Introduction
The endothelium lies in between the circulating
blood and vascular smooth muscle cells, which are responsible for
peripheral resistance (1). It may
be easily damaged and endothelial dysfunction occurs in the
pathogenesis of various cardiovascular complications, particularly
in hypertension (2–6). Damage to the endothelium can result
in a positive feedback mechanism as in arterial hypertension, as it
negatively affects the vascular tone and homeostasis upon damage.
Thus, it is an early independent predictor of cardiovascular events
(7–10). Endothelial dysfunction contributes
to an increase in large arterial stiffness in patients with
isolated systolic hypertension, resulting in impaired vascular
elasticity and compliance, and subsequent arterial hypertension
(11,12). Endothelial dysfunction increases
the risk of developing atherosclerotic lesions and related
cardiovascular events, even if the blood pressure of patients with
essential hypertension is controlled (13). Restoration of conduit artery
endothelial function is therefore a primary target in limiting
cardiovascular morbidity and mortality in patients with essential
hypertension (7).
Cilostazol, a type III phosphodiesterase inhibitor,
inhibits platelet aggregation and it is widely used in the
treatment of peripheral vascular diseases (14–16).
Previous studies have demonstrated that cilostazol serves a role in
the inhibition of endothelial cell apoptosis (14,17).
For example, it can suppress superoxide production and expression
of adhesion molecules in human endothelial cells (18). In addition, cilostazol may prevent
endothelial cell apoptosis by stimulating the extra-cellular
signal-regulated kinase (ERK) 1/2 and p38 MAPK signaling,
particularly in patients with hyperlipidemia and in pathological
tissue conditions, including ischemia, shock and sepsis (19,20).
Previous studies have suggested that the phosphoinositide 3 kinase
(PI3K)/Akt pathway serves an important role in preventing cell
apoptosis induced by numerous stimuli (21,22).
In endothelial cells, PI3K/Akt activation promotes cell survival
(2,23). Previous studies have demonstrated
that cilostazol produces a vasculo-angiogenic effect by
upregulating a broad signaling network that includes the
PI3K/Akt/endothelial nitric oxide synthase (eNOS) pathway (17). However, whether and how cilostazol
protects the endothelial function in patients with essential
hypertension remains unknown.
The aim of the present study was to determine
whether cilostazol suppresses endothelial cell apoptosis and
dysfunction in hypertension, and whether the PI3K/Akt pathway was
involved. Evidence from previous studies suggest that angiotensin
II (angII), a peptide of the rennin angiotensin system, exerts an
vasoconstrictive effect, induces intracellular reactive oxygen
species (ROS) production and causes vascular dysfunction and
arterial hypertension (24–30).
Therefore, Sprague Dawley (SD) rats were infused with angII to
generate hypertension. The effect of cilostazol on endothelial
function and apoptosis, as well as nitric oxide (NO) and superoxide
production were investigated. Additionally, the PI3K/Akt signaling
pathway was also investigated using human umbilical vein
endothelial cells (HUVECs).
Materials and methods
Chemicals
Cilostazol, acetylcholine (Ach), sodium
nitroprusside (SNP), angII and U46619 were gifts from Dr. Zhiqiang
Yan (Department of Neurosurgery, Urumqi General Hospital of Lanzhou
Military Command, Urumqi, China). LY294002 and
4′,6-diamidino-2-phenylindole (DAPI) were gifts from Dr. Wei Zhang
(Department of Cardiology, Tangdu Hospital, Fourth Military Medical
University, Xi'an, China). The HUVEC cell line was a gift from Mr.
Xiaofei Zhu (Department of Neurosurgery, Tangdu Hospital, Fourth
Military Medical University).
Animal model and experimental design
Male SD rats (weight, 200–220 g; age, 10–12 weeks)
were purchased from the Animal Center of The Fourth Military
Medical University (FMMU). They were maintained in a
temperature-controlled room (24°C), on a 12 h light/dark cycle and
given free access to food water. The experimental protocol was
approved by the Institutional Care and Use Committee of the FMMU,
which conforms to the Guidelines for the Care and Use of Laboratory
Animals of the US National Institutes of Health (NIH publication
no. 85–23, revised 1996) (31).
Rats were divided into four groups (n=10): (i) Saline-treated
group, rats treated with saline by intragastric administration
(IA); (ii) Saline + Cilo-treated group, rats treated with saline
and cilostazol (30 mg/kg/day) by IA; (iii) angII-treated group,
rats continuously infused with angII (1,000 ng/kg/min) by
subcutaneously implanted Alzet osmotic pumps (2004 model;
Cupertino, CA, USA) as previously described (32); and (iv) angII (1,000 ng/kg/min) +
Cilo-treated group, rats treated with cilostazol (30 mg/kg/day) in
addition to angII (1,000 ng/kg/min) infusion.
After 4 weeks of angII infusion and cilostazol
administration, rats were anesthetized with an intraperitoneal
injection of sodium pentobarbital (45 mg/kg; Shanghai Rongbai
Biological Technology Co., Ltd., Shanghai, China). Hemodynamic
measurements were obtained as previously described (33). In brief, the right carotid artery
of anesthetized rats was cannulated with a catheter connected to a
microtip pressure transducer (Chengdu Instrument Factory, Chengdu,
China) and the transducer was connected to a recording system
(Rm6280C, Biological Instruments). The systolic and diastolic blood
pressure (sBP and dBP, respectively) were then measured. Finally,
the rats were sacrificed by cervical vertebral dislocation. The
abdominal aortae were removed and divided into two sections by
transverse section. The first was incubated in 10% buffered
formalin (Sangon Biotech Co., Ltd., Shanghai, China) for 24 h and
then paraffin-embedded (Sangon Biotech Co., Ltd.) for further
assessment of apoptosis. The second was used for the detection of
endothelium-dependent and -independent vasorelaxation, and
assessment of superoxide anion and NO production.
Detection of endothelium-dependent and
independent vasorelaxation
Abdominal aortae were carefully dissected and
mounted as ring preparations of ~3 mm on hooks in individual organ
baths (Radnoti Glass Technology, Monrovia, CA, USA). Arterial
integrity was assessed first by stimulation of vessels with 120 mM
KCl, and then washed three times in physiological salt solution
(130 mM NaCl, 14.9 mM NaHCO3, 4.7 mM KCl, 1.18 mM
KH2PO4, 1.18 mM
MgSO47H2O, 1.56 mM CaCl2
H2O, 0.026 mM EDTA and 5.5 mM glucose). The temperature
was maintained at 37°C and 95% O2 and 5% CO2
was pumped into the physiological salt solution. Following washing
and stabilization, by contracting the segments with phenylephrine
(10 µmol/l), followed by relaxation with Ach (10
µmol/l). The contraction response was detected using an
organ chamber containing an isometric Mulvany-Halpern myograph
(model 610; DMT-USA, Inc., Marietta, GA, USA) and recorded using a
PowerLab 8/SP data acquisition system (ADInstruments Ltd., Colorado
Springs, CO, USA), as previously described (34–36).
Contractile responses of abdominal aortic rings were evoked with 30
nmol/l U46619. At the plateau of contraction, Ach
(1×10−8−1×10−4 mol/l) or SNP
(1×10−10−1×10−6 mol/l)were progressively
added to the organ bath to induce endothelium-dependent or
-independent relaxation.
Detection of superoxide anion
production
Superoxide anions from the aortae were measured
using flow injection chemiluminescence, as previously described
(37). The superoxide anion
concentration is reported as chemiluminescence intensity (CI) per
mg of tissue weight.
Detection of total NO production
The total NO production in aortae was determined by
measuring nitrite concentration, as previously described (37). The concentration of nitrite was
calculated from a nitrite standard curve.
Immunohistochemistry
Tissue samples were prepared from abdominal aortae.
After being paraffin-embedded, the abdominal aorta was exposed by a
transverse section and cut into 4 µm thick sections. The
cell nuclei of the sections were stained with DAPI as previously
described (26). The
immunofluorescence data were analyzed using an Eclipse Ni-E
microscope (Nikon, Tokyo, Japan) and NIS-elements imaging software
(Nikon).
