Introduction
Glutamate, one of twenty essential amino acids, is
well known to be the main physiological excitatory neurotransmitter
in the mammalian central nervous system (CNS). It has been reported
to regulate neurogenesis, neurite outgrowth, synaptogenesis and
neuron survival (1). However, in
various neurological disorders, including ischemia and stroke, a
high concentration of glutamate is considered to lead to
excitotoxicity and result in the death of neuronal cells. The
concentration of glutamate is strictly maintained in the CNS;
however, it is sharply and rapidly increased to 100–500 µM
and sequentially induces excitotoxic damage during cerebral
ischemia or trauma (2–4). Excessive glutamate production
activates several types of pre- and post-synaptic glutamate
receptor and then causes calcium overload. The accumulation of high
levels of intracellular calcium triggers a range of downstream
neurotoxic cascades, including energy deficiency, oxidative stress,
mitochondrial dysfunction and calcium overload, leading to
excitotoxic neuronal death (5).
Lysosomes, separated from the cytosol (which has a
neutral pH) by a tough single membrane (6), are intracellular organelles
characterized by an acidic environment with a pH of ~4.5. The pH
value within lysosomes is precisely regulated by ATP-dependent
proton pumps termed H+-ATPases, which actively pump
protons from the cytosol into the lysosomal lumen (7,8).
H+-ATPase is the key enzyme for physiologically
maintaining the acidic microenvironment within lysosomes and
regulating cellular pH. Thus, lysosomal destabilization not only
influences normal cellular activities but also affects cell
viability. Recently, it has been reported that partial rupture or
permeabilization of the lysosomal membrane induces apoptosis
through mitochondrial transmembrane potential loss or caspase
activation, while severe lysosomal rupture induces necrosis in
cancer cells (6,9). The leakage of cathepsins caused by
lysosomal membrane permeabilization (LMP) has been observed in the
brain tissues of various rodents and non-human primate stroke
models; for example, it was reported 1 h after global ischemia in
monkey brain (10,11), after transient focal ischemia in
mouse brain (12) and after a
5-min oxygen glucose deprivation in the rat hippocampal slices
(13). However, it remains unclear
whether LMP is involved in glutamate excitotoxic cascades, which
are regarded as the leading cause of neuronal death.
To the best of our knowledge, the present study
demonstrated for the first time that LMP is an early event in
glutamate excitotoxic cascades. It was inhibited following
treatment with the NMDA receptor antagonist MK-801 and the
Ca2+ chelator ethylene glycolbis (2-aminoethylether)-N,
N, N′, N′-tetraacetic acid (EGTA), and alleviated by treatment with
the antioxidant N-Acetyl-L-cysteine (NAC), in primary cultured
cortical neurons following glutamate treatment for 30 min.
Materials and methods
Materials
This study was approved by the ethics committee of
the Sun Yatsen University (Guangzhou, China). Newborn
Sprague-Dawley rats were provided by the Experimental Animal Center
of Sun Yatsen University (Guangzhou, China). Neurobasal A medium,
fetal bovine serum (FBS), B27 supplements, GlutaMAX, trypsin
solution, penicillin-streptomycin (P/S) and Fluo-4AM were purchased
from Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
The primary antibodies used in western blot analysis and
immunohistochemistry were as follows: Goat anti-rat polyclonal
cathepsin B (IgG; cat. no., sc-6493; dilution, 1:100) and goat
anti-rat polyclonal heat shock protein (Hsp)70.1 (IgG; cat. no.,
sc-1060; dilution, 1:200) and goat anti-rat monoclonal
anti-lysosomal-associated membrane protein 2 (LAMP2; IgG; cat. no.,
sc-8100; dilution, 1:100) were purchased from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA), and rabbit anti-rat
monoclonal anti-α fodrin (IgG; cat. no., ab75755; dilution,
1:1,000) from Abcam (Cambridge, MA, USA). The secondary antibodies
donkey anti-goat Alexa Fluor® 488 polyclonal IgG (cat.
no., A-11055; dilution, 1:1,000) and goat anti-rabbit Alexa Fluor
488 polyclonal IgG (cat. no., A-11008; dilution, 1:1,000) were
purchased from Invitrogen (Thermo Fisher Scientific, Inc.), and the
goat anti-rabbit IgG horseradish peroxidase (HRP)-conjugated
antibody (cat. no. 7074; dilution, 1:1,000) was purchased from Cell
Signaling Technology (Danvers, MA, USA) and the donkey anti-goat
IgG-HRP antibody (dilution, 1:5,000) from Santa Cruz Biotechnology,
Inc. (Stanta Cruz, CA, USA). Acridine orange (AO) EGTA and the
calpain inhibitors calpeptin and SJA6017 were purchased from
Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Cortical neurons were prepared from newborn
Sprague-Dawley rats as described previously with modifications
(14). Briefly, the cortex was
dissected and placed in ice-cold aseptic dissection solution (DS).
