexia, which is also
supported by the findings of the present study, in which the
expression of NF-κB p65 was increased in the PBMCs at the mRNA and
protein levels. Studies (36,37)
have also demonstrated that NF-κB is a downstream mediator of PPARα
and PPARγ signaling in the liver, which is supported by our
findings that increased expression levels of NF-κB p65 are
inhibited by pioglitazone, a specific agonist of PPARγ. Notably,
treatment of mice in the present study with L-carnitine alone
decreased the expression of NF-κB p65 in cancer cachexia, and this
effect of L-carnitine was reversed by GW9662, a selective inhibitor
of PPAR-γ, suggesting that L-carnitine inhibited the expression of
NF-κB p65 in a PPARγ-dependent manner. However, the exact role of
PPARα in the regulation of L-carnitine on the expression of NF-κB
p65 requires further investigated in the future.
The Cox-2/PGE2 pathway is important in regulating
oxidative stress and inflammation in the liver (38). Celecoxib, a specific inhibitor of
Cox-2, downregulates serum inflammatory cytokines in patients with
cancer cachexia (10). In the
present study, the increased levels of Cox-2 in the liver of mice
with cancer cachexia were decreased by L-carnitine, and this effect
was reversed by treatment with GW9662. This effect of GW9662 on
L-carnitine was restored by PDTC, a specific inhibitor of NF-κB
signaling. These results suggested that L-carnitine decreased the
expression of Cox-2 in the liver by PPARγ-dependent NF-κB
signaling.
Certain pro-inflammatory markers, including CRP,
PGE2, IL-6 and TNF-α, are well known to promote systemic
inflammation, thus aggravating the progression of cancer cachexia
(39). In particular, CRP may
induce IL-6 secretion, which is known to have a causative effect in
cancer cachexia (40,41). In the present study, it was found
that the elevation of the above-mentioned pro-inflammatory markers
were decreased by L-carnitine, and this inhibitory effect of
L-carnitine was reversed by GW9662, suggesting that PPARγ-dependent
NF-κB signaling is pivotal in the inflammatory response in cancer
cachexia.
One of the limitations of the present study was that
the role of PPARα in the regulation of liver inflammation by
L-carnitine was not investigated, although a previous study
demonstrated that it is PPARγ, rather than PPARα, which exerts
antioxidant and anti-inflammatory effects in the liver (19). However, other studies have
demonstrated that PPARα also exerts anti-inflammatory effects in
the liver following ischemia-reperfusion injury (37), and is a therapeutic target in
chronic obstructive pulmonary disease-induced cachexia, owing to
its anti-inflammatory effect (42). This discrepancy may be explained by
the different animal models used in these investigations.
Therefore, the role of PPARα in the amelioration of the liver
inflammatory response by L-carnitine in cancer cachexia requires
further investigation.
In conclusion, the present study demonstrated that
L-carnitine ameliorated liver inflammation and serum
pro-inflammatory markers in cancer cachexia via I-dependent PPARγ
signaling, including the downstream molecules of NF-κB p65 and
Cox-2. These results suggest that L-carnitine may be a candidate
for the amelioration of systemic inflammation in cancer
cachexia.
Acknowledgments
This study was supported by the Foundation of the
Science and Technology Committee of Shanghai Zhabei District of
China (grant no. 2012ZD04).
References
|
1
|
Trutschnigg B, Kilgour RD, Morais JA,
Lucar E, Hornby L, Molla H and Vigano A: Metabolic, nutritional and
inflammatory characteristics in elderly women with advanced cancer.
J Geriatr Oncol. 4:183–189. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Richards CH, Roxburgh CS, MacMillan MT,
Isswiasi S, Robertson EG, Guthrie GK, Horgan PG and McMillan DC:
The relationships between body composition and the systemic
inflammatory response in patients with primary operable colorectal
cancer. PLoS One. 7:e418832012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu S, Wu HJ, Zhang ZQ, Chen Q, Liu B, Wu
JP and Zhu L: L-carnitine ameliorates cancer cachexia in mice by
regulating the expression and activity of carnitine palmityl
transferase. Cancer Biol Ther. 12:125–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Guo C, Zhang D, Zhang J, Wang X
and Geng C: The altered tight junctions: An important gateway of
bacterial translocation in cachexia patients with advanced gastric
cancer. J Interferon Cytokine Res. 34:518–525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang LH, Yang XY, Mihalic K, Xiao W, Li D
and Farrar WL: Activation of estrogen receptor blocks
interleukin-6-inducible cell growth of human multiple myeloma
involving molecular cross-talk between estrogen receptor and STAT3
mediated by co-regulator PIAS3. J Biol Chem. 276:31839–31844. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonetto A, Aydogdu T, Kunzevitzky N,
Guttridge DC, Khuri S, Koniaris LG and Zimmers TA: STAT3 activation
in skeletal muscle links muscle wasting and the acute phase
response in cancer cachexia. PLoS One. 6:e225382011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martignoni ME, Dimitriu C, Bachmann J,
Krakowski-Rosen H, Ketterer K, Kinscherf R and Friess H: Liver
macrophages contribute to pancreatic cancer-related cachexia. Oncol
Rep. 21:363–369. 2009.PubMed/NCBI
|
|
8
|
Watchorn TM, Dowidar N, Dejong CH, Waddell
ID, Garden OJ and Ross JA: The cachectic mediator proteolysis
inducing factor activates NF-kappaB and STAT3 in human Kupffer
cells and monocytes. Int J Oncol. 27:1105–1111. 2005.PubMed/NCBI
|
|
9
|
Wang W, Andersson M, Lõnnroth C, Svanberg
E and Lundholm K: Prostaglandin E and prostacyclin receptor
expression in tumor and host tissues from MCG 101-bearing mice: A
model with prostanoid-related cachexia. Int J Cancer. 115:582–590.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mantovani G, Macciò A, Madeddu C, Serpe R,
Antoni G, Massa E, Dessì M and Panzone F: Phase II nonrandomized
study of the efficacy and safety of COX-2 inhibitor celecoxib on
patients with cancer cachexia. J Mol Med (Berl). 88:85–92. 2010.