Cell culture and treatment
HUVECs were provided by Dr. Xiaofei Zhu from the
Department of Neurosurgery, Tangdu Hospital, Fourth Military
Medical University (Xi'an, China). HUVEC monolayers were grown as
previously described (2,20). In brief, cells were plated into
dishes with Gibco Dulbecco's Modified Eagle's medium (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 1%
penicillin-streptomycin (Beyotime Institute of Biotechnology,
Shanghai, China) and 10% fetal bovine serum (Beyotime Institute of
Biotechnology), under 5% CO2 at 37°C, at a density of
1×105 cells/ml. Cells were supplemented with 5 U/ml
heparin (Sangon Biotech Co., Ltd.) and 100 ng/ml endothelial cell
growth substance (Collaborative Research Inc., Bedford, MA, USA).
When the cells reached confluence (90%), subcultures were prepared.
Cells in actively growing conditions between the third and fifth
passages were used for further experiments.
HUVECs were divided into four parallel groups:
Untreated cells (control group); cells treated with 10
µmol/l angII (angII-treated group); cells pretreated with 10
µmol/l cilostazol prior to incubation with angII (angII +
Cilo-treated group); and cells pretreated with a combination of
cilostazol and LY294002 (a PI3K inhibitor) prior to incubation with
angII (angII + Cilo + LY-treated group). The concentrations of
cilostazol and angII were selected on the basis of previous studies
(20,24).
Immunocytochemistry
Cultured HUVECs were fixed with 10% buffered
formalin and permeabilized with 0.5% Triton X-100 (Sangon Biotech
Co., Ltd.). The cell nuclei were stained with DAPI as previously
described (33).
Western blot analysis of HUVECs
Cultured HUVECs in lysis buffer containing 50 mmol/l
Tris-hydrochloride, 150 mmol/l sodium chloride, 1% Nonidet P-40,
0.25% superoxide dismutase, 1 mmol/l EDTA, 1 mmol/l NaF, 1 mmol/l
Na3VO3, 1 mM phenylmethylsulfonyl fluoride
and a proteinase inhibitor cocktail tablet (Roche Diagnostics,
Basel, Switzerland). Protein samples were assessed by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis, as described
previously (33). In brief, total
protein concentration of each sample was determined prior to
polyacrylamide gel electrophoresis, followed by the transfer of
proteins to polyvinylidene fluoride (PVDF) membranes (Sangon
Biotech Co., Ltd.). PVDF membranes were incubated with blocking
buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 h at room
temperature and subsequently immunoblotted with either of the
following primary antibodies: Polyclonal rabbit total Akt (1:1,000;
cat. no. SAB4500797; Sigma-Aldrich, St. Louis, MO, USA); polyclonal
rabbit phosphorylated-Akt (p-Akt) (1:1,000; cat. no. SAB4504017;
Sigma-Aldrich); cleaved and total monoclonal rabbit caspase-3
(1:1,000; cat. no. 9665; Cell Signaling Technology, Inc., Danvers,
MA, USA); and monoclonal mouse β-actin (1:10,000; cat. no. A5441;
Sigma-Aldrich). β-actin protein expression served as a loading
control. Following being immunoblotted with primary antibodies
overnight at 4°C, the PVDF membranes were washed three times (10
min each) in Tris-buffered saline containing Tween-20 and then
incubated with IRDye 680RD goat anti-rabbit IgG (1:5,000; cat. no.
925-68071; LI-COR Biosciences) or IRDye 800CW goat anti-mouse IgM