After mincing, the tissue was placed in DS containing 0.25% trypsin
(Thermo Fisher Scientific, Inc.) for 15 min at 37°C. Digestion was
terminated by DS containing 10% FBS followed by centrifuging for 5
min at 750 × g and the supernatant was discarded. The cell pellet
was resuspended in DS containing DNase I (5 mg/ml; (Sigma-Aldrich),
Mg2+ and FBS, and homogenized by pipetting up and down
~20 times. The supernatant was left for15 min and then centrifuged
for 5 min at 750 × g. The cell pellet was then collected and
resuspended in Neurobasal A medium supplemented with 10% FBS, 2%
B27, 0.25% GlutaMAX and 1% P/S. Cells were then seeded at a density
of 1×106 cells/ml into poly-l-lysine (0.5 mg/ml;
Sigma-Aldrich) coated dishes. Cells were incubated in a
CO2 chamber. The medium was replaced with non-serum
formula after seeding for 4 h and half of the medium was replaced
every 2–3 days. In the present study, the primary cultured cortical
neurons were exposed to 100, 200 or 400 µM glutamate for 30
min
AO redistribution assay
Cells were treated with different concentrations of
glutamate, glutamate and MK-801, or NAC. Cells were then incubated
with 5 µg/ml AO in Neurobasal medium A for 15 min at 37°C
and washed twice with phosphate-buffered saline (PBS) (Invitrogen;
Thermo Fisher Scientific, Inc.). Immunofluorescence images were
captured from randomly selected microscopic fields using a
fluorescent microscope equipped with a camera (IX71; magnification,
×200; Olympus Corp., Tokyo, Japan). During exposure to blue light,
AO-loaded cells showed red granular fluorescence and green diffuse
fluorescence. Once rupture of lysosomes occurs, AO is relocated
from lysosomes to the cytosol, this results in a decrease in the
granular (lysosomal) red fluorescence in combination with the
increased diffuse (cytosolic) green fluorescence.
Immunohistochemistry
Following glutamate exposure, cells were fixed with
4% paraformaldehyde for 15 min at room temperature and then washed
three times with PBS for 5 min per wash. Permeabilization was
performed with 0.3% Triton X-100 (Amresco LLC, OH, US) for 15 min.
The cells were then washed three times with PBS and incubated in
blocking buffer (ZSGB-BIO Company, Beijing, China) for 1 h,
followed by overnight incubation at 4°C with primary antibody
anti-cathepsin B and anti-LAMP2. The following day, cells were
washed with PBS three times, followed by 1 h incubation with
secondary antibody Alexa Fluor 488-conjugated donkey anti-goat IgG
for cathepsin B and goat anti-rabbit IgG for LAMP2 at room
temperature, and three washes with PBS. Finally, cells were
counterstained with DAPI (Sigma-Aldrich) for 3 min and rinsed with
PBS. Digital images were collected using a Zeiss LSM 710 confocal
microscope (Zeiss, Oberkochen, Germany).
Intracellular Ca2+
measurement
Intracellular calcium concentrations were measured
by the fluorescent cell-permeant calcium indicator Fluo-4AM. Cells
grown on black-walled 96-well plates were loaded with 5 µM
Fluo-4AM for 45 min in Hank's balanced salt solution (Invitrogen;
Thermo Fisher Scientific, Inc.) containing 0.02% Pluronic F-127
(Invitrogen; Thermo Fisher Scientific, Inc.) followed by 20 min of
de-esterification. Following loading, neurons were treated with 200
µM glutamate, 200 µM glutamate and 5 µM
MK-801, or 10 mM extracellular Ca2+ chelator EGTA for 30
min at 37°C in the dark. Then, neurons were washed with Hank's
balanced salt solution. Fluorescence with excitation at 488 nm and
emission at 506 nm was then read on a microplate fluorescence
reader (TECAN Group Ltd., Männedorf, Switzerland). Results are
presented as a fold change of the control.
Western blotting
Following treatment with glutamate for 30 min,
neurons were scraped and then resuspended in protein extraction
reagent. The cell lysate was centrifuged at 12,000 × g for 10 min
at 4°C and the supernatant was collected for electrophoresis. Prior
to electrophoresis, the concentration of protein was determined
using a bicinchoninic acid protein assay kit (Pierce Biotechnology,
Rockford, IL, USA) following the manufacturer's instructions. Equal
quantities of protein (40 µg) were separated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (CWBIO, Beijing,
China). Following electrophoresis, the proteins were transferred to
polyvinylidene difluoride membranes (F. Hoffmann-La Roche AG,
Basel, Switzerland), blocked with 5% skimmed milk in Tris-buffered
saline for 2 h, and washed three times with Tris-buffered saline.
Subsequently, the membranes were reacted with primary antibodies
overnight. The membranes were then washed three times with
Tris-buffered saline, followed by incubation with horseradish
peroxidase-labeled secondary antibody for 1 h at room temperature.