View Article : Google Scholar
|
|
11
|
Sue YM, Chou HC, Chang CC, Yang NJ, Chou Y
and Juan SH: L-carnitine protects against carboplatin-mediated
renal injury: AMPK- and PPARα-dependent inactivation of NFAT3. PLoS
One. 9:e1040792014. View Article : Google Scholar
|
|
12
|
Ishikawa H, Takaki A, Tsuzaki R, Yasunaka
T, Koike K, Shimomura Y, Seki H, Matsushita H, Miyake Y, Ikeda F,
et al: L-carnitine prevents progression of non-alcoholic
steatohepatitis in a mouse model with upregulation of mitochondrial
pathway. PLoS One. 9:e1006272014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Demiroren K, Dogan Y, Kocamaz H, Ozercan
IH, Ilhan S, Ustundag B and Bahcecioglu IH: Protective effects of
L-carnitine, N-acetylcysteine and genistein in an experimental
model of liver fibrosis. Clin Res Hepatol Gastroenterol. 38:63–72.
2014. View Article : Google Scholar
|
|
14
|
Vinci E, Rampello E, Zanoli L, Oreste G,
Pistone G and Malaguarnera M: Serum carnitine levels in patients
with tumoral cachexia. Eur J Intern Med. 16:419–423. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Breitkreutz R, Babylon A, Hack V, Schuster
K, Tokus M, Böhles H, Hagmüller E, Edler L, Holm E and Dröge W:
Effect of carnitine on muscular glutamate uptake and intramuscular
glutathione in malignant diseases. Br J Cancer. 82:399–403. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan SA, Ali A, Khan SA, Zahran SA,
Damanhouri G, Azhar E and Qadri I: Unraveling the complex
relationship triad between lipids, obesity and inflammation.
Mediators Inflamm. 2014:5027492014. View Article : Google Scholar
|
|
17
|
Silverio R, Laviano A, Rossi Fanelli F and
Seelaender M: L-Carnitine induces recovery of liver lipid
metabolism in cancer cachexia. Amino Acids. 42:1783–1792. 2012.
View Article : Google Scholar
|
|
18
|
Zambrano S, Blanca AJ, Ruiz-Armenta MV,
Miguel-Carrasco JL, Arévalo M, Vázquez MJ, Mate A and Vázquez CM:
L-Carnitine protects against arterial hypertension-related cardiac
fibrosis through modulation of PPAR-γ expression. Biochem
Pharmacol. 85:937–944. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
El-Sheikh AA and Rifaai RA: Peroxisome
proliferator activator receptor (PPAR)-γ ligand, but Not PPAR-α,
ameliorates cyclophosphamide-induced oxidative stress and
inflammation in rat liver. PPAR Res. 2014:6263192014. View Article : Google Scholar
|
|
20
|
Chen K, Li J, Wang J, Xia Y, Dai W, Wang
F, Shen M, Cheng P, Zhang Y, Wang C, et al: 15-Deoxy-γ
12,14-prostaglandin J2 reduces liver impairment in a model of
ConA-induced acute hepatic inflammation by activation of PPAR γ and
Reduction in NF-κB Activity. PPAR Res. 2014:2156312014.
|
|
21
|
Song S, Attia RR, Connaughton S, Niesen
MI, Ness GC, Elam MB, Hori RT, Cook GA and Park EA: Peroxisome
prolif-erator activated receptor alpha (PPARalpha) and PPARgamma
coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA
(CPT-1A) via independent gene elements. Mol Cell Endocrinol.
325:54–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kashinakunti SV, Kollur P, Kallaganada GS,
Rangappa M and Ingin JB: Comparative study of serum MDA and vitamin
C levels in non-smokers, chronic smokers and chronic smokers with
acute myocardial infarction in men. J Res Med Sci. 16:993–998.