(1:5,000; cat. no. 926-32280; LI-COR Biosciences) for 60 min at
room temperature, separately. The PVDF membranes were then washed
three times (10 min each) in phosphate-buffered saline with Tween
20. Bands were evaluated by densitometry using an Odyssey infrared
imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay of HUVECs and abdominal aorta
of endothelial cells
The TUNEL assay kit (Beyotime Institute of
Biotechnology) was used to detect apoptotic cells according to the
manufacturer's instructions. TUNEL-positive cells were detected by
microscopy (Eclipse Ni-E; Nikon). The apoptotic index is expressed
as the number of positively stained cells per total number of
endothelial cells.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. SPSS 13.0 software (SPSS, Inc, Chicago, IL, USA) was used
to analyze the data. One-way analysis of variance was used to
determine statistically significant differences among the four
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
AngII-infusion increases the sBP and dBP
levels in treated rats
Previous studies have demonstrated that angII
infusion at doses between 175 and 1,000 ng/kg/min may result in
hypertension in rats (32,38,39).
In order to demonstrate the hypertensive effect of angII, a dose of
1,000 ng/kg/min was infused into rats for the period of 4 weeks.
Following the infusion, the sBP and dBP levels were measured and
demonstrated to be significantly increased compared with the
saline-treated group (*P<0.05; Table I). Cilostazol was not identified to
exhibit an effect on angII-infusion-dependent hypertension
(Table I).
 | Table IComparison of blood pressure among
the four groups of treated rats. |
Table I
Comparison of blood pressure among
the four groups of treated rats.
| Group | sBP (mmHg) | dBP (mmHg) |
|---|
| Saline | 125.8±5.3 | 78.2±5.9 |
| Saline+Cilo | 125.7±6.0 | 81.3±2.3 |
| AngII | 192.5±4.6a | 111.8±6.3a |
| AngII+Cilo | 193.5±3.4a | 114.3±4.6a |
Cilostazol treatment inhibits
angII-induced dysfunction and apoptosis of endothelial cells
Evidence from previous studies has suggested that
angII induces aberrant oxidative stress in the vascular wall and,
therefore, intracellular ROS production, resulting in excessive
apoptosis and dysfunction of the epithelium and endothelium
(40–42). Whether cilostazol suppresses angII
induced endothelial dysfunction remains unknown. Thus, the
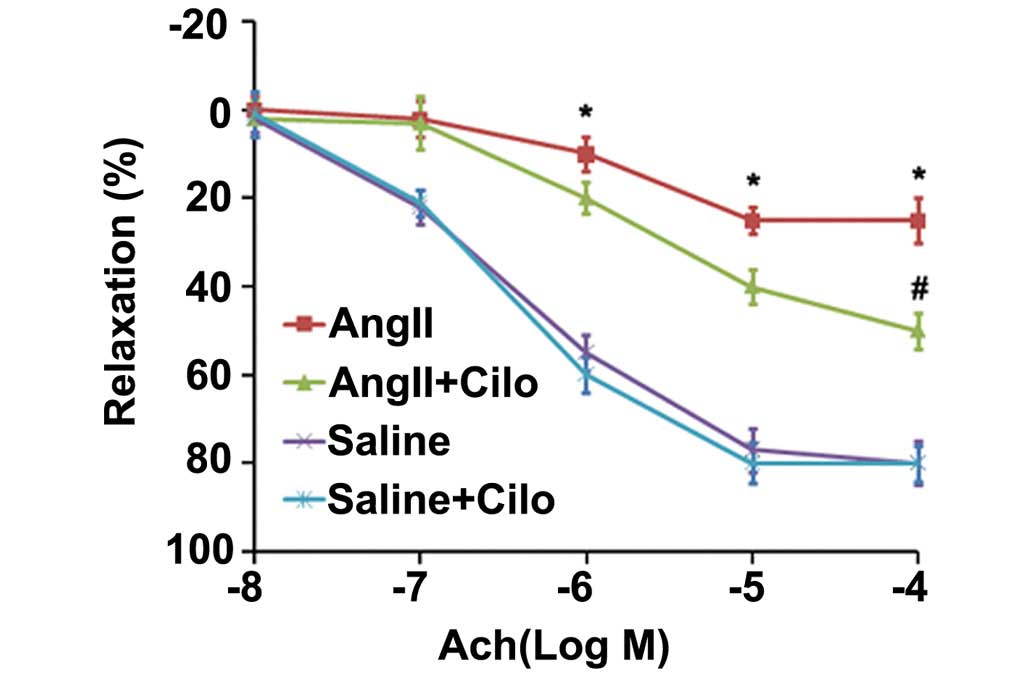
percentage of Ach-induced vascular relaxation was investigated
(Fig. 1). Compared with the
saline-treated rats, abdominal aorta rings from the angII-infused
rats demonstrated significantly impaired Ach-induced
endothelium-dependent relaxation (*P<0.05; Fig. 1). Cilostazol significantly reduced
the impairment in vasorelaxation in the angII +Cilo-treated group
compared with the angII-treated group (#P<0.05;
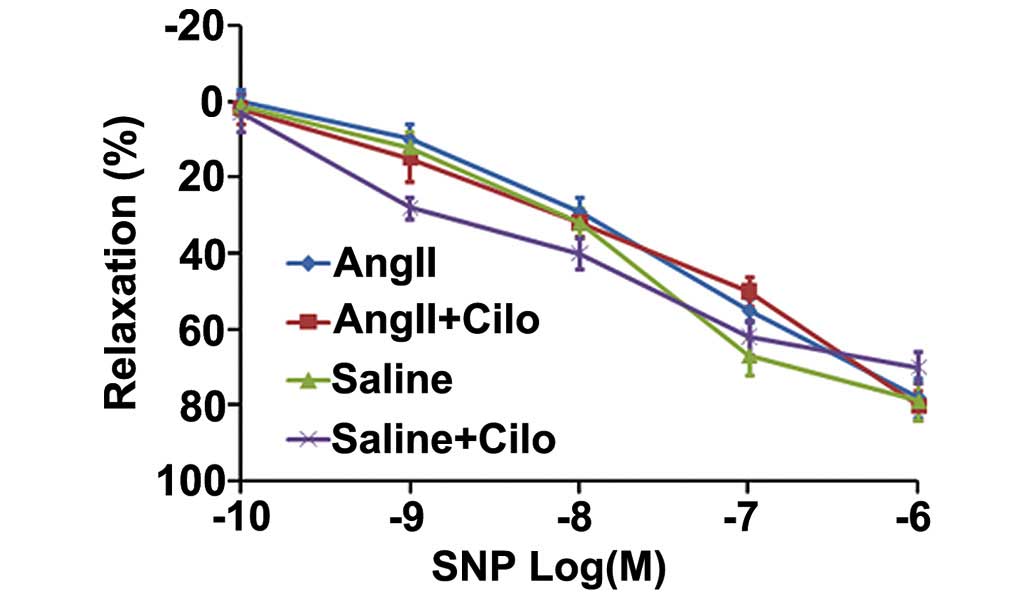
Fig. 1). No significant difference
among the four groups of rats was identified upon investigation of
SNP-induced endothelium-independent relaxation (Fig. 2).
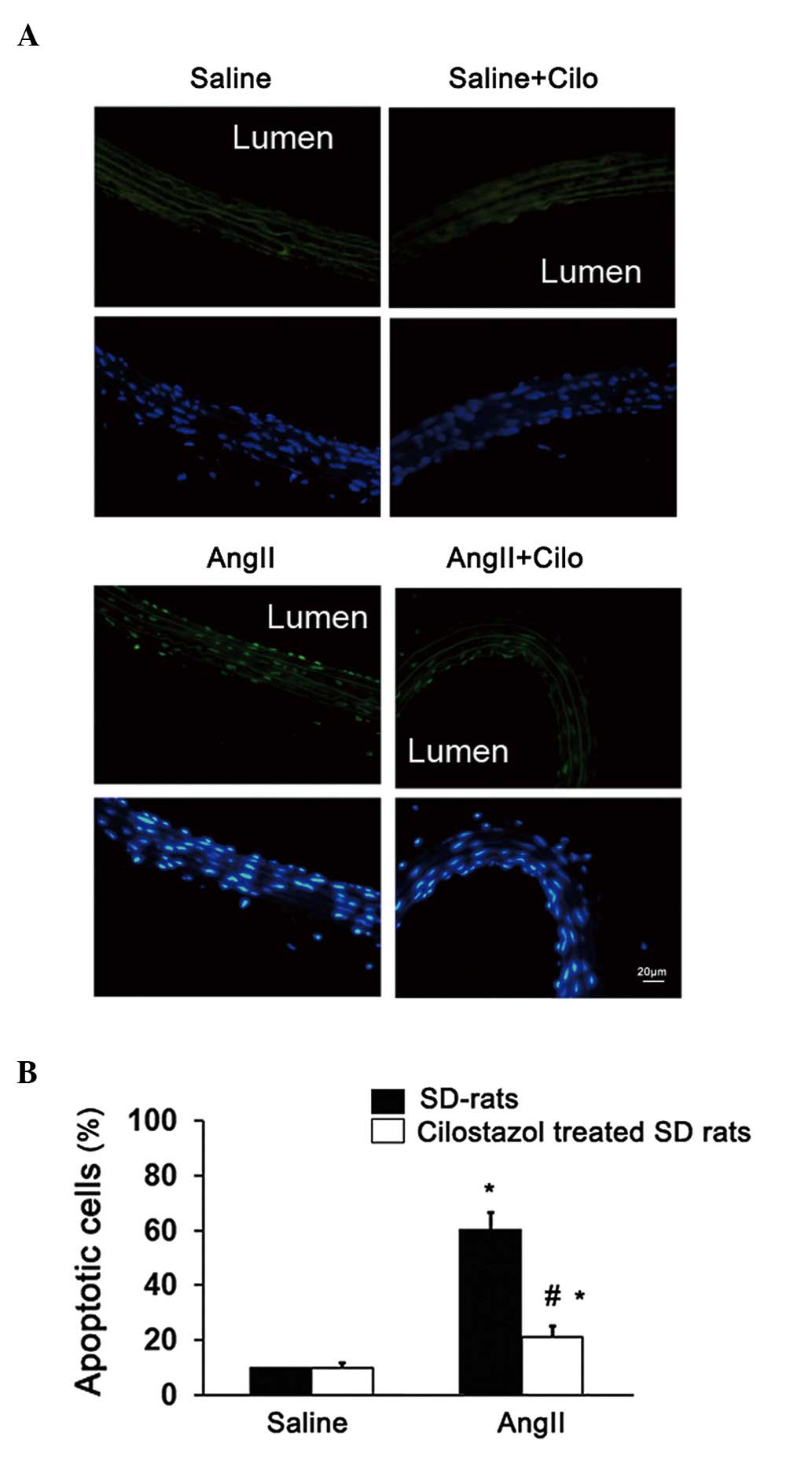
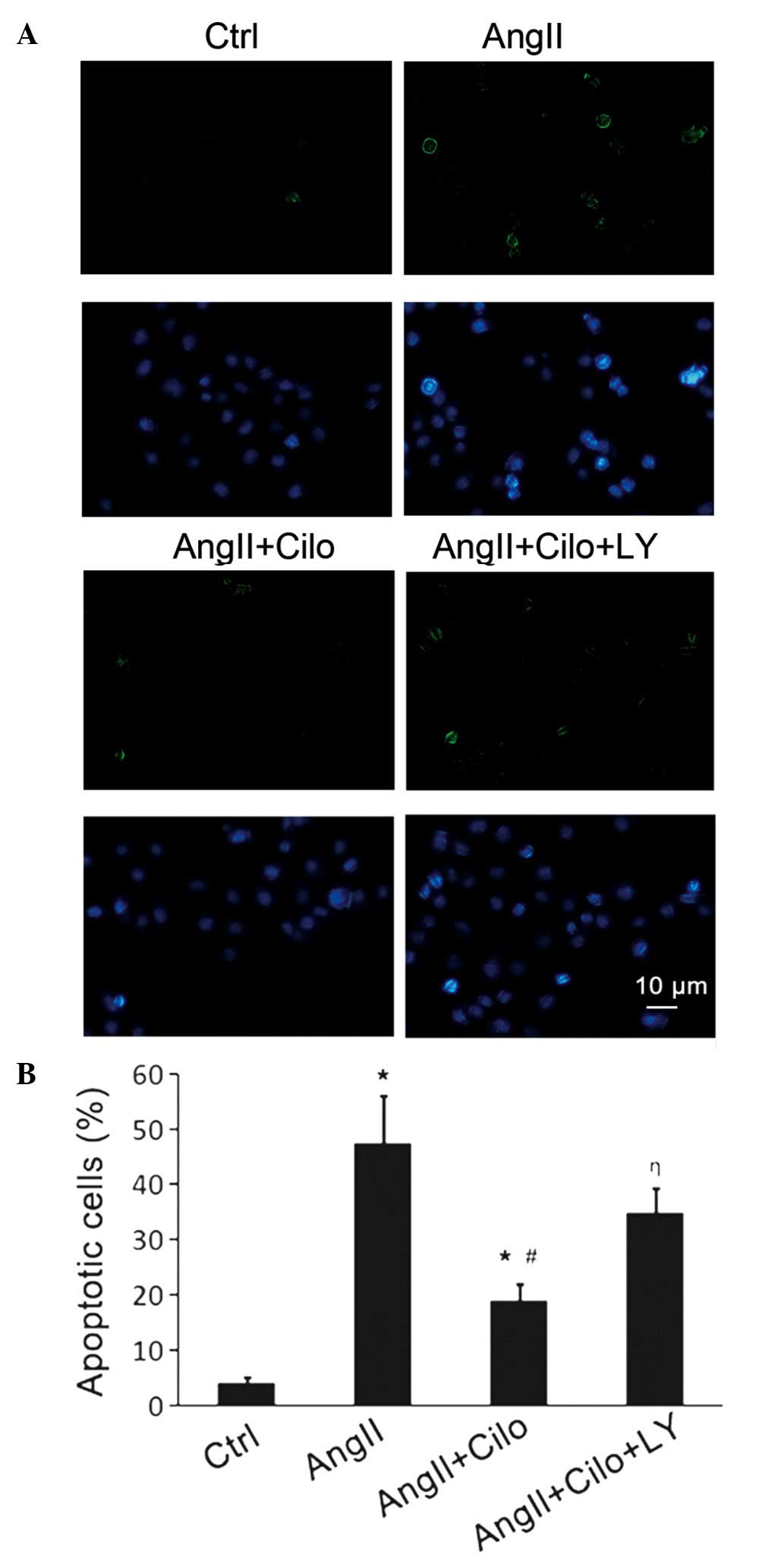
Furthermore, the apoptosis of endothelial cells was
investigated. Compared with the saline-treated group, endothelial
apoptosis was significantly increased in the angII-infused rats
(*P<0.05; Fig. 3).