The immune complexes were visualized using enhanced
chemiluminescence-detection reagents (Merck Millipore, Boston, MA,
USA) according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using SPSS 16.0
software (SPSS Inc., Chicago, IL, USA). Data are presented as the
mean ± standard deviation and were analyzed using one-way analysis
of variance followed by the Student-Newman-Keuls test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Lysosomal membrane permeabilization is
involved in early glutamate excitotoxicity
It has been reported that treatment with high
concentrations of glutamate (≥100 µM) for 24 h induces
excitotoxic cell death with notable morphologic changes in rat
primary cerebral cortical neurons. In the present study, the
primary cultured cortical neurons were exposed to 100, 200 or 400
µM glutamate for 30 min, and then the cells displayed
morphological body swelling and synapse injury in a
concentration-dependent manner compared with the control group
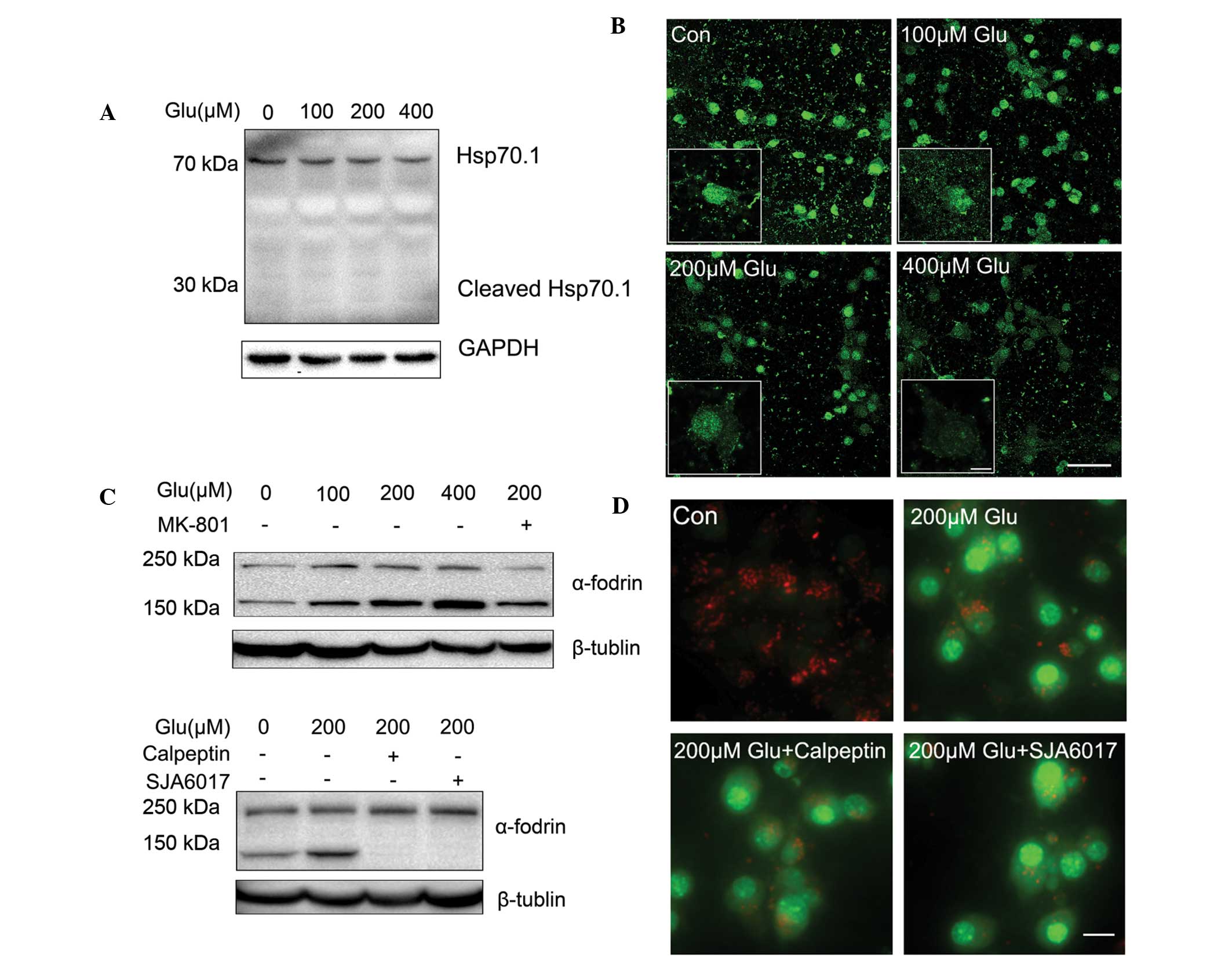
(Fig. 1A).