2011.
|
|
23
|
Hübscher SG: Histological assessment of
non-alcoholic fatty liver disease. Histopathology. 49:450–465.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jia XL, Li SY, Dang SS, Cheng YA, Zhang X,
Wang WJ, Hughes CE and Caterson B: Increased expression of
chondroitin sulphate proteoglycans in rat hepatocellular carcinoma
tissues. World J Gastroenterol. 18:3962–3976. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tisdale MJ: Mechanisms of cancer cachexia.
Physiol Rev. 89:381–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luna G, Florence L and Johansen K:
Hepatocellular carcinoma. A 5 year institutional experience. Am J
Surg. 149:591–594. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barreiro E, de la Puente B, Busquets S,
López-Soriano FJ, Gea J and Argiles JM: Both oxidative and
nitrosative stress are associated with muscle wasting in
tumour-bearing rats. FEBS Lett. 579:1646–1652. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng T, Zhang CL, Song FY, Zhao XL and Xie
KQ: CMZ reversed chronic ethanol-induced disturbance of PPAR-α
possibly by suppressing oxidative stress and PGC-1α acetylation and
activating the MAPK and GSK3β pathway. PLoS One. 9:e986582014.
View Article : Google Scholar
|
|
29
|
Al Rouq F and El Eter E: PPAR-γ activator
induces neuropro-tection in hypercholesterolemic rats subjected to
global cerebral ischemia/reperfusion injury: In vivo and in vitro
inhibition of oxidative stress. Exp Gerontol. 51:1–7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moore-Carrasco R, Figueras M, Ametller E,
López-Soriano FJ, Argilés JM and Busquets S: Effects of the
PPARgamma agonist GW1929 on muscle wasting in tumour-bearing mice.
Oncol Rep. 19:253–256. 2008.
|
|
31
|
Puigserver P, Rhee J, Lin J, Wu Z, Yoon
JC, Zhang CY, Krauss S, Mootha VK, Lowell BB and Spiegelman BM:
Cytokine stimulation of energy expenditure through p38 MAP kinase
activation of PPARgamma coactivator-1. Mol Cell. 8:971–982. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Asp Ml, Tian M, Kliewer KL and Belury MA:
Rosiglitazone delayed weight loss and anorexia while attenuating
adipose depletion in mice with cancer cachexia. Cancer Biol Ther.
12:957–965. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang J, Das SK, Jha P, Al Zoughbi W,
Schauer S, Claudel T, Sexl V, Vesely P, Birner-Gruenberger R,
Kratky D, et al: The PPARα agonist fenofibrate suppresses B-cell
lymphoma in mice by modulating lipid metabolism. Biochim Biophys
Acta. 1831:1555–1565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan K, Huang C, Fox J, Gaid M, Weaver A,
Li G, Singh BB, Gao H and Wu M: Elevated inflammatory response in
caveolin-1-deficient mice with Pseudomonas aeruginosa infection is
mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB). J
Biol Chem. 286:21814–21825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou W, Jiang ZW, Tian J, Jiang J, Li N
and Li JS: Role of NF-kappaB and cytokine in experimental cancer
cachexia. World J Gastroenterol. 9:1567–1570. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li CC, Yang HT, Hou YC, Chiu YS and Chiu
WC: Dietary fish oil reduces systemic inflammation and ameliorates
sepsis-induced liver injury by up-regulating the peroxisome
proliferator-activated receptor gamma-mediated pathway in septic
mice. J Nutr Biochem. 25:19–25. 2014. View Article : Google Scholar
|
|
37
|
Zuniga J, Cancino M, Medina F, Varela P,
Vargas R, Tapia G, Videla LA and Fernández V: N-3 PUFA
supplementation triggers PPAR-α activation and PPAR-alpha/NF-κB
interaction: Anti-inflammatory implications in liver
ischemia-reperfusion injury. PLoS One. 6:e285022011. View Article : Google Scholar
|
|
38
|
Ozturk H, Gezici A and Ozturk H: The
effect of celecoxib, a selective COX-2 inhibitor, on liver
ischemia/reperfusion-induced oxidative stress in rats. Hepatol Res.
34:76–83. 2006. View Article : Google Scholar
|
|
39
|
Onesti JK and Guttridge DC: Inflammation
based regulation of cancer cachexia. Biomed Res Int.
2014:1684072014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cossette E, Cloutier I, Tardif K,
DonPierre G and Tanguay JF: Estradiol inhibits vascular endothelial
cells pro-inflammatory activation induced by C-reactive protein.
Mol Cell Biochem. 373:137–147. 2013. View Article : Google Scholar :
|
|
41
|
White JP, Baltgalvis KA, Puppa MJ, Sato S,
Baynes JW and Carson JA: Muscle oxidative capacity during
IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp
Physiol. 300:R201–R211. 2011. View Article : Google Scholar :
|
|
42
|
Remels AH, Gosker HR, Langen RC and Schols
AM: The mechanisms of cachexia underlying muscle dysfunction in
COPD. J Appl Physiol (1985). 114:1253–1262. 2013. View Article : Google Scholar
|