Endothelial apoptosis was significantly decreased in angII-infused
rats treated with cilostazol compared with the angII-treated group
(#P<0.05; Fig. 3).
The cilostazol + saline-treatment had no effect on endothelial
apoptosis (Fig. 3) or the
Ach-induced vasorelaxation (Fig.
1). These results suggest that cilostazol alleviates the
endothelial dysfunction in angII-induced hypertension rats,
possibly through an anti-apoptotic effect.
Effect of cilostazol on the angII-induced
increase in superoxide anion production
AngII acts via the nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase-derived ROS to trigger endothelial cell
apoptosis and impair endothelium-dependent relaxation (34). Cilostazol may therefore alleviate
endothelial cell apoptosis and attenuate impairment in
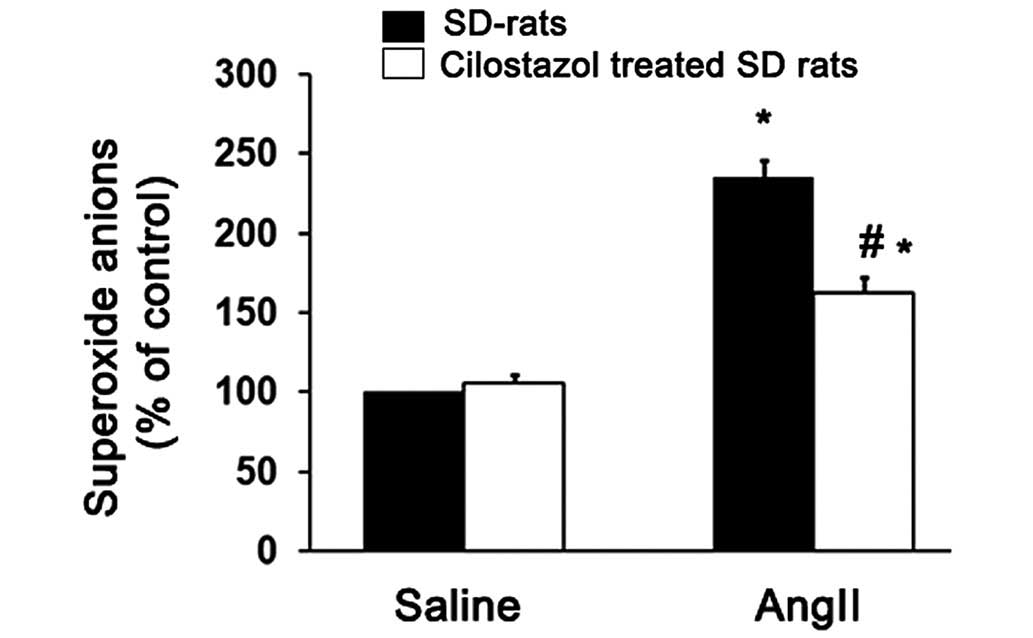
vasorelaxation by inhibiting superoxide production. Thus, the
superoxide levels produced in the aortic tissue from the four
groups of treated rats were determined and compared. AngII
significantly increased the superoxide production compared with the
saline-treated group (*P<0.05; Fig. 4). However, cilostazol significantly
suppressed the superoxide anion production compared with the
angII-only treated group (#P<0.05; Fig. 4).
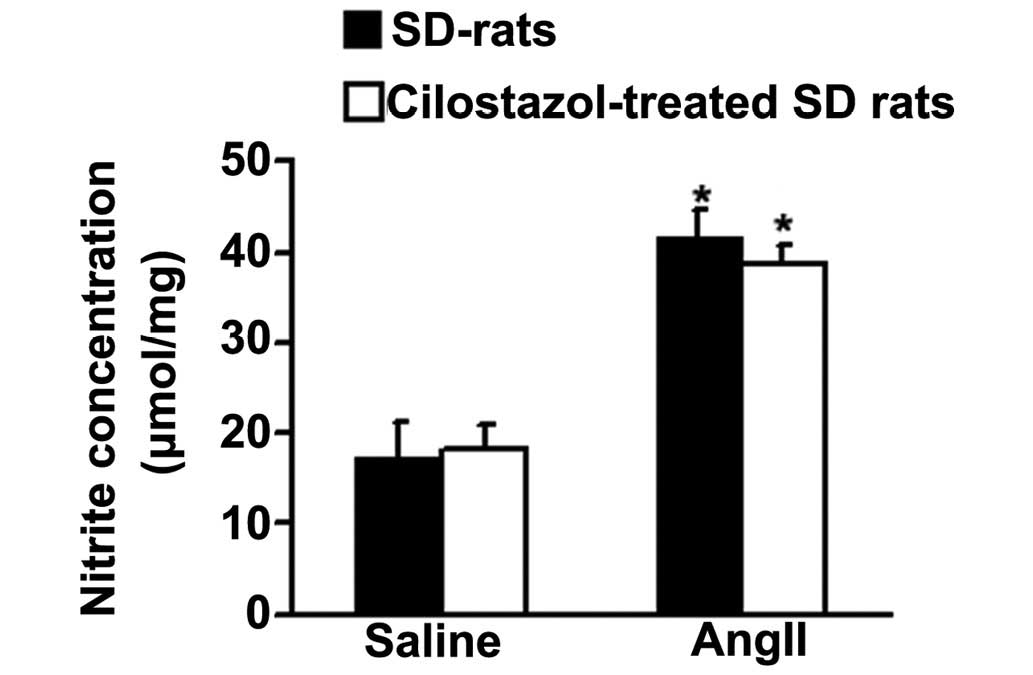
Effect of cilostazol on NO
production
To detect whether cilostazol has an effect on NO
production and thus improves vasorelaxation in response to Ach,
total NO production in aortae was determined from the concentration
of nitrite, a stable metabolite of NO in vitro (38). AngII significantly increased the
total NO production compared with the saline-treated, control group
(*P<0.05; Fig. 5).