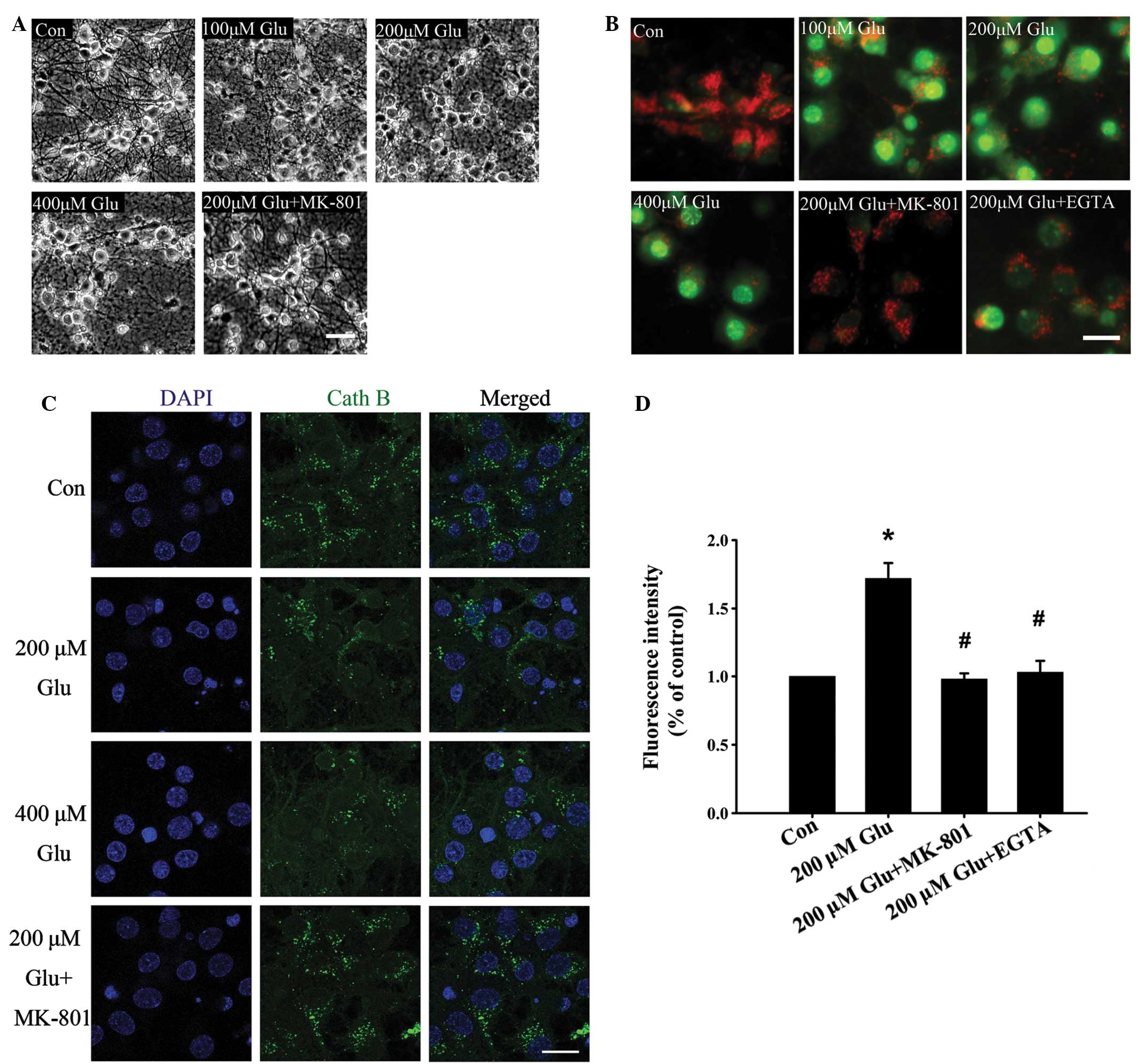 | Figure 1Effect of glutamate on lysosomal
membrane permeabilization. (A) Representative phase contrast images
of neurons left untreated, treated with different concentrations of
glutamate after 30 min or treated with glutamate and MK-801. Scale
bar, 50 µm. (B) Neurons were left untreated, treated with
different concentrations of glutamate alone, or treated with 200
µM glutamate with MK-801 and EGTA for 30 min. Neurons were
then stained with acridine orange and viewed under a microscope.
Scale bar, 20 µm. (C) Immunohistochemical analysis of
cathepsin B distribution following treatment with glutamate. In the
control group, staining appeared punctate, while cytosolic
cathepsin B increased following glutamate stimulation for 30 min.
Scale bar, 20 µm. (D) The effect of glutamate (200
µM) on intracellular Ca2+ was monitored by Fluo-4
AM. Data are presented as the mean ± standard deviation from three
independent experiments. *P<0.05, compared with the
control group; #P<0.05, compared with the 200
µM glutamate group. Glu, glutamine. |
The permeabilization of the lysosomal membrane
results in H+-ATPase dysfunction and redistribution of
cathepsins from lysosomes to the cytoplasm. As shown in Fig. 1B, following incubation with a
lysosome-specific fluorescence probe, AO (15), the neurons in control group
exhibited extensive lysosomal red fluorescence, while neurons
treated with different concentrations of glutamate exhibited an
increase in green fluorescence in the cytosol, indicating AO
redistribution caused by LMP. LMP induced by glutamate was further
confirmed by an immunofluorescence assay using antibodies against
the lysosome specific enzyme, cathepsin B. As shown in Fig. 1C, the fluorescence intensity of
cathepsin B in the cytoplasm decreased following glutamate
treatment, which indicated leakage caused by LMP.
It is now unanimously accepted that NMDA receptors
are important in excitotoxic neuronal death, predominantly owing to
their Ca2+ permeability. Influx of extracellular
Ca2+ is secondary to activation of the NMDA receptors by
glutamate, and the accumulation of high levels of intracellular
Ca2+ triggers a cascade of excitotoxic events. LMP of
neurons was mostly inhibited by the NMDA receptor antagonist MK-801
using AO (Fig. 1B) and cathepsin B
(Fig. 1C) staining. To further
confirm the effect of glutamate on lysosomal stability, the neurons
were switched to a Ca2+-free medium containing 10 mM
EGTA, which chelates extracellular Ca2+. As expected,
the glutamate-induced increase in intracellular Ca2+ was
abolished (Fig. 1D) and LMP was
markedly decreased (Fig. 1B),
indicating that the Ca2+ influx is critical in glutamate
induced-LMP. These results suggest that LMP is involved in early
glutamate excitotoxicity.
Calpain activation is not required for
LMP
It has been reported that calcium influx via the
NMDA receptor results in the activation of cytoplasmic proteases
(such as calpain), which hydrolyzes cytoskeletal and cellular
proteins, and leads to LMP (16,17).
Hsp70.1 and LAMP2, two substrates of calpain, have been reported to
be involved in calpain-mediated LMP (16,17).
Hsp70.1 is known to stabilize the lysosomal membrane by recycling
damaged proteins, and protecting cells from oxidative stress and
apoptotic stimuli (18). In the
case of ischemia, Hsp70.1 is carbonylated by oxidative stressors
and is more vulnerable to activated calpain (16,19).
However, in a rat retinal light damage model, degradation of LAMP2
rather than Hsp70.1 by calpain is also hypothesized to promote LMP
(17). Using western blot analysis
(Fig. 2A) and immunofluorescence
(Fig. 2B), it was found that in
primary cortical neurons treated with different concentrations of
glutamate, it was LAMP2 that was cleaved in different degrees.
These results are consistent with a previous study of the model of
light-induced retinal degeneration (17), suggesting that calpain-LAMP2
cascades are key in the induction of glutamate-mediated LMP.
It was further investigated whether the activation
of calpain induced LMP following treatment with glutamate. Cleavage
of α-fodrin leading to the formation of 145/150 kDa fragments is a
well-recognized marker for calpain-generated protein breakdown
(20–22). As shown in Fig. 2C, treatment with glutamate led to a
significant increase of α-fodrin 145/150 kDa fragmentation. This
effect was blocked by MK801, indicating that calpain was activated
by overloaded intracellular Ca2+ via NMDA receptors in
neurons following treatment with glutamate. Calpeptin and SJA6017
are two cell-permeable calpain inhibitors. It was found that
calpeptin and SJA6017 abolished the 145/150 kDa fragmentation of
α-fodrin (Fig. 2C), indicating
that the glutamate-induced calpain activation was inhibited.
However, AO staining suggested that the calpain inhibitors did not
alleviate the permeabilization of lysosomal membrane induced by 200
µM glutamate (Fig. 2D).
Therefore, these results suggest that calpain activation is not
required for LMP caused by glutamate in primary cultured cortical
neurons.