Cilostazol had no effect on the angII-induced NO production.
Inhibition of angII-induced HUVEC
apoptosis by cilostazol
AngII treatment (10 µmol/l) resulted in a
significant increase in the number of TUNEL-positive (apoptotic)
HUVECs compared with the control group (*P<0.05;
Fig. 6). Pretreatment with
cilostazol (10 µmol/l) reduced the number of TUNEL-positive
HUVECs produced on exposure to angII, compared with the
angII-treated group (#P<0.05; Fig. 6). LY294002, a specific inhibitor of
PI3K, was used to detect whether the PI3K/Akt pathway was involved
in the effect of the cilostazol treatment. Compared with the angII
+ Cilo-treated group, HUVECs pretreated with a combination of
cilostazol and LY294002 demonstrated increased numbers of
TUNEL-positive cells (ηP<0.05; Fig. 6).
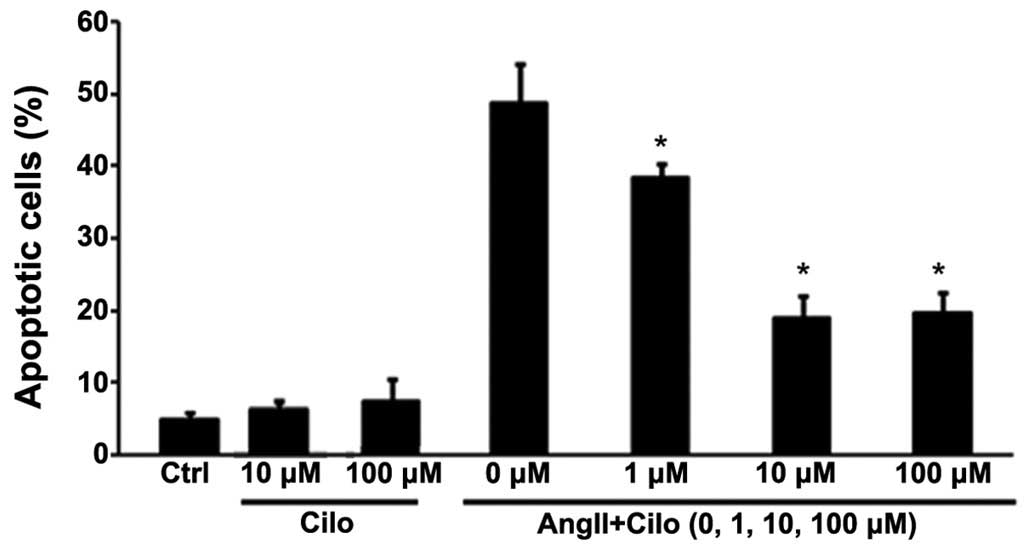
Cilostazol (1, 10 and 100 µmol/l) was
administered to HUVECs to investigate the effect concentration.
Cilostazol at 1 µmol/l mildly inhibited angII-induced HUVEC
apoptosis, compared with concentrations of 10 or 100 µmol/l
which significantly attenuated apoptosis, to similar levels, in the
angII treated group (*P<0.05; Fig. 7).
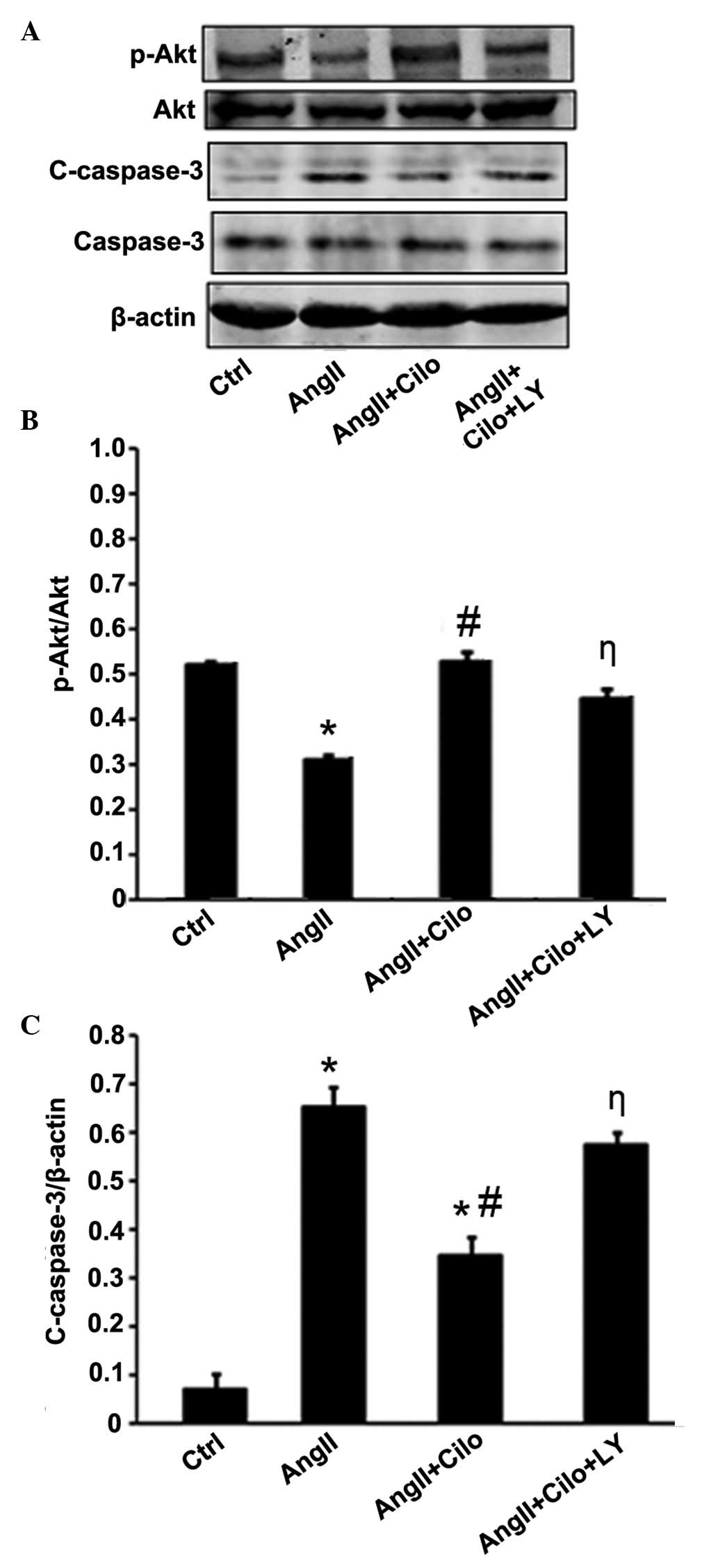
Effect of cilostazol on Akt and cleaved
caspase-3 protein expression levels in HUVECs
To elucidate the mechanism underlying the
cilostazol-dependent reduction of angII-induced apoptosis, the
effect of cilostazol on Akt phosphorylation was examined using
western blot analysis (Fig. 8A).
As demonstrated in Figure 8B,
angII significantly reduced the phosphorylation of Akt compared
with the control group (*P<0.05) and cilostazol
attenuated the reduction of Akt phosphorylation compared with the
angII-treated group (#P<0.05). Furthermore, the
effect of cilostazol on Akt phosphorylation was reduced by
combinational treatment with LY294002 compared with the angII +
Cilo-treated group (ηP<0.05; Fig. 8B).
AngII treatment upregulated the cleaved caspase-3
protein expression levels compared with the control group
(*P<0.05; Fig. 8C)
and cilostazol treatment suppressed this effect compared with the
angII-treated group (#P<0.05; Fig. 8C). LY294002 attenuated the effect
of cilostazol on cleaved caspase-3 protein expression compared with
the angII + Cilo-treated group (ηP<0.05; Fig. 8C).