ROS are mediators of lysosomal
permeabilization
It has been reported that the activation of calpain
and the generation of free radicals markedly contribute to neuronal
death that is induced by glutamate excitotoxicity. Free radicals,
particularly ROS, damage cell function by oxidative modification of
the proteins, lipids, carbohydrates and nucleic acids of
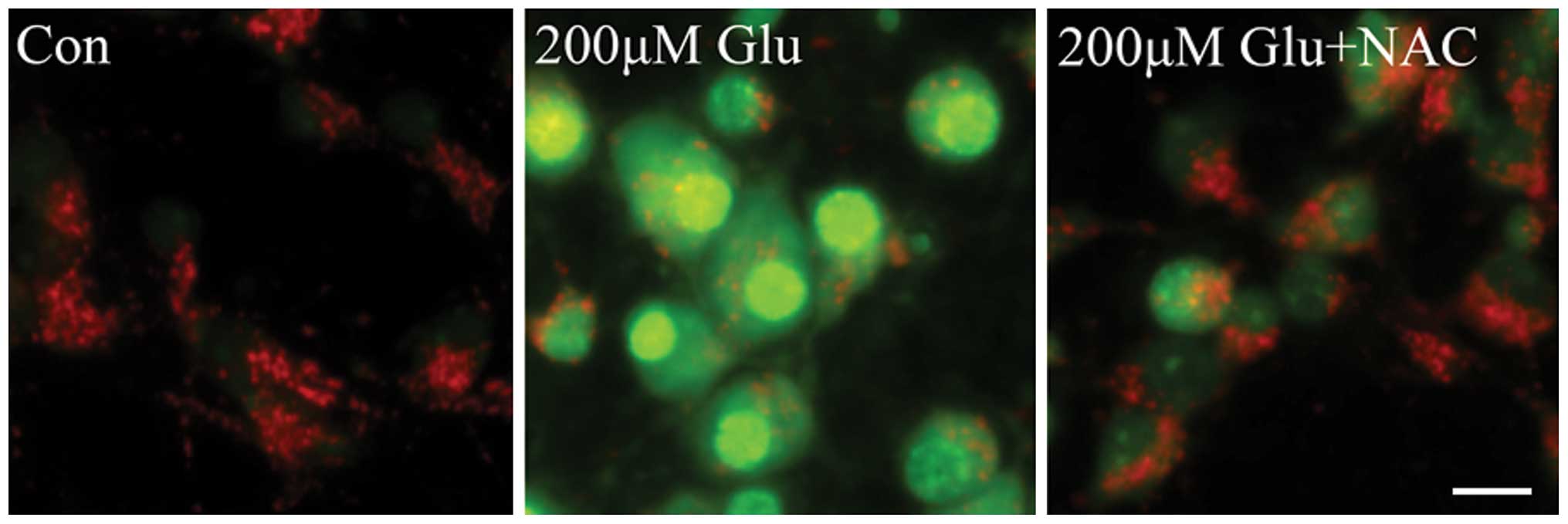
intracellular organelles. To determine the role of ROS in LMP, the
effect of the antioxidant NAC on glutamate-induced permeabilization
of lysosomes was investigated. As shown in Fig. 3, LMP was significantly increased in
neurons following treatment with 200 µM glutamate, which was
markedly inhibited by NAC, as evidenced by decreased AO green
signals and increased red fluorescence. These results indicate that
ROS is a mediator of lysosomal permeabilization downstream of the
NMDA receptor in glutamate excitotoxicity.
Discussion
Glutamate excitotoxicity is considered as one of the
most important pathological mechanisms of neuron loss in acute and
chronic CNS diseases, including acute ischemic stroke.
Intra-arterial fibrinolytic therapy within a 3–6 h window following
the onset of large artery cerebral thrombotic occlusions provides
benefits for rescuing ischemic brain penumbra. This suggests that
the irreversible biochemical processes activated by glutamate, for
example abnormal proteolysis of calpain, contribute to neuronal
death.
The results demonstrated that LMP was increased 30
min after treatment with glutamate, which was abolished by the NMDA
receptor antagonist MK-801 or calcium chelator EGTA. Kubota et
al (23) reported that the
fluorescence signals of LysoSensor and AO dye, and the
immunostaining signals of cathepsin D were unaltered following
treatment with glutamate for 8 h in HT22 cells (an immortalized
mouse hippocampal cell line) indicating that the organization and
membrane integrity of lysosomes was stable. These inconsistencies
may be due to the distinction between oxidative glutamate toxicity
in HT22 and the excitotoxicity in primary cultured cortical neurons
in which glutamate indirectly causes a depletion of intracellular
glutathione through blocking the cystine uptake, which is mediated
by cystine/glutamate antiporter (24).
Moreover, the present study demonstrated that
glutamate-induced LMP was not dependent upon calpain activation.
Involvement in early cascade events of glutamate excitotoxicity
strongly implied another key role of ROS-mediated LMP in
irreversible neural injury. Brain tissue possesses a number of
important endogenous defenses against ischemic injury, including
glutathione, and the enzymatic superoxide dismutase. However,
during injury, these natural antioxidant defenses can be quickly
overwhelmed and followed by energy impairment, leading to increased
production of superoxide radicals, nitric oxide and hydrogen
peroxide. Development of oxidative stress can rapidly lead to
serious disturbances in cerebral function via damage to proteins,
lipids, carbohydrates and nucleic acids (25), while treatment with antioxidant NAC
decreases the extent these injuries in different models of brain
ischemia (26,27). Lysosomes contain a large number of
acidic hydrolases and the pH inside lysosomes is ~4.5. Once rupture
or permeabilization occurs, large quantities of acidic hydrolases,
such as cathepsin B, are released into cytoplasm. This results in
intracellular acidosis and promotes the irreversible degradation of
proteins and lipids (8). Data from
the present study demonstrated that inhibition of ROS by NAC
significantly rescued lysosomes from permeabilization, 30 min after
glutamate treatment. This suggested that the LMP-cathepsin
activation mediated by ROS was an early irreversible injury to
neural cells following glutamate excitotoxicity, which was
consistent with the observations of a strong protective effect of
cathepsin B inhibitors in monkey (10) and mouse (12) ischemia models. These findings,
along with those of previous studies (10–12),
provide further evidence that LMP may be a promising target for
neuronal protection.