Discussion
The results of the present study demonstrated that
cilostazol suppressed the endothelial cell apoptosis induced by
angII, in vivo and in vitro. In vivo,
cilostazol suppressed the angII-induced endothelial dysfunction and
increase in superoxide production, without affecting the total NO
production. In vitro, cilostazol suppressed the
angII-induced upregulation of cleaved caspase-3 protein levels and
increase in apoptosis of HUVECs, and attenuated the angII-induced
reduction of Akt phosphorylation. The effect of cilostazol on
apoptotic HUVECs was blunted by LY294002, a PI3K inhibitor.
A previous study demonstrated that patients with
essential hypertension suffer from endothelial dysfunction,
particularly in the conduit arteries, due to damage from abnormal
blood pressure (43). In turn, a
damaged endothelium has a negative effect on the vascular tone,
homeostasis and arterial stiffness. The endothelial dysfunction
elevates the risk of cardiovascular events in patients with
essential hypertension, whether the blood pressure is controlled or
not (13). Endothelial cell
apoptosis can induce endothelium dysfunction, therefore, its
prevention may improve endothelial function and decrease the risk
of cardiovascular disease (41).
AngII is important in the pathogenesis of hypertension (44) and increased levels of angII promote
the formation of atherosclerotic lesions (45). Previous experimental studies have
indicated that atherosclerotic lesion-prone vascular regions are
characterized by a high endothelial cell turnover, which has been
attributed to an increased rate of endothelial cell apoptosis
(30,46). Thus, angII-induced endothelial cell
apoptosis may serve an essential role in endothelial dysfunction in
patients with essential hypertension. In the present study, angII
was validated as a useful tool to induce hypertension and
endothelial apoptosis in rats. The results of the present study
demonstrated that angII treatment led to hypertension, endothelial
dysfunction, an increase in superoxide production and endothelial
apoptosis, consistent with previous studies (39,47–50).
Cilostazol is a type III phosphodiesterase inhibitor
and serves a role in the inhibition of endothelial cell apoptosis
(14,17). Thus, patients suffering from
hypertension may benefit from administration of this drug, although
the efficacy and mechanism of action in patients with hypertension
have not yet been determined. In the present study, the effect of
cilostazol on angII-induced (hypertensive) endothelial apoptosis
and endothelial function was investigated. Cilostazol treatment
suppressed the angII-induced endothelial dysfunction and apoptosis
in vivo without affecting the blood pressure.
Vascular relaxation critically depends on the
balance between superoxide and NO production by the vascular
endothelium (51). Therefore, the
superoxide anion and NO production was detected in the aortae of
treated rats. Cilostazol attenuated the angII-induced increase in
superoxide anion production, however had no effect on NO
production. It may be considered controversial that angII increased
the NO production and suppressed endothelial function, compared
with cilostazol treatment which improved the endothelial function
without affecting the NO production. However, these results may be
due to the actions of different NO synthases (NOS), as endothelial
NOS (eNOS) and inducible NOS (iNOS) serve different roles in the
pathophysiology of cardiovascular diseases (52–54).
Relatively low concentrations of NO appear to favor cell
proliferation and anti-apoptotic responses compared with higher
levels of NO which favor pathways inducing cell cycle arrest,
mitochondrial respiration and apoptosis (55). Under pathological conditions
increased amounts of NO are produced, resulting in stimulation of
iNOS expression, and possibly endothelial dysfunction (56,57).
Further research is required to assess this effect.
In order to further investigate the mechanisms
involved in the protective effects of cilostazol against
endothelial apoptosis, HUVECs were utilized as an experimental
tool. In vitro, cilostazol significantly reduced the
angII-induced HUVEC apoptosis. Additionally, cilostazol attenuated
the angII-induced reduction in Akt phosphorylation, and this
protective effect of cilostazol on HUVEC apoptosis was inhibited by
LY294002. The PI3K/Akt pathway is considered to be an important
pathway for cell survival (58,59),
particularly in endothelial cells (2). Caspase-3 serves as a central member
of the apoptotic cascade and can be activated to cleave the
inhibitor of endonuclease, which cuts the DNA and induces the final
stage of apoptosis. The present study demonstrated that angII
treatment led to an upregulation of cleaved caspase-3 and further
treatment with cilostazol downregulated the cleaved caspase-3 in
angII-treated cells.
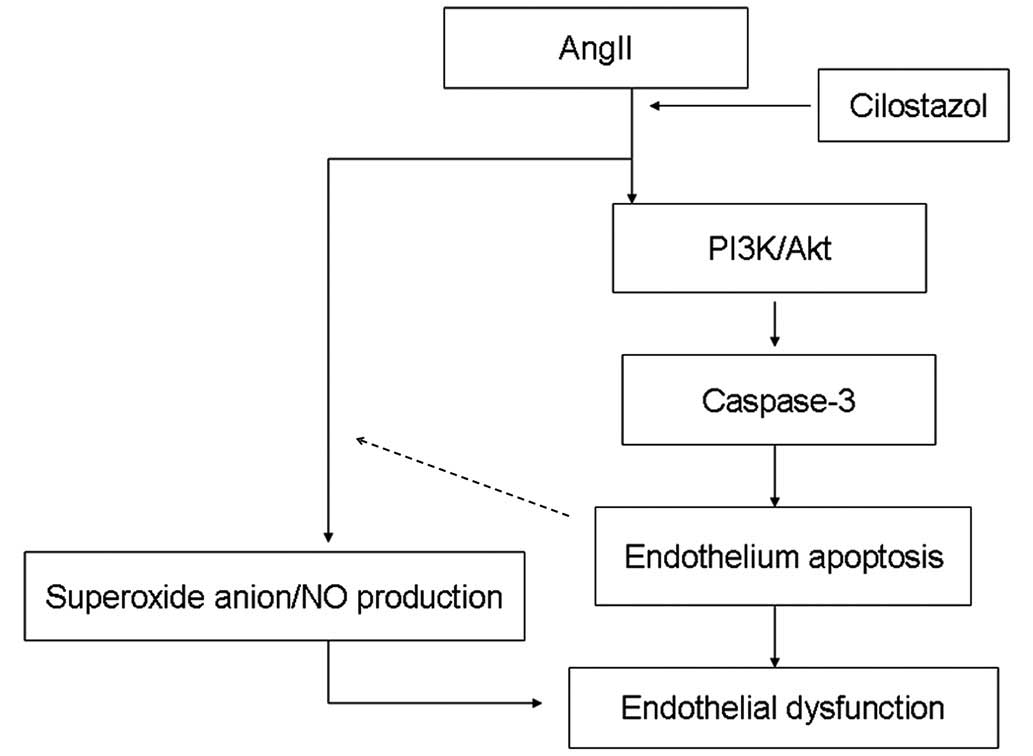
In conclusion, cilostazol protects HUVECs from
apoptosis by stimulating the PI3K/Akt pathway and inhibiting the
caspase pathway. As indicated in Figure 9, the results of the current study
suggest that cilostazol demonstrated a protective role against
endothelial apoptosis by affecting the PI3K/AKt pathway and the
superoxide anion/NO balance in animals suffering from angII-induced
hypertension. Cilostazol may therefore represent a novel
therapeutic agent for patients with essential hypertension.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300077) and the
2012 Graduate Students Creativity Foundation of Tangdu Hospital, at
The Fourth Military Medical University, China (grant no.
00543).
References
|
1
|
Spieker LE, Flammer AJ and Lüscher TF: The
vascular endothelium in hypertension. Handbook Exp Pharmacol.
176:249–283. 2006. View Article : Google Scholar
|
|
2
|
Zhang W, Wang R, Han SF, Bu L, Wang SW, Ma
H and Jia GL: Alpha-linolenic acid attenuates high glucose-induced
apoptosis in cultured human umbilical vein endothelial cells via
PI3K/Akt/eNOS pathway. Nutrition. 23:762–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fraga-Silva RA, Costa-Fraga FP, Murça TM,
Moraes PL, Martins Lima A, Lautner RQ, Castro CH, Soares CM, Borges
CL, Nadu AP, et al: Angiotensin-converting enzyme 2 activation
improves endothelial function. Hypertension. 61:1233–1238. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun GB, Qin M, Ye JX, Pan RL, Meng XB,
Wang M, Luo Y, Li ZY, Wang HW and Sun XB: Inhibitory effects of
myricitrin on oxidative stress-induced endothelial damage and early
athero-sclerosis in ApoE−/− mice. Toxicol Appl Pharmacol.