In order to determine the detailed molecular
mechanisms underlying ROS-mediated LMP caused by glutamate further
investigation is required. Increasing evidence suggests that LMP
may be governed by several distinct mechanisms in a stimulus- and
cell-type-dependent manner (6,
9, 10,12,13,16,18).
Different from other studies which demonstrated that calpain
promoted lysosomal membrane destabilization during neuronal death
with different stimuli, such as transient focal ischemia, oxygen
glucose deprivation or global ischemia (10,13,28,29),
the results of the present study revealed that glutamate-induced
LMP was not mediated by calpain. It was hypothesized that activated
calpain cleaves oxidative stress-induced carbonylated Hsp70.1 at
the lysosomal membrane, which results in lysosomal
rupture/permeabilization. In the present study, the degradation of
Hsp70.1 was not detectable when the neurons had been subjected to
glutamate treatment. Another substrate of calpain, LAMP2, was
significantly degraded following glutamate treatment. This was
consistent with a previous study, which demonstrated that
degradation of LAMP2 mediated LMP in a model of light-induced
retinal degeneration (17).
However, inhibition of calpain activation had no effect on
alleviating LMP, indicating that calpain is not required for LMP in
early glutamate excitotoxicity.
It was hypothesized that ROS may compromise the
integrity of lysosomes via intralysosomal iron-mediated
peroxidation of membrane lipids (30). This may suggest a nonspecific
mechanism for lysosomal permeabilization, such as pore formation or
limited membrane damage, rather than selective transport across the
membrane, which would likely be specific for a single group of
biomolecules. Monomers of LAMP2, a type 1 transmembrane protein on
the lysosomal membrane, can associate with other proteins to form a
multi-protein channel required for delivery of proteins to
lysosomes for degradation during chaperone-mediated autophagy
(17). The present results suggest
the possibility of cleavage of LAMP2 by other proteases (which are
activated by free radical accumulation but not calpain) is involved
in glutamate-mediated LMP in primary cultured cortical neurons.
In conclusion, LMP is a process that occurs at early
stages of glutamate excitotoxicity in primary cultured cortical
neurons, suggesting that LMP inhibitors may aid in minimizing
ischemic neural injury.
Acknowledgments
This study was supported by the Leading talent
project in science and technology of Guangzhou Development District
(grant no. 2013 L-p090), the Introduction of Innovative R&D
Team Program of Guangdong Province (grant no. 2013Y104), the
National Natural Science Foundation of China (grant no. 81470162)
and the National Major Scientific and Technological Special Project
for “Significant New Drugs Development” of the Ministry of Science
and Technology of China (project no. 20155ZX09J15106-001).
References
|
1
|
Mattson MP: Glutamate and neurotrophic
factors in neuronal plasticity and disease. Ann N Y Acad Sci.
1144:97–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choi DW and Rothman SM: The role of
glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu
Rev Neurosci. 13:171–182. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Beal MF: Mechanisms of excitotoxicity in
neurologic diseases. FASEB J. 6:3338–3344. 1992.PubMed/NCBI
|
|
4
|
Yang JL, Sykora P, Wilson DM III, Mattson
MP and Bohr VA: The excitatory neurotransmitter glutamate
stimulates DNA repair to increase neuronal resiliency. Mech Ageing
Dev. 132:405–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehta A, Prabhakar M, Kumar P, Deshmukh R
and Sharma PL: Excitotoxicity: Bridge to various triggers in
neurodegenerative disorders. Eur J Pharmacol. 698:6–18. 2013.
View Article : Google Scholar
|
|
6
|
Guicciardi ME, Leist M and Gores GJ:
Lysosomes in cell death. Oncogene. 23:2881–2890. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishi T and Forgac M: The vacuolar
(H+)-atpases-nature's most versatile proton pumps. Nat Rev Mol Cell
Bio. 3:94–103. 2002. View
Article : Google Scholar
|
|
8
|
Syntichaki P, Samara C and Tavernarakis N:
The vacuolar H+-ATPase mediates intracellular acidification
required for neurodegeneration in C elegans. Curr Biol.
15:1249–1254. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tardy C, Codogno P, Autefage H, Levade T
and Andrieu-Abadie N: Lysosomes and lysosomal proteins in cancer
cell death (new players of an old struggle). Biochim Biophys Acta.
1765:101–125. 2006.PubMed/NCBI
|
|
10
|
Yamashima T, Kohda Y, Tsuchiya K, Ueno T,
Yamashita J, Yoshioka T and Kominami E: Inhibition of ischaemic
hippocampal neuronal death in primates with cathepsin B inhibitor
CA-074: A novel strategy for neuroprotection based on
'calpain-cathepsin hypothesis'. Eur J Neurosci. 10:1723–1733. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamashima T, Tonchev AB, Tsukada T, Saido
TC, Imajoh-Ohmi S, Momoi T and Kominami E: Sustained calpain
activation associated with lysosomal rupture executes necrosis of
the postischemic CA1 neurons in primates. Hippocampus. 13:791–800.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kilinc M, Gürsoy-Ozdemir Y, Gürer G,
Erdener SE, Erdemli E, Can A and Dalkara T: Lysosomal rupture,
necroapoptotic interactions and potential crosstalk between
cysteine proteases in neurons shortly after focal ischemia.
Neurobiol Dis. 40:293–302. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Windelborn JA and Lipton P: Lysosomal
release of cathepsins causes ischemic damage in the rat hippocampal
slice and depends on NMDA-mediated calcium influx, arachidonic acid
metabolism and free radical production. J Neurochem. 106:56–69.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abramov AY, Canevari L and Duchen MR:
Beta-amyloid peptides induce mitochondrial dysfunction and
oxidative stress in astrocytes and death of neurons through
activation of NADPH oxidase. J Neurosci. 24:565–575. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zdolsek JM, Olsson GM and Brunk UT:
Photooxidative damage to lysosomes of cultured macrophages by
acridine orange. Photochem Photobiol. 51:67–76. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sahara S and Yamashima T: Calpain-mediated
Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys
Res Commun. 393:806–811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Villalpando Rodriguez GE and Torriglia A:
Calpain 1 induce lysosomal permeabilization by cleavage of
lysosomal associated membrane protein 2. Biochim Biophys Acta.
1833:2244–2253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nylandsted J, Gyrd-Hansen M, Danielewicz
A, Fehrenbacher N, Lademann U, Høyer-Hansen M, Weber E, Multhoff G,
Rohde M and Jäättelä M: Heat shock protein 70 promotes cell
survival by inhibiting lysosomal membrane permeabilization. J Exp
Med. 200:425–435. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stetler RA, Gan Y, Zhang W, Liou AK, Gao
Y, Cao G and Chen J: Heat shock proteins: Cellular and molecular
mechanisms in the central nervous system. Prog Neurobiol.
92:184–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martin SJ, O'Brien GA, Nishioka WK,
McGahon AJ, Mahboubi A, Saido TC and Green DR: Proteolysis of
fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem.
270:6425–6428. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pike BR, Zhao X, Newcomb JK, Posmantur RM,
Wang KK and Hayes RL: Regional calpain and caspase-3 proteolysis of
alpha-spectrin after traumatic brain injury. Neuroreport.
9:2437–2442. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dutta S, Chiu YC, Probert AW and Wang KK:
Selective release of calpain produced alphalI-spectrin
(alpha-fodrin) breakdown products by acute neuronal cell death.
Biol Chem. 383:785–791. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kubota C, Torii S, Hou N, Saito N,
Yoshimoto Y, Imai H and Takeuchi T: Constitutive reactive oxygen
species generation from autophagosome/lysosome in neuronal
oxidative toxicity. J Biol Chem. 285:667–674. 2010. View Article : Google Scholar :
|
|
24
|
Li Y, Maher P and Schubert D:
Phosphatidylcholine-specific phospholipase C regulates
glutamate-induced nerve cell death. Proc Natl Acad Sci USA.
95:7748–7753. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Warner DS, Sheng H and Batinić-Haberle I:
Oxidants, antioxidants and the ischemic brain. J Exp Biol.
207:3221–3231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jatana M, Singh I, Singh AK and Jenkins D:
Combination of systemic hypothermia and N-acetylcysteine attenuates
hypoxic-ischemic brain injury in neonatal rats. Pediatr Res.
59:684–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sekhon B, Sekhon C, Khan M, Patel SJ,
Singh I and Singh AK: N-Acetyl cysteine protects against injury in
a rat model of focal cerebral ischemia. Brain Res. 971:1–8. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yap YW, Whiteman M, Bay BH, Li Y, Sheu FS,
Qi RZ, Tan CH and Cheung NS: Hypochlorous acid induces apoptosis of
cultured cortical neurons through activation of calpains and
rupture of lysosomes. J Neurochem. 98:1597–1609. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamashima T, Saido TC, Takita M, Miyazawa
A, Yamano J, Miyakawa A, Nishijyo H, Yamashita J, Kawashima S, Ono
T and Yoshioka T: Transient brain ischaemia provokes Ca2+, PIP2 and
calpain responses prior to delayed neuronal death in monkeys. Eur J
Neurosci. 8:1932–1944. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Johansson AC, Appelqvist H, Nilsson C,
Kågedal K, Roberg K and Ollinger K: Regulation of
apoptosis-associated lysosomal membrane permeabilization.
Apoptosis. 15:527–540. 2010. View Article : Google Scholar : PubMed/NCBI
|