271:114–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guan Q, Zhang Y, Yu C, Liu Y, Gao L and
Zhao J: Hydrogen sulfide protects against high-glucose-induced
apoptosis in endothelial cells. J Cardiovasc Pharmacol. 59:188–193.
2012. View Article : Google Scholar
|
|
6
|
Park HS, Cho K, Park YJ and Lee T: Chronic
nicotine exposure attenuates proangiogenic activity on human
umbilical vein endothelial cells. J Cardiovasc Pharmacol.
57:287–293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bellien J, Iacob M, Remy-Jouet I, Lucas D,
Monteil C, Gutierrez L, Vendeville C, Dreano Y, Mercier A, Thuillez
C, et al: Epoxyeicosatrienoic acids contribute with altered nitric
oxide and endothelin-1 pathways to conduit artery endothelial
dysfunction in essential hypertension. Circulation. 125:1266–1275.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bellien J, Joannides R, Richard V and
Thuillez C: Modulation of cytochrome-derived epoxyeicosatrienoic
acids pathway: A promising pharmacological approach to prevent
endothelial dysfunction in cardiovascular diseases? Pharmacol Ther.
131:1–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Modena MG, Bonetti L, Coppi F, Bursi F and
Rossi R: Prognostic role of reversible endothelial dysfunction in
hypertensive post-menopausal women. J Am Coll Cardiol. 40:505–510.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Virdis A, Ghiadoni L, Versari D,
Giannarelli C, Salvetti A and Taddei S: Endothelial function
assessment in complicated hypertension. Curr Pharm Des.
14:1761–1770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wallace SM, Yasmin, McEniery CM,
Mäki-Petäjä KM, Booth AD, Cockcroft JR and Wilkinson IB: Isolated
systolic hypertension is characterized by increased aortic
stiffness and endothelial dysfunction. Hypertension. 50:228–233.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Y, Yabluchanskiy A, Lindsey ML and
Chilton RJ: Is isolated systolic hypertension worse than combined
systolic/diastolic hypertension? J Clin Hypertens (Greenwich).
14:808–809. 2012. View Article : Google Scholar
|
|
13
|
Bellien J, Remy-Jouet I, Iacob M, Blot E,
Mercier A, Lucas D, Dreano Y, Gutierrez L, Donnadieu N, Thuillez C,
et al: Impaired role of epoxyeicosatrienoic acids in the regulation
of basal conduit artery diameter during essential hypertension.
Hypertension. 60:1415–1421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim KY, Shin HK, Choi JM and Hong KW:
Inhibition of lipopolysaccharide-induced apoptosis by cilostazol in
human umbilical vein endothelial cells. J Pharmacol Exp Ther.
300:709–715. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ge J, Han Y, Jiang H, Sun B, Chen J, Zhang
S and Du Z; RACTS (Randomized Prospective Antiplatelet Trial of
Cilostazol Versus Ticlopidine in Patients Undergoing Coronary
Stenting) Trial Investigators: RACTS: a prospective randomized
antiplatelet trial of cilostazol versus ticlopidine in patients
undergoing coronary stenting: long-term clinical and angiographic
outcome. J Cardiovasc Pharmacol. 46:162–166. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Azuma M, Houchi H, Mizuta M, Kinoshita M,
Teraoka K and Minakuchi K: Inhibitory action of cilostazol, a
phosphodiesterase III inhibitor, on catecholamine secretion from
cultured bovine adrenal chromaffin cells. J Cardiovasc Pharmacol.
41(Suppl 1): S29–S32. 2003.PubMed/NCBI
|
|
17
|
Chao TH, Tseng SY, Li YH, Liu PY, Cho CL,
Shi GY, Wu HL and Chen JH: A novel vasculo-angiogenic effect of
cilostazol mediated by cross-talk between multiple signalling
pathways including the ERK/p38 MAPK signalling transduction
cascade. Clin Sci (Lond). 123:147–159. 2012. View Article : Google Scholar
|
|
18
|
Park SY, Lee JH, Kim CD, Lee WS, Park WS,
Han J, Kwak YG, Kim KY and Hong KW: Cilostazol suppresses
superoxide production and expression of adhesion molecules in human
endothelial cells via mediation of cAMP-dependent protein
kinase-mediated maxi-K channel activation. J Pharmacol Exp Ther.
317:1238–1245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin HK, Kim YK, Kim KY, Lee JH and Hong
KW: Remnant lipoprotein particles induce apoptosis in endothelial
cells by NAD (P)H oxidase-mediated production of superoxide and
cytokines via lectin-like oxidized low-density lipoprotein
receptor-1 activation: Prevention by cilostazol. Circulation.
109:1022–1028. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lim JH, Woo JS and Shin YW: Cilostazol
protects endothelial cells against lipopolysaccharide-induced
apoptosis through ERK1/2- and P38 MAPK-dependent pathways. Korean J
Intern Med. 24:113–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Franke TF, Kaplan DR and Cantley LC: PI3K:
Downstream AKTion blocks apoptosis. Cell. 88:435–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McGee MA and Abdel-Rahman AA: Enhanced
vascular PI3K/Akt-NOX signaling underlies the peripheral
NMDAR-mediated pressor response in conscious rats. J Cardiovasc
Pharmacol. 63:395–405. 2014. View Article : Google Scholar :
|
|
23
|
Xu MC, Shi HM, Wang H and Gao XF:
Salidroside protects against hydrogen peroxide-induced injury in
HUVECs via the regulation of REDD1 and mTOR activation. Mol Med
Rep. 8:147–153. 2013.PubMed/NCBI
|
|
24
|
Yang HY, Bian YF, Zhang HP, Gao F, Xiao
CS, Liang B, Li J, Zhang NN and Yang ZM: Angiotensin- (1-7)
treatment ameliorates angiotensin II-induced apoptosis of human
umbilical vein endothelial cells. Clin Exp Pharmacol Physiol.
39:1004–1010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang XS, Ren JH, Lu JP and Fan Y:
Atorvastatin protects against angiotensin II-induced injury and
dysfunction in human umbilical vein endothelial cells through
bradykinin 2 receptors. J Cardiovasc Pharmacol. 56:171–176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Q, Zhang M, Ding Y, Wang Q, Zhang W,
Song P and Zou MH: Activation of NAD (P)H oxidase by
tryptophan-derived 3-hydroxykynurenine accelerates endothelial
apoptosis and dysfunction in vivo. Circ Res. 114:480–492. 2014.
View Article : Google Scholar :
|
|
27
|
Urso C and Caimi G: Oxidative stress and
endothelial dysfunction. Minerva Med. 102:59–77. 2011.In Italian.
PubMed/NCBI
|
|
28
|
Xu Y, Ruan S, Xie H and Lin J: Role of
LOX-1 in Ang II-induced oxidative functional damage in renal
tubular epithelial cells. Int J Mol Med. 26:679–690. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee YH, Marquez AP, Mungunsukh O and Day
RM: Hepatocyte growth factor inhibits apoptosis by the profibrotic
factor angiotensin II via extracellular signal-regulated kinase 1/2
in endothelial cells and tissue explants. Mol Biol Cell.
21:4240–4250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Riwanto M, Rohrer L, Roschitzki B, Besler
C, Mocharla P, Mueller M, Perisa D, Heinrich K, Altwegg L, von
Eckardstein A, et al: Altered activation of endothelial anti- and
proapoptotic pathways by high-density lipoprotein from patients
with coronary artery disease: Role of high-density
lipoprotein-proteome remodeling. Circulation. 127:891–904. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
National Research Council: Guide for the
Care and Use of Laboratory Animals. 8th edition. National Academy
Press; Washington, DC: 1996
|
|
32
|
Cassis LA, Marshall DE, Fettinger MJ,
Rosenbluth B and Lodder RA: Mechanisms contributing to angiotensin
II regu-lation of body weight. Am J Physiol. 274:E867–E876.
1998.PubMed/NCBI
|
|
33
|
Su F, Shi M, Yan Z, Ou D, Li J, Lu Z and
Zheng Q: Simvastatin modulates remodeling of Kv4.3 expression in
rat hypertrophied cardiomyocytes. Int J Biol Sci. 8:236–248. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu
C, Viollet B, Yan D and Zou MH: AMPKalpha2 deletion causes aberrant
expression and activation of NAD (P)H oxidase and consequent
endothelial dysfunction in vivo: Role of 26S proteasomes. Circ Res.
106:1117–1128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Giachini FR, Osmond DA, Zhang S, Carneiro
FS, Lima VV, Inscho EW, Webb RC and Tostes RC: Clopidogrel,
independent of vascular P2Y12 receptor, improves the arterial
function in small mesenteric arteries from Ang II-hypertensive
rats. Clin Sci (Lond). 118(7): 463–71. 2010. View Article : Google Scholar
|
|
36
|
Sanchez M, Lodi F, Vera R, Villar IC,
Cogolludo A, Jimenez R, Moreno L, Romero M, Tamargo J,
Perez-Vizcaino F, et al: Quercetin and isorhamnetin prevent
endothelial dysfunction, superoxide production, and overexpression
of p47phox induced by angiotensin II in rat aorta. J Nutr.
137:910–915. 2007.PubMed/NCBI
|
|
37
|
Li R, Wang WQ, Zhang H, Yang X, Fan Q,
Christopher TA, Lopez BL, Tao L, Goldstein BJ, Gao F, et al:
Adiponectin improves endothelial function in hyperlipidemic rats by
reducing oxidative/nitrative stress and differential regulation of
eNOS/iNOS activity. Am J Physiol Endocrinol Metab. 293:E1703–E1708.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gonzalez AA, Green T, Luffman C, Bourgeois
CR, Navar LG and Prieto MC: Renal medullary cyclooxygenase-2 and
(pro)renin receptor expression during angiotensin II-dependent
hypertension. Am J Physiol Renal Physiol. 307(8): F962–70. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pezeshki Z, Eshraghi-Jazi F and
Nematbakhsh M: Vascular response to graded angiotensin II infusion
in offspring subjected to high-salt drinking water during
pregnancy: The effect of blood pressure, heart rate, urine output,
endothelial permeability, and gender. Int J Vasc Med.
2014:8765272014.PubMed/NCBI
|
|
40
|
Xu Z, Lu G and Wu F: Simvastatin
suppresses homocysteine-induced apoptosis in endothelial cells:
Roles of caspase-3, cIAP-1 and cIAP-2. Hypertens Res. 32:375–380.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dimmeler S and Zeiher AM: Endothelial cell
apoptosis in angiogenesis and vessel regression. Circ Res.
87:434–439. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vergeade A, Mulder P, Vendeville C,
Ventura-Clapier R, Thuillez C and Monteil C: Xanthine oxidase
contributes to mitochondrial ROS generation in an experimental
model of cocaine-induced diastolic dysfunction. J Cardiovasc
Pharmacol. 60:538–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dharmashankar K and Widlansky ME: Vascular
endothelial function and hypertension: Insights and directions.
Curr Hypertens Rep. 12:448–455. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schäfer SC, Pellegrin M, Wyss C, Aubert
JF, Nussberger J, Hayoz D, Lehr HA and Mazzolai L: Intravital
microscopy reveals endothelial dysfunction in resistance arterioles
in Angiotensin II-induced hypertension. Hypertens Res. 35:855–861.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Daugherty A, Manning MW and Cassis LA:
Angiotensin II promotes atherosclerotic lesions and aneurysms in
apolipoprotein E-deficient mice. J Clin Invest. 105:1605–1612.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Caplan BA and Schwartz CJ: Increased
endothelial cell turnover in areas of in vivo Evans Blue uptake in
the pig aorta. Atherosclerosis. 17:401–417. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu T, Shen D, Xing S, Chen J, Yu Z, Wang
J, Wu B, Chi H, Zhao H, Liang Z, et al: Attenuation of exogenous
angiotensin II stress-induced damage and apoptosis in human
vascular endothelial cells via microRNA-155 expression. Int J Mol
Med. 31:188–196. 2013.
|
|
48
|
Marampon F, Gravina GL, Scarsella L,
Festuccia C, Lovat F, Ciccarelli C, Zani BM, Polidoro L, Grassi D,
Desideri G, et al: Angiotensin-converting-enzyme inhibition
counteracts angiotensin II-mediated endothelial cell dysfunction by
modulating the p38/SirT1 axis. J Hypertens. 31:1972–1983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nako H, Kataoka K, Koibuchi N, Dong YF,
Toyama K, Yamamoto E, Yasuda O, Ichijo H, Ogawa H and Kim-Mitsuyama
S: Novel mechanism of angiotensin II-induced cardiac injury in
hypertensive rats: The critical role of ASK1 and VEGF. Hypertens
Res. 35:194–200. 2012. View Article : Google Scholar
|
|
50
|
Chen J, Chen W, Zhu M, Zhu Y, Yin H and
Tan Z: Propofol attenuates angiotensin II-induced apoptosis in
human coronary artery endothelial cells. Br J Anaesth. 107:525–532.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kröller-Schön S, Jansen T, Schüler A,
Oelze M, Wenzel P, Hausding M, Kerahrodi JG, Beisele M, Lackner KJ,
Daiber A, et al: Peroxisome proliferator-activated receptor γ,
coactivator 1α deletion induces angiotensin II-associated vascular
dysfunction by increasing mitochondrial oxidative stress and
vascular inflammation. Arterioscler Thromb Vasc Biol. 33:1928–1935.
2013. View Article : Google Scholar
|
|
52
|
Gealekman O, Abassi Z, Rubinstein I,
Winaver J and Binah O: Role of myocardial inducible nitric oxide
synthase in contractile dysfunction and beta-adrenergic
hyporesponsiveness in rats with experimental volume-overload heart
failure. Circulation. 105:236–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen F, Wu JL, Fu GS, Mou Y and Hu SJ:
Chronic treatment with qiliqiangxin ameliorates aortic endothelial
cell dysfunction in diabetic rats. J Cardiovasc Pharmacol Ther.
20:230–40. 2015. View Article : Google Scholar
|
|
54
|
Kossmann S, Hu H, Steven S, Schönfelder T,
Fraccarollo D, Mikhed Y, Brähler M, Knorr M, Brandt M, Karbach SH,
et al: Inflammatory monocytes determine endothelial nitric-oxide
synthase uncoupling and nitro-oxidative stress induced by
angiotensin II. J Biol Chem. 289:27540–27550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Napoli C, Paolisso G, Casamassimi A,
Al-Omran M, Barbieri M, Sommese L, Infante T and Ignarro LJ:
Effects of nitric oxide on cell proliferation: Novel insights. J Am
Coll Cardiol. 62:89–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Laskin DL and Pendino KJ: Macrophages and
inflammatory mediators in tissue injury. Annu Rev Pharmacol
Toxicol. 35:655–677. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang W, Fu F, Tie R, Liang X, Tian F,
Xing W, Li J, Ji L, Xing J, Sun X, et al: Alpha-linolenic acid
intake prevents endothelial dysfunction in high-fat diet-fed
streptozotocin rats and underlying mechanisms. Vasa. 42:421–428.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yu J, Li M, Qu Z, Yan D, Li D and Ruan Q:
SDF-1/CXCR4-mediated migration of transplanted bone marrow stromal
cells toward areas of heart myocardial infarction through
activation of PI3K/Akt. J Cardiovasc Pharmacol. 55:496–505.
2010.PubMed/NCBI
|
|
59
|
Zheng H, Fu G, Dai T and Huang H:
Migration of endothelial progenitor cells mediated by stromal
cell-derived factor-1alpha/CXCR4 via PI3K/Akt/eNOS signal
transduction pathway. J Cardiovasc Pharmacol. 50:274–280. 2007.
View Article : Google Scholar : PubMed/NCBI
|